

Abstract
Long-term use of combination therapy against human immunodeficiency virus type (HIV-1) provides strong selective pressure on the virus, and HIV-1 variants that are resistant to multiple inhibitors have been isolated. HIV-1 variants containing amino acid substitutions within the coding region of HIV-1 reverse transcriptase (RT), such as the 3′-azido-3′-deoxythymidine (AZT)-resistant variant AZT-R (M41L/D67N/K70R/T215Y/K219Q) and a variant containing an insertion in the fingers domain (S69SGR70/T215Y), are resistant to the nucleoside RT inhibitor (NRTI) AZT because of an increase in the level of excision of AZT monophosphate (AZTMP) from the primer. While rare, variants have also been isolated which contain deletions in the RT coding region. One such virus, described by Imamichi et al. (J. Virol 74:10958-10964, 2000; J. Virol. 74:1023-1028, 2000; J. Virol. 75:3988-3992, 2001), contains numerous amino acid substitutions and a deletion of codon 67, which we have designated the Δ67 complex of mutations. We have expressed and purified HIV-1 RT containing these mutations. We compared the polymerase and pyrophosphorolysis (excision) activity of an RT with the Δ67 complex of mutations to wild-type RT and the two other AZT-resistant variants described above. All of the AZT-resistant variants we tested excise AZTMP and 9-[2-(R)-(phosphonomethoxy)propyl]adenine (PMPA [tenofovir]) from the end of a primer more efficiently than wild-type RT. Although the variant RTs excised d4TMP less efficiently than AZTMP and PMPA, they were able to excise d4TMP more efficiently than wild-type RT. HIV-1 RT containing the Δ67 complex of mutations was not able to excise as broad a range of NRTIs as the fingers insertion variant SSGR/T215Y, but it was able to polymerize efficiently with low concentrations of deoxynucleoside triphosphates and seems to be able to excise AZTMP and PMPA at lower ATP concentrations than AZT-R or SSGR/T215Y, suggesting that a virus containing the Δ67 complex of mutations would replicate reasonably well in quiescent cells, even in the presence of AZT.
Drug resistance remains a major problem in the treatment of human immunodeficiency virus type 1 (HIV-1) infections. Nucleoside analog reverse transcriptase inhibitors (NRTIs) are widely used in combination therapy; mutations in the HIV-1 reverse transcriptase (RT) reduce the effectiveness of these inhibitors. Resistant RT variants have an increased ability to discriminate between the normal deoxynucleoside triphosphate (dNTP) and the NRTI. This discrimination can occur at the level of incorporation; i.e., the RT variant does not incorporate the NRTI as efficiently as the wild-type HIV-1 RT. Alternatively, the discrimination can occur after the incorporation of the NRTI by increasing the removal (excision) of the NRTI blocking the 3′ end of the primer. Excision occurs by pyrophosphorolysis by means of either pyrophosphate itself or a pyrophosphate donor such as ATP. Pyrophosphorolysis is, mechanistically, the reverse of polymerization. During the chemical step of polymerization, the 3′ end of the primer is located in what has been designated as the priming or P site, and the incoming dNTP is bound in the nucleotide binding or N site (3-5). This is the ternary complex (RT plus template-primer [T/P] plus dNTP). For excision to be able to remove an NRTI blocking the 3′ end of the primer, the end of the primer must be the N site in a binary complex (RT plus T/P). A pyrophosphate donor, PPi or ATP, reacts with the NRTI at the 3′ end of the primer, yielding an unblocked primer and an NRTI triphosphate or a dinucleoside tetraphosphate, depending on which pyrophosphate donor is used (24). In vitro, PPi is an effective pyrophosphate donor, leading to the removal of 3′-azido-3′-deoxythymidine 5′-monophosphate (AZTMP) from the end of a primer (1, 3-5, 21, 24-28, 30). However, the RT variants that are associated with resistance to 3′-azido-3′-deoxythymidine (AZT) usually excise AZTMP less efficiently with PPi than wild-type RT. There is enhanced excision by the AZT-resistant RT variants, relative to wild-type RT, when ATP is the pyrophosphate donor, suggesting that ATP is the main pyrophosphate donor in vivo (3-5, 21, 24-27, 29, 30), although it has been proposed that PPi-based excision may play a role in vivo (30). AZT resistance has been strongly associated with the presence of the mutations T215(Y/F) (see reference 34 for a review). We proposed that aromatic interactions of tyrosine or phenylalanine side chains at the 215 position with the adenine ring of the ATP contribute to improved ATP binding and, as a consequence, to improved ATP-based pyrophosphorolysis of NRTI-terminated T/Ps. During pyrophosphorolysis, the phosphates of the ATP are oriented toward the polymerase active site, with the β and γ phosphates of ATP occupying positions similar to those of the γ and β phosphates of a dNTP bound at the N site (3-6, 13, 25, 33). The T215(Y/F) mutations are usually associated with a suite of mutations that modulate the excision reaction. An RT variant containing classic AZT resistance mutations (M41L, D67N, K70R, T215Y, K219Q) is able to excise AZTMP more efficiently than an RT carrying T215Y alone when ATP is the pyrophosphate donor but shows only a minimal increase in the excision of ddTMP or ddAMP (3, 24, 25). There are resistant variants that have T215Y and, in some cases, additional AZT resistance mutations associated with amino acid insertions in the fingers subdomain (for a review, see reference 34). One of these RT variants, S69SGR70/T215Y, was analyzed previously. The fingers insertion appears to destabilize the ternary complex, leading to enhanced excision not only of AZTMP but also of a number of other NRTIs (4). Other investigators have reported similar results with RT variants containing related mutations (21, 27). There are other mutations that lead to resistance to multiple NRTIs (34). Some of these RT variants include an unusual and infrequent deletion of codon 67 (Δ67) (10, 11, 14-18, 22, 31). Amino acid 67 is located in the fingers subdomain of HIV-1 RT. Imamichi et al. (14-16) isolated virus from a patient undergoing long-term antiviral therapy which contained the mutation Δ67 and a set of mutations (M41L/Δ67/T69G/K70R/L74I/K103N/T215Y/K219Q), hereafter designated the Δ67 complex of mutations. The T69G and Δ67 mutations, as well as amino acid substitutions associated with AZT resistance, first emerged under AZT-didanosine (ddI) therapy. After delavirdine was combined with the AZT-ddI therapy, the mutations L74I and K103N appeared. The locations of the amino acid substitutions are shown in Fig. 1. The virus containing the Δ67 complex of mutations is highly resistant to nevirapine and AZT but is sensitive to ddI, despite the presence of the L74I mutation (14-16). The interplay among the various amino acid substitutions appears to be intricate. The presence of the Δ67 mutation by itself had little effect on AZT sensitivity, but the addition of T69G and other mutations associated with AZT resistance [K70R and T215(F/Y), and K219Q] caused a significant increase in AZT resistance (14-16). The addition of other mutations (K103N and L74I) further increased resistance to AZT in vivo. Virus containing the T69G mutation without the Δ67 mutation were resistant to ddI but at the expense of viral replication capacity. The subsequent development of the Δ67 mutation improved the replication of the virus and also increased the level of AZT resistance. However, as described above, resistance to ddI was lost (14-16). We have isolated and characterized the properties of a purified HIV-1 RT containing the Δ67 complex of mutations and compared it to two AZT-resistance variants, AZT-R and SSGR/T215Y, and wild-type RT. Although the RT carrying the Δ67 complex of mutations is not as efficient as SSGR/T215Y in its ability to excise a broad spectrum of NRTIs, the Δ67 complex of mutations is particularly efficient in its ability to excise AZTMP at low ATP concentrations.
FIG. 1.

Model of the HIV-1 RT polymerase active site. The three aspartic acid residues (D110, D185, and D186) located at the active site are shown in purple. The subdomains of HIV-1 near the active site are color coded (palm, red; thumb, green; fingers, blue). A T/P is shown bound to the RT with the 3′ terminus of the primer in the P site. A dNTP is modeled in the N site, with the β and γ phosphates labeled. The nonnucleoside RT inhibitor (NNRTI) binding site is shown; the putative binding site for ATP located near the T215Y residue. The residues described in the text are shown in gray and yellow space-filling models; the amino acid residues in the figure are those normally found in wild-type HIV-1 RT. The labels indicate the amino acid substitutions found in the Δ67 complex.
MATERIALS AND METHODS
Preparation of HIV-1 RT.
The open reading frames encoding wild-type HIV-1 RT and each of the RT mutants were cloned into a plasmid containing the HIV-1 protease (PR) open reading frame as previously described (3). The plasmid is based on the expression vector pT5m and was introduced into the Escherichia coli strain BL21(DE3)pLysE. After induction with isopropyl β-d-thiogalactopyranoside, the plasmid expresses both the p66 form of HIV-1 RT (either wild-type or a mutant) and HIV-1 PR. Approximately 50% of the overexpressed p66 RT is converted to the p51 form by HIV-1 PR, and p66/p51 heterodimers accumulate in E. coli. The p66/p51 heterodimers were purified by metal chelate chromatography (3).
Polymerase assays.
The polymerase assays were done as previously described (3). For each sample, 0.25 μg of single-stranded M13mp18 DNA (New England Biolabs) was hybridized to 0.5 μl of a 1.0-optical density/ml solution of the −47 sequencing primer (New England Biolabs). T/P was suspended in 100.0 μl of 25 mM Tris (pH 8.0); 75 mM KCl; 8.0 mM MgCl2; 100.0 μg of bovine serum albumin (BSA) per ml; 10.0 mM CHAPS (3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate); 2.0 mM dithiothreitol (DTT); 10.0 μM (each) dATP, dTTP, and dGTP; 5.0 μM dCTP; 2.0 μM [α-32P]dCTP; and the indicated concentration of inhibitor. Extension was initiated by the addition of 1.0 μg of wild-type RT or RT variant. The mixture was incubated for 30 min at 37°, and then the reaction was halted by the addition of 3 ml of ice-cold trichloroacetic acid. Precipitated DNA was collected by suction filtration through Whatman GF/C glass filters. The amount of incorporated radioactivity was determined by liquid scintillation counting. Wild-type RT was considered to have 100% activity; the activity of the RT variants was normalized to wild-type RT. The inhibition of polymerization by AZT triphosphate (AZTTP) or PPi was done in a similar manner, except that various concentrations of AZTTP (0.0, 0.1, 0.2, 0.5, and 1.0 μM) or NaPPi (0.0, 0.2, 0.5, and 1.0 mM) were added to the reaction. The activity of the enzyme was considered to be 100% in the absence of the AZTTP or PPi; the decreased level of incorporated radioactivity was normalized to this value.
Processivity assay.
The processivity assay has been previously described (3). In brief, for each sample to be assayed, a 0.5-μl/ml solution of culture at an optical density of 1.0 of the −47 sequencing primer (New England Biolabs) was 5′ end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. After purification, the labeled primer was annealed to single-stranded M13mp18 DNA (1.0 μl of a DNA stock at a concentration of 0.25 μg/μl for each sample to be assayed) by heating and slow cooling. The labeled T/P was resuspended in 25 mM Tris (pH 8.0), 75 mM KCl, 8.0 mM MgCl2, 100.0 μg of BSA per ml, 10.0 mM CHAPS, and 2.0 mM DTT. One microgram of wild-type RT or an RT variant was added to each tube and allowed to bind to the labeled T/P for 2 min. Extension was initiated by the addition of dNTPs, to a final concentration of 10.0 μM each, plus 0.5 U of poly(rC) · oligo(dG) per ml, and the reactions were allowed to proceed at 370 for 10 m. The poly(rC) · oligo(dG) prevents RT from rebinding to the labeled primer by binding the RT after it disassociates from the labeled T/P.
Low dNTP extension assay.
The low dNTP extension assay has been described previously (3). Briefly, −47 sequencing primer (New England Biolabs) was 5′ end-labeled with [γ-32P]ATP and T4 polynucleotide kinase. After purification, the labeled primer was annealed to single-stranded M13mp18 DNA (New England Biolabs) by heating and slow cooling. For each sample, 1.0 μg of wild-type RT or RT variant was added to the labeled T/P in 25 mM Tris-Cl (pH 8.0), 75 mM KCl, 8.0 mM MgCl2, 2.0 mM DTT, 100 μg of BSA per ml, and 10.0 mM CHAPS. The reaction mixture was supplemented with 0.1, 0.2, 0.5, or 1.0 μM (each) dATP, dCTP, dGTP, and dTTP. The reactions were allowed to proceed at 370 for 15 min and were then halted by phenol-chloroform extraction. The samples were precipitated by the addition of 1 volume of isopropanol and fractionated by electrophoresis on a 6.0% polyacrylamide gel, and the gel was autoradiographed.
Pyrophosphorolysis.
ATP- and NaPPi-dependent pyrophosphorolysis analysis was done as previously described (3). A synthetic DNA oligonucleotide (5′-GTCACTGTTCGAGCACCA-3′; Biosource, Camarillo, Calif.) was 5′ end-labeled and then annealed to the template (5′-AATCAGTGTAGACAATCCCTAGCANTGGTGCTCGAACAGTGAC-3′). The N residue was varied, depending on which NRTI was being examined. The 3′ end of the primer was then blocked by the addition of AZTMP (or other NRTI monophosphate). After purification of the blocked T/P, the T/P was incubated with HIV-1 RT or the RT variant in the presence of various concentrations (described in the figure legends) of either NaPPi or ATP for 15 min. The reactions were halted by the addition of EDTA: the salts and nucleosides were removed by passage through a CentriSep column (Princeton Separations), and the T/P was precipitated by the addition of isopropyl alcohol. The products were fractionated on a 15% polyacrylamide sequencing gel. The total amount of T/P (blocked and unextended plus deblocked and extended) and the amount of full-length product (deblocked and extended to the end of the template) were determined by using a PhosphorImager.
RESULTS
Polymerase assays.
Wild-type HIV-1 RT, the RT variants AZT-R (M41L/D67N/K70R/T215Y/K219Q), the Δ67 complex of mutations (M41L/Δ67/T69G/K70R/L74I/K103N/T215Y/K219Q), and SSGR/T215Y (T69S and K70R mutations with a serine and a glycine inserted between them, combined with T215Y) were analyzed for their polymerase activity on an M13mp18 template annealed to a sequencing primer as described in Materials and Methods. The positions of the relevant amino acids in HIV-1 RT are shown in Fig. 1. AZT-R had 72% of the polymerase activity of wild-type RT, while SSGR/T215Y had 75% of the polymerase activity of wild-type RT. This is similar to values that have been previously reported (4). With this substrate, the Δ67 complex of mutations had 115% of the activity of wild-type RT. We then analyzed the polymerase activity of the RT variants with a processivity assay, which also uses single-stranded M13mp18, this time annealed to a 5′ end-labeled primer. However, in the processivity assay, the addition of an unlabeled poly(rC) · oligo(dG) “trap” limits the RT to one cycle of polymerization, since once the RT disassociates from the labeled substrate, it binds to the poly(rC) · oligo(dG), which is present in excess. As shown in Fig. 2, wild-type HIV-1 RT has greater processivity than any of the RT variants tested. The strong band at approximately 350 nucleotides probably represents the RT pausing at a stem-loop structure in the M13mp18 template that is necessary for M13 replication. When RT is paused at secondary structures, it is more likely to disassociate from the T/P (7). Of the RT variants examined, AZT-R had the best processivity, followed by SSGR/T215Y and then the Δ67 complex of mutations (Fig. 2). This is the reverse of the polymerase activity described above, where the Δ67 complex of mutations had the most activity, while AZT-R had the least. The same T/P was used in both cases. The difference lies in the fact that the first assay allows multiple rounds of disassociation and rebinding of the RT to the substrate while the processivity assay measures one cycle of polymerization. Processivity depends not only on the polymerase activity of the RT, but also on how well the RT binds the T/P. These data suggest that AZT-R has a slower rate of polymerization than does the Δ67 complex of mutations but that it does not disassociate from the T/P as readily as the Δ67 complex. Gel shift assays also indicate that the Δ67 complex does not bind a T/P as strongly as wild-type HIV-1 RT (data not shown).
FIG. 2.
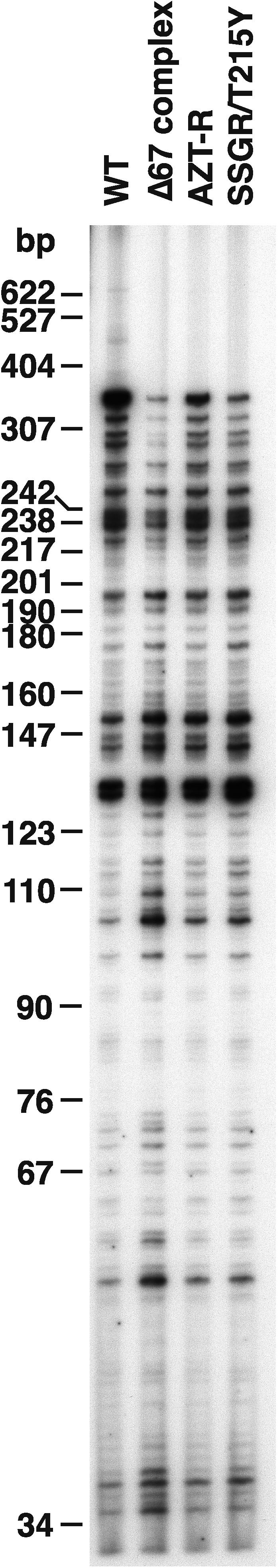
Processivity of the wild-type and mutant RTs. As described in Materials and Methods, a 5′ end-labeled primer was annealed to single-stranded M13mp18 DNA and then extended with wild-type HIV-1 RT or an RT variant in the presence of a 10.0 μM concentration of each dNTP and unlabeled poly(rC) · oligo(dG). The cold trap limits extension to one round of polymerization. The location of the size marker bands is shown on the left.
We also measured the ability of the RT variants to extend a primer annealed to single-stranded M13mp18 DNA in the presence of low concentrations of dNTPs (Fig. 3). Normally, the polymerase assays are done in the presence of a 10.0 μM concentration of each dNTP. For all dNTP concentrations used, the RT variant SSGR/T215Y had the greatest difficulty extending the primer, followed by AZT-R. At the lowest dNTP concentrations (a 0.1 or 0.2 μM concentration of each dNTP), the Δ67 complex was somewhat better at extending the primer than was wild-type RT. At a 0.5 or 1.0 μM concentration of each dNTP, the wild-type HIV-1 RT had a greater ability to extend the primer than the Δ67 complex (Fig. 3).
FIG. 3.

Extension of the primer in the presence of low dNTP concentrations. A 5′ end-labeled primer was annealed to single-stranded M13mp18 DNA and extended with wild-type RT or an RT variant in the presence of 0.1, 0.2, 0.5, and 1.0 μM concentrations of each dNTP. The location of the size marker bands is shown on the left.
Excision and extension of primers blocked with nucleoside analogs.
NRTI resistance mutations can either decrease the level of NRTI that is incorporated or increase the level of NRTI excision. To determine if any of the RT variants had a reduced ability to incorporate AZTTP, inhibition assays were done as previously described (3). As described in the Materials and Methods, an M13mp18 DNA template was annealed to a primer and extended with normal dNTPs, [α-32P]dCTP as a marker, variable amounts of AZTTP, and either wild-type RT or an RT variant. The amount of [α-32P]dCTP incorporated in the presence of the various concentrations of AZTTP was measured to determine the degree to which the various RTs were inhibited by AZTTP. The RT variants were inhibited to the same extent as wild-type RT, indicating that these variants do not cause AZT resistance by enhancing discrimination between dTTP and AZTTP during the incorporation step (data not shown). This suggests that resistance to AZT engendered by the Δ67 complex is probably due to an enhanced ability to excise AZTMP from the primer, similar to what has been described for AZT-R and SSGR/T215Y.
Excision requires that the NRTI at the 3′ end of the primer be located in the N site and that a pyrophosphate donor be present. We examined the abilities of wild-type RT and the RT variants to excise AZTMP by using ATP as the pyrophosphate donor in the presence of high levels of dNTPs. The high concentrations of dNTPs favor the formation of the ternary complex, which decreases the level of excision (see Fig. 7). The excision-extension assay measures both the ability of the enzyme to bind and use the pyrophosphate donor to excise the blocking NRTI and the ability to extend the unblocked primer to the end of the template strand. As previously described (4), the AZT-R and SSGR/T215Y RT variants were more efficient at unblocking the AZTMP-blocked primer than was wild-type RT when ATP was used as the pyrophosphate donor (Fig. 4A). The Δ67 complex was as efficient at excising the AZTMP as AZT-R and SSGR/T215Y when 10.0 mM ATP was present in the reaction. Interestingly, the Δ67 complex had a relatively high efficiency of excision at the lower ATP concentrations (2.0 and 5.0 mM) where AZT-R and SSGR/T215Y were less efficient. To determine if the Δ67 complex was able to excise AZTMP at even lower ATP concentrations, the experiments were repeated with ATP concentrations at 0.2, 0.5, and 1.0 mM. Since there is evidence to suggest that when dNTPs are present at high concentrations they can act as pyrophosphate donors (unpublished observations), the concentrations of dNTPs were also decreased. AZT-R and SSGR/T215Y were only slightly more efficient at removing the AZTMP than was wild-type RT at low ATP concentrations (Fig. 4B). The Δ67 complex was relatively efficient at excising AZTMP at ATP concentrations as low as 0.5 mM.
FIG. 7.

Diagram of the equilibria affecting the concentration of the excision complex (RT with the T/P bound in the N site and an appropriately bound pyrophosphate donor). Factors that can affect the equilibria, such as dNTPs favoring the formation of the ternary complex or the azido group of AZT favoring the formation of the binary complex, are shown near the appropriate arrow. Some factors, such as the presence of M184V or the base composition near the 3′ end of the primer, are known to affect the overall efficiency of excision (4), but it is not clear which of the equilibria are affected. These are listed below the diagram.
FIG. 4.

Excision and extension at high and low ATP concentrations. As described in Materials and Methods, a 5′ end-labeled primer was annealed to a template. The 3′ end of the primer was then blocked by the addition of the appropriate NRTI. An excision-extension assay using ATP as the pyrophosphate donor was done as described in Materials and Methods. All experiments were done at least twice at different times. When experiments were repeated with different batches of enzymes, the results were reproducible. (A) Excision and extension of an AZTMP-blocked primer at high levels of dNTPs (a 100.0 μM concentration of each dNTP) to favor the ternary complex and various concentrations of ATP (2.0, 5.0, and 10.0 mM). (B) Excision and extension of AZTMP at low levels of dNTPs (a 10.0 μM concentration of each dNTP) and ATP (0.2, 0.5, and 1.0 mM). (C) Excision and extension of d4TMP at high levels of dNTPs and various concentrations of ATP (2.0, 5.0, and 10.0 mM). (D) Excision and extension of d4TMP at moderate levels of dNTPs (a 25.0 μM concentration of each dNTP) and various concentrations of ATP.
There is evidence to suggest that AZT resistance mutations cause low-level resistance to stavudine (d4T), which can be important clinically (19, 23, 32). We used the excision-extension assay to examine the ability of wild-type HIV-1 RT and the RT variants to excise d4TMP. In the presence of high levels of dNTPs, a condition which favors the formation of the stable ternary complex and disfavors excision, wild-type HIV-1 RT was not able to excise the d4TMP blocking group from the primer (Fig. 4C). The Δ67 complex and AZT-R were able to excise d4TMP more efficiently than wild-type RT; SSGR/T215Y excised d4TMP more efficiently than the Δ67 complex or AZT-R RT (Fig. 4C). The difference between wild-type RT and either AZT-R or the Δ67 complex does not appear to be large, but in our assay these RT variants excise d4TMP three- to fivefold more efficiently than wild-type RT. However, even a small change (1.4-fold) in d4T susceptibility is associated with failure to achieve a significant reduction in the viral load in patients (35), suggesting that AZT-R and the Δ67 complex would give clinical resistance to d4T. The difference is more striking when the amount of the dNTPs present in the reaction is decreased to a 25.0 μM concentration of each, decreasing the amount of ternary complex and favoring excision. At this dNTP concentration, all the RT variants excise d4TMP more efficiently than wild-type RT (Fig. 4D).
We tested other nucleoside analogs in the excision-extension assay to determine whether the Δ67 complex behaved like AZT-R or was similar to SSGR/T215Y, which can excise a broad range of nucleoside analogs. When the primer was blocked with ddAMP, SSGR/T215Y was able to excise the analog, although with lower efficiency than AZTMP or d4TMP; AZT-R and the Δ67 complex were somewhat more efficient at the excision of ddAMP than wild-type RT (Fig. 5A). Similar results were obtained when ddTTP was used to block the primer (Fig. 5B). It should be noted that SSGR/T215Y was able to excise ddTMP from the primer more efficiently than ddAMP (approximately a threefold difference), which is similar to what has been previously reported (4).
FIG. 5.



Excision and extension with ddAMP-, ddTMP-, and PMPA-blocked primers. Excision-extension assays were performed with high levels of each dNTP (a 100.0 μM concentration of each dNTP) and various concentrations of ATP (2.0, 5.0, and 10.0 mM). All experiments were done at least twice at different times. Some experiments were done with different batches of enzymes, and the results are reproducible. Excision and extension of a ddAMP-blocked primer (A), a ddTMP-blocked primer (B), and a PMPA-blocked primer (C).
The last NRTI examined was 9-[2-(R)-(phosphonomethoxy)propyl]adenine (PMPA [tenofovir]). As shown in Fig. 5C, all of the RT variants tested were able to efficiently excise PMPA from the primer, with SSGR/T215Y being the most efficient. The Δ67 complex was able to excise PMPA efficiently even at the lowest ATP concentration (2.0 mM); this is similar to the result obtained when AZTMP was the blocking group.
Wild-type RT and the RT variants were also tested for their ability to use pyrophosphate as the donor. As described in the Discussion, excision by means of PPi is not significantly influenced by the presence of the T215Y mutation and, because PPi-dependent pyrophosphorolysis is the reverse reaction of normal polymerization, PPi-dependent excision is more dependent than ATP-dependent excision on the overall polymerase activity of the enzyme and on how tightly PPi is bound at the active site. When the primer was blocked by an AZTMP and PPi was the pyrophosphate donor, the Δ67 complex was significantly more efficient at excision and extension of the AZTMP-blocked primer than the wild-type enzyme or SSGR/T215Y, which are similar in their excision-extension efficiency (Fig. 6A). AZT-R was the enzyme least able to excise the blocking group and extend the primer. When ddTMP and d4TMP were the blocking NRTIs on the primer and PPi was the pyrophosphate donor, the Δ67 complex was again the most efficient at excision and extension, while SSGR/T215Y was somewhat better than wild-type HIV-1 RT; AZT-R was the least efficient at PPi-dependent excision (Fig. 6B and C). All of the RT variants and wild-type RT were very efficient at excising PMPA from the primer; the Δ67 complex was able to efficiently excise PMPA even at the lowest concentration of NaPPi (Fig. 6D). In contrast, none of the enzymes were efficient at excising ddAMP from the primer, although the Δ67 complex was the most active (Fig. 6E).
FIG. 6.





Excision-extension assays using NaPPi. The excision-extension assays were performed with NRTI-blocked primers in the presence of high levels of each dNTP (a 100.0 μM concentration of each) and various concentrations of NaPPi (50.0, 100.0, and 200.0 μM). All experiments were done at least twice at different times. Similar results were obtained when experiments were repeated with different batches of enzyme. Excision and extension of an AZTMP-blocked primer (A), a ddTMP-blocked primer (B), a d4TMP-blocked primer (C), a ddAMP-blocked primer (D), and a PMPA-blocked primer (E).
We further examined the ability of the various RTs to bind PPi by testing the ability of PPi to inhibit with the normal polymerization reaction (Materials and Methods). The ability of wild-type RT or the RT variant SSGR/T215Y to extend a primer was unaffected by PPi concentrations up to 1.0 mM. The polymerization by AZT-R was somewhat affected by the presence of PPi, but the Δ67 complex was affected to the greatest extent of the RTs tested (data not shown).
DISCUSSION
Excision of an NRTI by pyrophosphorolysis is a relatively simple reaction chemically, but the efficiency of the reaction is influenced by a number of factors (Fig. 7). Ultimately, the efficiency of the excision reaction depends on the amount of the relevant enzyme-substrate complex, i.e., RT with the T/P bound in the N site and an appropriately bound pyrophosphate donor (Fig. 7). Factors that influence the concentration of this complex affect the efficiency of excision. For example, binding a dNTP at the N site (the ternary complex) blocks the excision reaction because the T/P does not have access to the N site (3-5, 24, 25, 27, 33, 36), while the presence of the azido group on AZT interferes with dNTP binding, destabilizing the ternary complex and favoring excision of AZTMP from the primer (3-5, 33, 36). Nucleoside analog-resistant RT variants with insertions in the fingers subdomain, such as SSGR/T215Y, also destabilize the ternary complex, allowing the efficient excision of a wider range of NRTIs (4, 26). The presence of the T215(Y/F) mutation is important for the increased efficiency of ATP binding. Previous work has modeled the adenine ring of ATP interacting with the aromatic ring of the tyrosine or phenylalanine (3-6, 25, 33). This interaction will tether the adenine ring of ATP to HIV-1 RT, but the β and γ phosphates of the ATP must be able to reach the polymerase active site to excise AZTMP, or other nucleoside analogs, from the end of the primer at the N site.
We have characterized the properties of a purified HIV-1 RT containing the Δ67 complex of mutations and compared these properties to wild-type RT and two classes of previously characterized AZT resistance mutations (AZT-R and SSGR/T215Y). While the Δ67 complex is slightly more active (115%) than wild-type RT (100%), AZT-R (72%), or SSGR/T215Y (75%) in reactions that involve multiple rounds of polymerization, the Δ67 complex is less processive than wild-type RT or the other RT variants. This suggests that the Δ67 complex does not remain bound to the T/P as long as the other RTs. In polymerase reactions with low concentrations of dNTPs (0.1 and 0.2 μM concentrations of each dNTP), the Δ67 complex is slightly more efficient at polymerization than wild-type RT and significantly more efficient than AZT-R and SSGR/T215Y. It is only at somewhat higher dNTP concentrations (0.5 and 1.0 μM concentrations of each dNTP) where wild-type RT is the most efficient enzyme. AZT-R and SSGR/T215Y are the least efficient at all dNTP concentrations tested. The exact mechanism by which the Δ67 complex of mutations enhances the efficiency of the enzyme at very low dNTP concentrations is not known, but it is possible that it is related to the ability to carry out excision at relatively low concentrations of ATP or PPi (discussed below).
The Δ67 complex of mutations was similar to AZT-R in the range of nucleoside analogs that could be excised by using ATP as the pyrophosphate donor. AZTMP was efficiently excised by both mutants, but ddTMP and ddAMP were not. In contrast, SSGR/T215Y, which destabilizes the ternary complex, efficiently excises all three nucleoside analogs. The Δ67 complex was distinctly different from AZT-R and SSGR/T215Y in that it was able to excise AZTMP efficiently even at low ATP concentrations. A similar effect was seen when PMPA was tested (discussed below). Efficient excision with ATP actually involves two processes: how well ATP binds to the RT variant and how readily the ATP is used in the excision reaction. As previously proposed, enhanced binding of ATP involves the T215(Y/F) mutations (3-5). The addition of the mutations M41L, D67N, K70R, and K219Q to T215Y further enhanced the excision of AZTMP compared to T215Y alone (3). Both AZT-R and the Δ67 complex contain M41L, K70R, T215Y, and K219Q. However, the Δ67 complex also contains additional mutations (T69G, L74I, and K103N) and a deletion of codon 67 rather than D67N. The Δ67 complex is, in the context of a virus, more resistant to AZT than viruses with the classical AZT resistance mutations (14). It is likely that some of these other amino acid changes in the Δ67 complex further enhance the binding of ATP and/or enhance the use of ATP in the excision reaction; however, at this time, it is not clear which amino acid substitution(s) is responsible for these effects.
The Δ67 complex was also tested for its ability to excise PMPA from the primer. PMPA differs from the other NRTIs that we tested in that it is an acyclic nucleoside lacking both the 2′ and 3′ carbon groups. While the Δ67 complex can efficiently excise PMPA at lower ATP concentrations than AZT-R or SSGR/T215Y, all of the AZT-resistant RT variants we tested can excise PMPA. This contrasts with the results of Naeger et al. (28) but agrees with recent PMPA excision data obtained with the use of more highly resistant RT variants (38). While some clinical data suggest that PMPA does not readily select RT variants with high levels of resistance (20), other data suggest that, in viral replication assays, high-level resistance (>10-fold) to PMPA is associated with AZT-resistance mutations and with insertions in the fingers subdomain (12). This agrees with the data presented here. Since all of the AZT-resistant mutants can efficiently excise PMPA, it is possible that, like AZTMP, the presence of PMPA at the end of the primer may interfere with the formation of a stable ternary complex. The crystal structure of a T/P bound to HIV-1 RT with PMPA at the end of the primer in the P site indicates that the PMPA nucleotide appears to be in at least two conformations. These multiple conformations are predicted to result from the lack of a complete sugar ring (37). These multiple conformations could affect the equilibrium between the N site form, which favors excision, and the P site form, which does not. It could also interfere with the binding of the incoming dNTP, which would decrease the level of the ternary complex.
The planar sugar ring of d4T (d4T has a double bond between C′2 and C′3) appears to make it a relatively poor substrate for excision compared to AZT and PMPA. However, the level of resistance needed to have a clinical effect against d4T is small (1.4-fold) (32), and it has been reported that mutations causing resistance to AZT can cause cross-resistance to d4T (12, 19, 23, 32, 35). At high dNTP concentrations, which favor the formation of the ternary complex and inhibit the excision reaction, SSGR/T215Y excises d4TMP to a much greater extent than wild-type RT. AZT-R and the Δ67 complex excise d4TMP less well than SSGR/T215Y but are more efficient than wild-type RT. In our assay, the difference between excision by wild-type RT and excision by AZT-R or the Δ67 complex at 5.0 mM ATP is approximately three- to fivefold, while SSGR/T215Y is at least 20-fold more efficient. Since even a small increase in susceptibility to d4T (1.4-fold difference) can lead to clinical failure (35), the excision data support the data showing that these RT variants cause clinical resistance to d4T (12, 19, 23, 32, 35). The difference is even more pronounced if the level of dNTPs is decreased.
Since the Δ67 complex can use dNTPs and ATP at much lower levels than the other RT variants, it is possible that the Δ67 complex could also bind PPi more tightly. When PPi is the pyrophosphate donor, excision is the reverse of polymerization, which is not the case when ATP is the donor. For all NRTIs that were tested with PPi as the donor, AZT-R was significantly less efficient at excision than wild-type RT. This reflects the difference in polymerase activity measured for AZT-R compared to wild-type RT (72 versus 100%) and is similar to data that has previously been reported (3-5). SSGR/T215Y, which is slightly more active than AZT-R, is close to wild-type RT in excision efficiency with PPi. We had previously proposed that PPi was unlikely to be the pyrophosphate donor in vivo because the AZT-resistant RTs had no advantage relative to wild-type RT in the efficiency of PPi-dependent pyrophosphorolysis; this is supported by the data shown here. However, the Δ67 complex, which is only slightly more active than wild-type RT (115% of the activity of wild-type), had significantly more PPi-dependent excision than wild-type RT with almost all the NRTIs we tested (the exception is ddATP). This increase is larger than can be explained by a simple increase in polymerase activity. This may indicate that the Δ67 complex does not release the PPi as readily as the other RT variants we tested and/or that there is an effect on the N site-P site translocation equilibrium. This is the first AZT-resistant RT variant we have analyzed that shows enhanced PPi-dependent excision and extension compared to wild-type RT. Mutations that cause resistance to foscarnet, an analog of PPi, decrease unblocking of the primer by pyrophosphorolysis. The proposed mechanism is that the foscarnet-resistant RT variants have a decreased ability to bind PPi, foscarnet, and ATP or that the position of the substrates may be altered when they are bound (27). The opposite results are seen with the Δ67 complex. The polymerase activity of the Δ67 complex is inhibited by concentrations of PPi that do not affect wild-type RT, again suggesting that the Δ67 complex binds PPi better than wild-type RT.
In summary, the Δ67 complex is similar to AZT-R in the range of NRTIs that can be effectively excised when ATP is the pyrophosphate donor and does not have the very broad spectrum of excision seen for SSGR/T215Y. Based on the processivity and gel shift assays, the Δ67 complex remains bound to the T/P less well than the other RT variants and wild-type RT. However, the Δ67 complex can efficiently use dNTPs at low concentrations and seems to use ATP (and PPi) more efficiently when excising AZTMP and PMPA. In quiescent peripheral blood lymphocytes, the level of dNTPs is low, and viral DNA synthesis is slow and inefficient. When peripheral blood lymphocytes are activated, the levels of dNTPs are greatly increased, and the production of full-length viral DNA is stimulated (9). It has also been shown that resting T lymphocytes have twofold less purines (ATP and GTP) and up to eightfold less pyrimidines (UTP and CTP) compared to phytohemagglutinin-stimulated T lymphocytes (2, 8). The ability to synthesize ribonucleotides may also be impaired in HIV-1-infected T lymphocytes (2). A virus containing the Δ67 complex of mutations may be better able than the wild-type virus to replicate in cells where the levels of dNTPs are low. Furthermore, the Δ67 complex, with its ability to excise AZTMP from the primer at low ATP concentrations, may allow a virus containing these mutations to replicate in quiescent cells even when AZTTP is present. This also brings up the possibility that, in contrast to the RT variants that do not show enhanced excision of AZTMP or PMPA with PPi, excision with PPi might play a role in resistance to these compounds by viruses carrying the Δ67 complex of mutations.
Acknowledgments
We thank Pat Clark for preparing purified enzymes and Hilda Marusiodis for help in preparing the manuscript. PMPA diphosphate was the kind gift of Gilead Sciences, Inc., and we are grateful to Mike Miller and Kirsten White for helpful discussions.
Research in S.H.H.'s laboratory was supported by the National Cancer Institute and NIGMS.
REFERENCES
- 1.Arion, D., N. Kaushik, S. McCormick, G. Borkow, and M. A. Parniak. 1998. Phenotypic mechanism of HIV-1 resistance to 3′-azido-3′-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry 37:15908-15917. [DOI] [PubMed] [Google Scholar]
- 2.Bofill, M., L. D. Fairbanks, K. Ruckemann, M. Lipman, and H. A. Simmonds. 1995. T-Lymphocytes from AIDS patients are unable to synthesize ribonucleotides de novo in response to mitogenic stimulation. J. Biol. Chem. 270:29690-29697. [PubMed] [Google Scholar]
- 3.Boyer, P. L., S. G. Sarafianos, E. Arnold, and S. H. Hughes. 2001. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol. 75:4832-4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyer, P. L., S. G. Sarafianos, E. Arnold, and S. H. Hughes. 2002. Nucleoside analog resistance caused by insertions in the fingers of human immunodeficiency virus type 1 reverse transcriptase involves ATP-mediated excision. J. Virol. 76:9143-9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyer, P. L., S. G. Sarafianos, E. Arnold, and S. H. Hughes. 2002. The M184V mutation reduces the selective excision of zidovudine 5′-monophosphate (AZTMP) by the reverse transcriptase of human immunodeficiency virus type 1. J. Virol. 76:3248-3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamberlain, P. P., J. Ren, C. E. Nichols, L. Douglas, J. Lennerstrand, B. A. Larder, D. I. Stuart, and D. K. Stammers. 2002. Crystal structures of zidovudine- or lamivudine-resistant human immunodeficiency virus type 1 reverse transcriptases containing mutations at codons 41, 184, and 215. J. Virol. 76:10015-10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coffin, J. M., S. H. Hughes, and H. E. Varmus (ed.). 1997. Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [PubMed]
- 8.Fairbanks, L. D., M. Bofill, K. Ruckemann, and H. A. Simmonds. 1995. Importance of ribonucleotide availability to proliferating T-lymphocytes from healthy humans. J. Biol. Chem. 270:29682-29689. [PubMed] [Google Scholar]
- 9.Gao, W.-Y., A. Cara, R. C. Gallo, and F. Lori. 1993. Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. USA 90:8925-8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giri, J., H. J. Rueda, A. Monticelli, and N. Planes. 2000. Case report of a novel amino acid deletion in codon 67 and T69G substitution in the reverse transcriptase of HIV-1. Antivir. Ther. 5:227-228. [PubMed] [Google Scholar]
- 11.Giri, J., R. H. Jaureguli, A. Monticelli, D. A. Pirola, and N. Planes. 2001. Infrequent deletion in 67 codon in HIV reverse transcriptase in antiretroviral treatment failure. Medicina (B. Aires) 61:193-195. [PubMed] [Google Scholar]
- 12.Harrigan, P. R., M. D. Miller, P. McKenna, Z. L. Brumme, and B. A. Larder. 2002. Phenotypic susceptibilities to tenofovir in a large panel of clinically derived human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 46:1067-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang, H., R. Chopra, G. F. Verdine, and S. C. Harrison. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669-1675. [DOI] [PubMed] [Google Scholar]
- 14.Imamichi, T., S. C. Berg, H. Imamichi, J. C. Lopez, J. A. Metcalf, J. Falloon, and H. C. Lane. 2000. Relative replication fitness of a high-level 3′-azido-3′-deoxythymidine-resistant variant of human immunodeficiency virus type 1 possessing an amino acid deletion at codon 67 and a novel substitution (Thr → Gly) at codon 69. J. Virol. 74:10958-10964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imamichi, T., T. Sinha, H. Imamichi, Y.-M. Zhang, J. A. Metcalf, J. Falloon, and H. C. Lane. 2000. High-level resistance to 3′-azido-3′-deoxythymidine due to a deletion in the reverse transcriptase gene of human immunodeficiency virus type 1. J. Virol. 74:1023-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imamichi, T., M. A. Murphy, H. Imamichi, and H. C. Lane. 2001. Amino acid deletion at codon 67 and Thr-to-Gly change at codon 69 of human immunodeficiency virus type 1 reverse transcriptase confer novel drug resistant profiles. J. Virol. 75:3988-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaliki, V., N. K. Day, E. Dinglasan, M. James-Yarish, R. Hitchcock, D. Skapura, A. Chinta, L. Johnson, A. Andreopoulos, A. Rey, R. A. Good, and S. Haraguchi. 2000. Emergence of HIV-1 variants containing codon insertions and deletions in the β3-β4 hairpin loop domain of reverse transcriptase. Immunol. Letts. 74:173-175. [DOI] [PubMed] [Google Scholar]
- 18.Kim, E.-Y., L. Vrang, B. Öberg, and T. C. Merigan. 2001. Anti-HIV type 1 activity of 3′-fluoro-3′deoxythymidine for several different multi-drug-resistant mutants. AIDS Res. Hum. Retrovir. 17:401-407. [DOI] [PubMed] [Google Scholar]
- 19.Lennerstrand, J., D. K. Stammers, and B. A. Larder. 2001. Biochemical mechanism of human immunodeficiency virus type 1 reverse transcriptase resistance to stavudine. Antimicrob. Agents Chemother. 45:2144-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Margot, N. A., E. Isaacson, I. McGowan, A. Cheng, and M. D. Miller. 2003. Extended treatment with tenofovir disproxil fumarate in treatment-experienced HIV-1-infected patients: genotypic, phenotypic, and rebound analyses. J. Acquir. Immune Defic. Syndr. 33:15-21. [DOI] [PubMed] [Google Scholar]
- 21.Mas, A., M. Parera, C. Briones, V. Soriano, M. A. Martínez, E. Domingo, and L. Menéndez-Arias. 2000. Role of a dipeptide insertion between codons 69 and 70 of HIV-1 reverse transcriptase in the mechanism of AZT resistance. EMBO J. 19:5752-5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masciari, R., L. Cosco, M. C. Diaco, D. N. Della, T. Ferraro, T. Raimondi, B. Ruperti, and E. Santandrea. 2002. HIV-1: a case of RT67 deletion in a multi-treated non responder patient. New Microbiol. 25:83-88. [PubMed] [Google Scholar]
- 23.Maxeiner, H.-G., W. Keulen, R. Schuurman, M. Bijen, L. de Graaf, A. van Wijk, N. Back, M. W. Kline, C. A. B. Boucher, and M. Nijhuis. 2002. Selection of zidovudine resistance mutations and escape of human immunodeficiency virus type 1 from antiretroviral pressure in stavudine-treated pediatric patients. J. Infect. Diseases 185:1070-1076. [DOI] [PubMed] [Google Scholar]
- 24.Meyer, P. R., S. E. Matsuura, A. G. So, and W. A. Scott. 1998. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 95:13471-13476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer, P. R., S. E. Matsuura, A. A. Tolun, I. Pfeifer, A. G. So, J. W. Mellors, and W. A. Scott. 2002. Effects of specific zidovudine resistance mutations and substrate structure on nucleotide-dependent primer unblocking by human immunodeficiency type 1 reverse transcriptase. Antimicrob. Agents Chemother. 46:1540-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meyer, P. R., J. Lennerstrand, S. E. Matsuura, B. A. Larder, and W. A. Scott. 2003. Effects of dipeptide insertions between codons 69 and 70 of human immunodeficiency virus type 1 reverse transcriptase on primer unblocking, deoxynucleoside triphosphate inhibition, and DNA chain elongation. J. Virol. 77:3871-3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer, P. R., S. E. Matsuura, D. Zonarich, R. R. Chopra, E. Pendarvis, H. Z. Bazmi, J. W. Mellors, and W. A. Scott. 2003. Relationship between 3′-azido-3′-deoxythymine resistance and primer unblocking activity in foscarnet-resistant mutants of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 77:6127-6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naeger, L. K., N. A. Margot, and M. D. Miller. 2001. Tenofovir (PMPA) is less susceptible to pyrophosphorolysis and nucleotide-dependent chain-terminator removal than zidovudine or stavudine. Nucleosides Nucleotides Nucleic Acids 20:635-639. [DOI] [PubMed] [Google Scholar]
- 29.Naeger, L. K., N. A. Margot, and M. D. Miller. 2002. ATP-dependent removal of nucleoside reverse transcriptase inhibitors by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother. 46:2179-2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ray, A. S., E. Murakami, A. Basavapathruni, J. A. Vaccaro, D. Ulrich, C. K. Chu, R. F. Schinazi, and K. S. Anderson. 2003. Probing the molecular mechanisms of AZT drug resistance mediated by HIV-1 reverse transcriptase using a transient kinetic analysis. Biochemistry 42:8831-8841. [DOI] [PubMed] [Google Scholar]
- 31.Ross, L., M. Johnson, R. G. Ferris, S. A. Short, L. R. Boone, T. E. Melby, R. Lanier, M. Shaefer, and M. St. Clair. 2000. Deletions in the β3-β4 hairpin loop of HIV-1 reverse transcriptase are observed in HIV-1 isolated from subjects during long-term antiviral therapy. J. Hum. Virol. 3:144-149. [PubMed] [Google Scholar]
- 32.Ross, L., A. Scarsella, S. Raffanti, K. Henry, S. Becker, R. Fisher, Q. Liao, A. Hirani, N. Graham, M. St. Clair, and J. Hernandez. 2001. Thymidine analog and multinucleoside resistance mutations are associated with decreased phenotypic susceptibility to stavudine in HIV type 1 isolated from zidovudine-naive patients experiencing viremia on stavudine-containing regimens. AIDS Res. Hum. Retrovir. 17:1107-1115. [DOI] [PubMed] [Google Scholar]
- 33.Sarafianos, S. G., A. D. Clark, Jr., K. Das, S. Tuske, J. J. Birktoft, P. Ilankumaran, A. R. Ramesha, J. M. Sayer, D. M. Jerina, P. L. Boyer, S. H. Hughes, and E. Arnold. 2002. Structures of HIV-1 reverse transcriptase with pre- and post-translocation AZTMP-terminated DNA. EMBO J. 21:6614-6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schinazi, R. F., B. A. Larder, and J. W. Mellors. 2001. Mutations in retroviral genes associated with drug resistance: 2000-2001 update. Int. Antivir. News 8:65-91. [Google Scholar]
- 35.Shulman, N. S., M. D. Hughes, M. A. Winters, R. W. Schafer, A. R. Zolopa, N. D. S. Hellman, M. Bates, J. M. Whitcomb, and D. A. Katzenstein. 2002. Subtle decreases in stavudine phenotypic susceptibility predict poor virologic response to stavudine monotherapy in zidovudine-experienced patients. J. Acquir. Immune Defic. Syndr. 31:121-127. [DOI] [PubMed] [Google Scholar]
- 36.Tong, W., C.-D. Lu, S. K. Sharma, S. Matsuura, A. G. So, and W. A. Scott. 1997. Nucleotide-induced stable complex formation by HIV-1 reverse transcriptase. Biochemistry 36:5749-5757. [DOI] [PubMed] [Google Scholar]
- 37.Tuske, S., S. G. Sarafiarios, A. D. Clark, Jr., J. Ding, L. K. Naeger, K. L. White, M. D. Miller, C. S. Gibbs, P. L. Boyer, P. Clark, G. Wang, B. L. Gaffney, R. A. Jones, D. M. Jerina, S. H. Hughes, and E. Arnold. 2004. Structures of HIV-1 RT-DNA complexes before and after the incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 11:469-474. [DOI] [PubMed] [Google Scholar]
- 38.White, K. L., J. M. Chen, N. A. Margot, T. Wrin, C. J. Petropoulos, L. K. Naeger, S. Swaminathan, and M. D. Miller. 2004. Molecular mechanisms of resistance to tenofovir by HIV-1 reverse transcriptase containing a diserine insertion after residue 69 and multiple thymidine analog-associated mutations. Antimicrob. Agents Chemother. 48:992-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]