

Abstract
A major cause of tuberculosis (TB) resistance to the aminoglycoside kanamycin (KAN) is the Mycobacterium tuberculosis (Mtb) acetyltransferase Eis. Upregulation of this enzyme is responsible for inactivation of KAN through acetylation of its amino groups. A 123 000-compound high-throughput screen (HTS) yielded several small-molecule Eis inhibitors that share an isothiazole S,S-dioxide heterocyclic core. These were investigated for their structure–activity relationships. Crystal structures of Eis in complex with two potent inhibitors show that these molecules are bound in the conformationally adaptable aminoglycoside binding site of the enzyme, thereby obstructing binding of KAN for acetylation. Importantly, we demonstrate that several Eis inhibitors, when used in combination with KAN against resistant Mtb, efficiently overcome KAN resistance. This approach paves the way toward development of novel combination therapies against aminoglycoside-resistant TB.
Graphical Abstract
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a major global health threat. In 2013, approximately 9.0 million people developed TB, and nearly 1.5 million died from the disease.1 With the emergence of both multidrug-resistant TB [defined as TB with resistance to at least isoniazid and rifampin] (MDR-TB; ~ 10% of new TB cases in 20131) and extensively drug-resistant TB [defined as MDR-TB with added resistance to at least a fluoroquinolone and an injectable drug (i.e., kanamycin (KAN), capreomycin, or amikacin)] (XDR-TB), the need for novel strategies to combat drug resistant TB is urgent.2
Among mechanisms of clinically important transmissible drug resistance is the recently identified inactivation of an MDR-TB therapeutic, the aminoglycoside kanamycin, through its acetylation by an upregulated acetyltransferase, the Eis (enhanced intracellular survival) enzyme.3 The unique multiacetylation mechanism by which Eis inactivates KAN and other aminoglycosides has been thoroughly investigated.4–13
One strategy to combat drug resistance arising as a result of drug-modifying enzymes is to use a combination therapy that includes an antibiotic along with an inhibitor of its associated resistance enzyme.14–18 In Mtb, including MDR-TB, the combination of the β-lactamase inhibitor clavulanate and the β-lactam meropenem was demonstrated to overcome resistance to β-lactam antibiotics.14 With this strategy in mind, efforts have also been made toward identifying inhibitors of aminoglycoside acetyltransferases (AACs) present in nonmycobacteria, with limited preclinical progress, but these are not applicable for the mechanistically and structurally distinct Mtb Eis acetyltransferase. For example, aminoglycoside-acetyl coenzyme A bisubstrate inhibitors were found to potently inhibit AAC(6′) enzymes in vitro, but were not effective in cell-based assays.19–22 Numerous membrane-active cationic peptides that inhibit AAC(6′) enzymes in vitro were previously identified.23 However, these peptides displayed no synergy with aminoglycosides against resistant bacterial strains due to the inability of these membrane binding peptides to permeate into the cytoplasm. Finally, the natural product aranorosin was found to be an inhibitor of the bifunctional AAC(6′)-Ie/APH(2″)-Ia enzyme, and its combination with the aminoglycoside arbekacin was shown to stop the growth of a methicillin-resistant Staphylococcus aureus strain.24
We set out to identify inhibitors of Eis that could be coadministered with KAN in order to prevent inactivation by Eis in Mtb. Here, we report the identification as well as the biochemical and biological characterization of several potent inhibitors of Eis that are able to restore the antibacterial activity of KAN in a KAN-resistant strain, Mtb K204. To clarify in atomic detail the mode of action of these compounds and their structure–activity relationships, we also determined crystal structures of EisC204A in complex with CoA and two of these inhibitors.
RESULTS
High-Throughput Screening Leads to the Identification of a Promising Eis Inhibitor Scaffold
In order to identify Mtb Eis inhibitors, we carried out a HTS of molecular libraries comprising 123 000 structurally diverse small molecules using a previously reported Eis acetylation assay in the miniaturized format (Figure 1a).25 The initial HTS assay was performed using the aminoglycoside neomycin B (NEO) as a substrate, which was selected over KAN to maximize the signal-to-noise ratio under the HTS conditions. However, KAN was used in all post-HTS assays, since it is the clinically relevant aminoglycoside. The HTS yielded 617 hits containing an isothiazole S,S-dioxide heterocyclic core that were retested (Figure S1 and Table S1). Of these 617 molecules, 133 showed reproducible inhibition of Eis enzymatic activity in the same single-point HTS assay. Thirty-six of these 133 compounds were chosen for further testing in dose–response assays, based on the potency of their inhibition of Eis in the HTS assay and the structural diversity of their chemical substituents (R1 and R2 groups). In a previous study of Eis inhibitors evaluating fewer molecules,25 we demonstrated that active compounds contained (i) a group that may become positively charged, usually an amine, or (ii) an aromatic group. The presence of either of these groups was, therefore, also a criterion for selection of compounds for dose–response testing. Additionally, we included four analogues that were not a part of the original library, but which contained the same structural core (Figure 1b). This process yielded a group of 40 molecules, for which we determined IC50 values against purified Eis. We also measured the effect of these compounds on KAN MIC for KAN-susceptible (H37Rv) and KAN-resistant (K204) Mtb strains (Table 1), as described in the two following sections.
Figure 1.
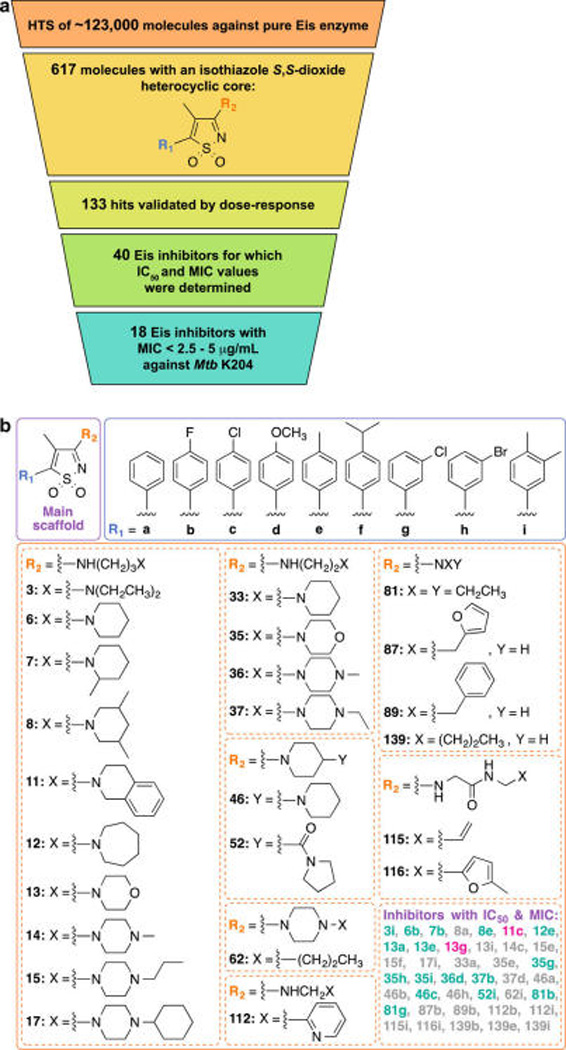
(a) Schematic representation of the winnowing of ~123 000 small organic molecules to 18 showing inhibition of both Eis enzymatic activity and growth of Mtb K204 in the presence of KAN. (b) Structures of the 41 Eis inhibitors (with an isothiazole S,S-dioxide heterocyclic scaffold) for which IC50 against pure Eis enzyme and MIC values against strains H37Rv and K204 of Mtb have been determined. Note: Compounds highlighted in turquoise are those for which MIC values of KAN against Mtb K204 were found to be <2.5–5 µg/mL. Compounds highlighted in gray are those for which MIC values of KAN against Mtb K204 were found to be >5–10 µg/mL. Compounds highlighted in fuchsia are those for which the X-ray structure in complex with EisC204A and CoA has been determined.
Table 1.
Eis Inhibition IC50 Values of Tested Compounds and the Effect of the Compounds on Kanamycin A MIC Values against H37Rv and K204 Mtb Strains
| Cpd | IC50 (µM)a | Concentration tested (µM)b |
H37Rv MICKAN (µg/ mL)c |
K204 MICKAN (µg/ mL)d |
|---|---|---|---|---|
| 1.25 | 10 | |||
| 3i | 0.120 ± 0.035 | 12 | 0.625 | 1.25 |
| 6b | 0.152 ± 0.045 | 15.2 | 0.625 | 2.5 |
| 7b | 0.102 ± 0.034 | 10.2 | 0.625 | 2.5 |
| 8a | 2.85 ± 0.26 | 184.5 | 0.625–1.25 | 5 |
| 8e | 0.200 ± 0.036 | 20 | 0.625 | 2.5–5 |
| 11c | 0.238 ± 0.089 | 23.8 | 0.625 | 1.25 |
| 12e | 0.183 ± 0.043 | 18.3 | 0.625 | 2.5 |
| 13a | 2.82 ± 0.21 | 282.3 | 0.625 | 2.5 |
| 13e | 0.331 ± 0.071 | 33.1 | 0.625 | 2.5 |
| 13g | 0.234 ± 0.060 | 23.4 | 0.625 | 2.5–5 |
| 13i | 0.054 ± 0.002 | 5.42 | 0.625 | 5 |
| 14c | 0.112 ± 0.008 | 11.2 | 0.625 | 5 |
| 15e | 0.535 ± 0.045 | 53.5 | 0.625–1.25 | 5 |
| 15f | 0.148 ± 0.016 | 14.8 | 0.625–1.25 | 10 |
| 17i | 0.232 ± 0.038 | 23.2 | 0.625–1.25 | 5 |
| 33a | 0.123 ± 0.023 | 13.2 | 0.625 | 5 |
| 35e | > 200 | 20 | 0.625 | 5 |
| 35g | 2.49 ± 0.49 | 100 | 0.625 | 2.5 |
| 35h | 3.23 ± 0.99 | 100 | 0.625 | 2.5 |
| 35i | 0.985 ± 0.190 | 98.5 | 0.625 | 2.5 |
| 36d | 0.152 ± 0.031 | 15.3 | 0.625 | 2.5–5 |
| 37b | 0.657 ± 0.105 | 65.7 | 0.625 | 2.5–5 |
| 37d | > 200 | 20 | 0.625–1.25 | 5–10 |
| 46a | 0.432 ± 0.057 | 43.2 | 0.625–1.25 | 10 |
| 46b | 0.092 ± 0.021 | 9.21 | 0.625–1.25 | 5 |
| 46c | 0.109 ± 0.022 | 10.9 | 0.625 | 2.5 |
| 46h | 0.135 ± 0.031 | 13.5 | 0.625 | 5 |
| 52i | 0.580 ± 0.096 | 58 | 0.625 | 2.5 |
| 62i | 0.386 ± 0.080 | 38.6 | 0.625 | 5 |
| 81b | 1.25 ± 0.22 | 124.8 | 0.625 | 2.5–5 |
| 81g | 1.42 ± 0.42 | 142.3 | 0.625 | 2.5 |
| 87b | > 200 | 20 | 1.25 | 10 |
| 89b | > 200 | 20 | 1.25 | 10 |
| 112b | > 200 | 20 | 0.625 | 10 |
| 112i | 0.621 ± 0.229 | 62.1 | 0.625 | 5 |
| 115i | > 200 | 20 | 0.625 | 10 |
| 116i | > 200 | 20 | 1.25 | 10 |
| 139b | > 200 | 20 | 0.625 | 10 |
| 139e | > 200 | 20 | 1.25 | 10 |
| 139i | > 200 | 20 | 1.25 | 10 |
The IC50 values in the Eis acetyltransferase assay.
At these concentrations, these compounds did not inhibit the growth of H37Rv or that of K204 Mtb, when tested in the absence of KAN.
KAN MIC values for H37Rv Mtb in the absence (first line) and in the presence of each compound at the specified concentrations.
Same as c, but for K204 Mtb.
Structure–Activity Relationship Studies
Various R1 and R2 side-chains in the identified isothiazole S,S-dioxide heterocyclic scaffold (Figure 1b, Figure S1, and Table S1) were explored to improve anti-Eis potency in vitro. Based on the 617 initial HTS hit molecules, we first drew several unambiguous conclusions about the desirable structure of the R2 side-chain. Most of the compounds for which R2 contained an aromatic ring, a branched alkyl group, a cyclohexyl moiety, or a benzyl functionality did not efficiently inhibit Mtb Eis (as indicated by × highlighted in orange in Table S1). Among the 40 compounds that were pursued beyond the dose–response assay, compounds containing R2 groups 8, 13, 15, 35, 46, and 81 inhibited Eis in vitro when combined with multiple R1 substituents (as indicated by two checks and an × and three checks, highlighted in dark yellow and dark green, respectively, in Table S1), while other R2 groups were shown to inhibit Eis in dose–response assays when in combination with only one of the possible R1 substituents (6b, 7b, 11c, 12e, 14c, 17i, 33a, 36d, 37b, 52i, 62i, and 112i). Overall, two large families of R2 substituents emerged as potential potent inhibitors of Eis: (i) compounds with R2 containing two nitrogen atoms separated by three carbon atoms (structures 3, 6–8, 11–15, and 17), particularly compounds with large bulky groups or a cyclohexyl ring attached to the extended nitrogen atom were most often inhibitory, and (ii) compounds containing an R2 group with two nitrogen atoms separated by two carbon atoms also efficiently inhibited Eis (structures 33, 35–37, and 112). In this series of molecules, only compounds with a nitrogen atom located in a cyclohexyl ring were inhibitory.
We next explored the effect of the R1 substituents on Eis inhibition. Upon initial inspection of the 617 HTS hits, the identity of the R1 substituent appeared to have little effect on their Eis inhibitory activity. However, when analyzing these side chains statistically, patterns emerged. The p-fluorophenyl group (b) had the highest percentage (34%) of compounds inhibiting Eis in the initial HTS; this was followed closely by the p-methylphenyl group (e, 33%) and the m,p-dimethylphenyl group (i, 30%), suggesting that these three groups as R1 substitutions have a better chance of contributing positively to Eis inhibition. The next best R1 substituent was the p-chlorophenyl group (c) with 27% of the compounds within this group displaying inhibition of Eis. The m-chlorophoenyl group (g) and p-anisole group (d) were next with 19% and 18% of their compounds displaying Eis inhibition, respectively. Finally, those with 13%, 12%, and 9% of compounds showing Eis inhibition were compounds containing the p-isopropylphenyl (f), the m-bromophenyl (h), and the phenyl (a) group as R1, respectively.
Having established general trends for the R1 and R2 substituents by analyzing the data from Table S1, we next focused on the measured in vitro potency (IC50) of the 40 selected compounds (Table 1). Several trends consistent with those established above emerged from these quantitative data. With the exception of compound 112i, monosubstituted R2 amine substituents comprised of a straight alkyl chain (139b, 139e, 139i), an aromatic ring (87b, 89b, 112b), or an amide functionality (115i, 116i) did not inhibit purified Eis. Compounds with R2 substituents containing a diamine separated by two carbon atoms with the second amine present in a cyclohexyl ring all displayed good to moderate Eis inhibition. However, no conclusion could be formed as to what type of cyclohexyl ring was best, an unsubstituted (33a), a morpholino (35e, g, h, and i), or a piperazinyl (36d or 37b) ring, as most IC50 values were similar for these compounds. Compound 13i with a diamine separated by three carbons at the R2 position and the m,p-dimethylphenyl group at the R1 position was found to be the most potent Eis inhibitor (IC50 = 0.054 ± 0.002 µM). Interestingly, compound 46b with a cyclohexyl ring directly attached to the core scaffold followed by a second piperazine ring in the para position was found to be the second best Eis inhibitor (IC50 = 0.092 ± 0.021 µM). These results indicate that either a rotationally free alkyl chain (as in compound 13i) or cyclically restricted alkyl linkers could be useful in designing Mtb Eis inhibitors.
To establish whether the inhibitors identified were specific to Eis over AACs from other families, compounds 46b and 46c were tested against AAC(6′)-Ie/APH(2″)-Ia from Staphylococcus aureus,26,27 AAC(3)-IV from E. coli,28 and AAC(2′)-Ic from Mtb.29,30 Under optimal reaction conditions for each enzyme, no inhibition by either compound was observed with any of these AACs (Table S2). These data demonstrate that the compounds are exquisitely selective for Mtb Eis over other AACs.
Eis Inhibitors Abolish Eis-Mediated Resistance of Mtb to KAN
Having identified inhibitors of the purified Mtb Eis enzyme, we next determined their activity in cellulo. Mycobacteria are notorious for their waxy cell wall that is difficult to penetrate for small molecules. Therefore, we anticipated that not all of the compounds that exhibited inhibition of purified Eis would be effective in cell cultures. We measured the effect of the 40 selected compounds (at a concentration of 100-fold higher than their IC50 values when possible) on KAN MIC values against Mtb K204, a KAN-resistant strain bearing a clinically observed eis promoter mutation known to upregulate Eis production (Table 1). In the KAN-susceptible H37Rv strain, the MIC value of KAN is 1.25 µg/mL, whereas in the KAN-resistant K204 strain, the MIC value of KAN is 10 µg/mL. As expected, compounds that did not inhibit purified Mtb Eis (35e; 37d; 87b; 89b; 112b; 115i; 116i; and 139b, 139e, and 139i) did not overcome resistance to KAN in Mtb K204. Twenty-eight of the remaining 30 Eis inhibitors resulted in at least a 2-fold reduction in KAN MIC against Mtb K204, with two of these compounds, 3i and 11c, lowering KAN MIC 8-fold, down to the level of the KAN-susceptible H37Rv strain. Notably, these compounds did not have any effect on the growth of either mycobacterial strain in the absence of KAN. Therefore, these compounds act synergistically with KAN against KAN-resistant Mtb. The compounds that reduced the MIC value of the KAN-resistant Mtb strain 4-fold had IC50 values that ranged from 0.102 ± 0.034 to 3.23 ± 0.99 µM. Generally, in vitro (Eis inhibition) and in cellulo (KAN-sensitization) activities were correlated among the compounds tested in both assays. This correlation, together with the lack of inhibition of cell growth in the absence of KAN, serve as strong evidence of inhibition of KAN acetylation by Eis as the principal mechanism of action in the mycobacterial cell. Not unexpectedly, among very potent inhibitors of the enzymatic activity of Eis in vitro, a small fraction of the compounds had little or no effect at sensitizing the resistant Mtb strain K204 to KAN. The lack of activity in the cells was likely due to the inability of these compounds to go through the cell envelope or due to their aggregation in the cell culture media at the concentrations that were much higher than those used in the enzymatic assay.
Crystal Structures of Eis-Inhibitor Complexes Reveal the Inhibition Mechanism
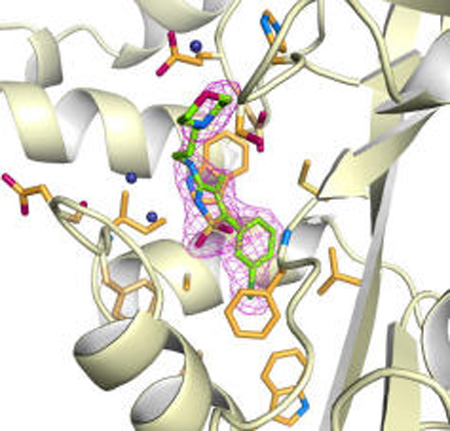
To elucidate the mechanism of Eis inhibition by the isothiazole S,S-dioxide heterocyclic core-containing compounds, rationalize the observed SAR, and guide future rational inhibitor design, we determined 2.2-Å-resolution crystal structures of Eis in complex with CoA and inhibitors 13g and 11c (Figure 2a,c), which displayed both potent inhibition of Eis acetylation in vitro and the KAN resistance abolishing effect in the Mtb cultures. Both inhibitors and their chemical features were clearly distinguishable and modeled into strong Fo−Fc electron density (Figure 2b,e). The only exception was the 1,2,3,4-tetrahydroisoquinoline group of 11c, for which only partial electron density was observed (Figure 2e), likely due to a somewhat dynamic behavior of this highly hydrophobic group in a partially polar environment, as allowed by the flexible diamine linkage. No other significant difference electron density that could be attributed to a bound inhibitor was observed elsewhere. Both inhibitors exhibited high steric complementarity with the Eis surface. The binding site for both inhibitors overlapped with the aminoglycoside substrate-binding site, as was recently established by the structure of Mtb Eis in complex with CoA and tobramycin (Figure 2c,f; Figure S2).10 In addition, the R2 groups of both inhibitors reached the C-terminal carboxyl group of Eis, which was proposed to serve as the general base in the acetyl transfer reaction. Therefore, these crystal structures indicate that the inhibitors block access of aminoglycosides to the active site of the enzyme. Explaining the critical role of the isothiazole S,S-dioxide heterocyclic core, this group is bound in the same location and orientation in both Eis-inhibitor structures (Figure 2). Remarkably, despite the nearly identical positions of this group, there are differences in how this group interacts with the protein for the two inhibitors. These differences are due to significant conformational changes in the region spanning residues 26–31 of Eis (Figure 2b,e), which uniquely adapts to bind the two structurally different R2 groups. For inhibitor 13g (Figure 2a,b,c), one of the sulfonyl oxygens forms a direct hydrogen bond with the main chain amide nitrogen of Phe84, and the ring nitrogen forms a water-mediated hydrogen bond with the main chain amide nitrogen of Ile28. In this case, the Eis conformation resembles that observed in a previously reported crystal structure with the aminoglycoside-binding pocket unoccupied.4 Inhibitor 11c does not make any water-mediated contacts due to a small positional shift of the inhibitor and a conformational change of Eis. Instead, the two sulfonyl oxygens are at a distance of ~4 Å, consistent with the stabilizing van der Waals and electrostatic interactions. The isothiazole ring is sterically sandwiched between the side chains of Ile28 and Ser83 in one direction and between the side chains of Phe24 and Trp36 in the orthogonal direction. The R1 groups of the two compounds are almost coplanar and fit neatly into a nearly fully nonpolar environment of side chains Leu63, Trp13, and Ala33 and the aliphatic stems of Arg37, Met65, and Phe84, explaining the nonpolar phenyl ring with small substitutions as effective R1 groups (Figure 1). Such an environment, devoid of hydrogen bond donors or acceptors, is appropriate for accommodating halogen substituents, as is the case with 11c and 13g. The halogens can be accommodated in the para and/or one, but not both, meta positions. Substitutions at both meta positions would not be accommodated, as reflected in the list of potent inhibitors (Figure 1), since one of them would clash sterically with the side chain of Met65. In contrast with R1, the conformations of the R2 groups of the two inhibitors are drastically different with one exception—the positively charged ring nitrogen of R2 occupies nearly the same location where it forms a salt bridge with the carboxyl group of Glu401 in both structures. Besides this interaction, the R2 group of compound 13g makes steric and weak electrostatic interactions with His119, Ser121, Glu401, and the C-terminal carboxyl group (weakly electrostatically interacting with the stem N) on one side and makes water-mediated steric interactions with region 26–31 on the other side. In contrast, for the R2 of the compound 11c, while the interactions with one face of this group are made with the same amino acid residues, the other face interacts directly through hydrophobic and steric interactions with the region 26–31, which is in a different conformation. Most prominently, the side chain of Asp26 is flipped to interact with the stem, and Phe24 orthogonally stacks against the double ring. Because the R1 group abuts the hydrophobic wall, whereas the R2 group points toward the more extended end of the substrate binding channel, there is more variability in both the identity and the size of R2 substituents, as exhibited in the list of effective R2 groups (Figure S1).
Figure 2.
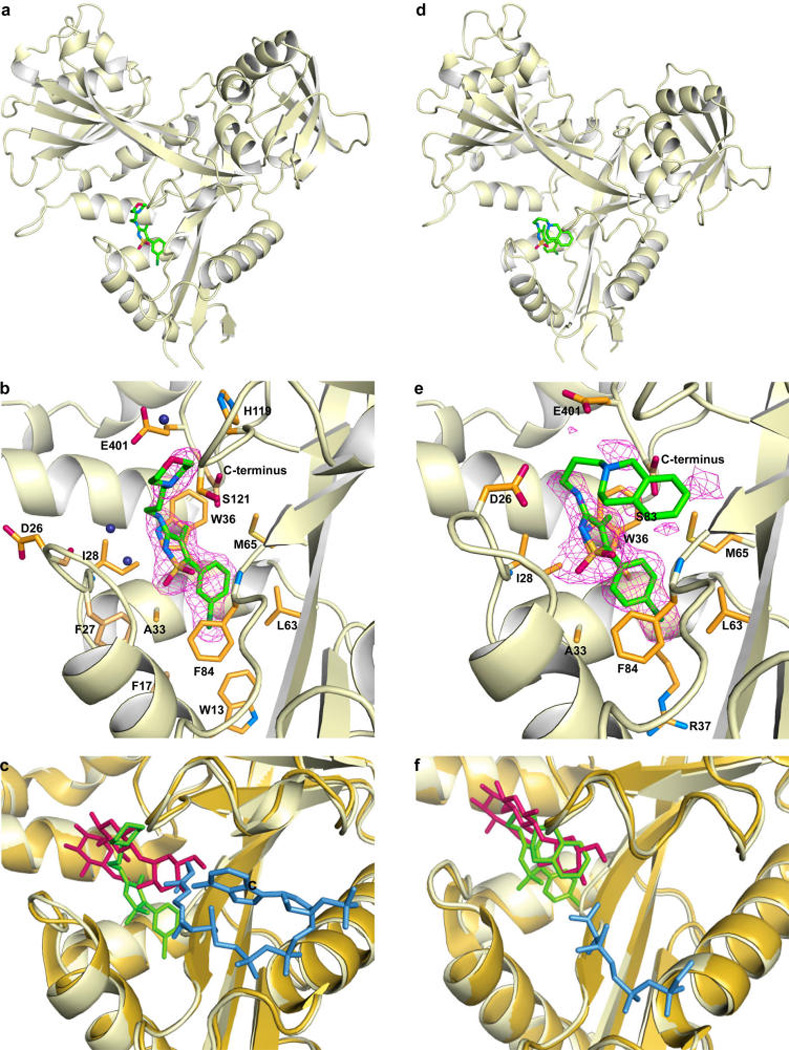
Crystal structures of EisC204A from Mtb in complex with 13g (a and b) and 11c (d and e). The overall views of the Eis monomer with the inhibitors bound (a and c) and zoom-in views of the active site (b and e) show that the inhibitors (green sticks with atoms labeled as follow: C = green, N = blue, O = red, S = orange, and Cl = dark green) occupy a site overlapping with the aminoglycoside substrate-binding site. The Fo−Fc omit map contoured at 3σ is shown by the purple mesh. Inhibitor interacting residues are shown as orange sticks (with N and O in blue and red, respectively). Water molecules mediating inhibitor binding are shown as navy blue spheres. The side chain of Asp26 and the backbone of this residue and its neighbors adopt different conformations in the two complexes. The CoA molecule is not shown in panels a, b, d, and e to simplify the view. Superposition of these respective inhibitor structures (in green) with the previously reported crystal structure of EisC204A (pale orange) in complex with CoA and tobramycin is shown in panels c and f. The tobramycin is shown in red, and the ordered part of CoA (from the structures of Eis-inhibitor-CoA complexes) is in blue. The CoA molecule from the complex of Eis with tobramycin is bound similarly and is not shown.
DISCUSSION
While it has been met with initial skepticism, target-based rational drug design is now gaining momentum and is widely used in industrial and academic drug discovery pipelines, due to advances in HTS technology, robust assay development practices, and availability of large and diverse chemical libraries. Prominent examples of successful rational drug design are HIV protease inhibitors ritonavir and the anticancer drug Gleevec. Here, we report the discovery and initial preclinical development of novel first-in-class isothiazole S,S-dioxide heterocyclic inhibitors of a unique acetyltransferase Eis from Mtb as agents that overcome one mechanism of resistance to KAN in Mtb. Among the diverse pool of compounds tested, we found agents that we validated as potent inhibitors of Eis both in the test tube and in the relevant Mtb cultures. Using a ~5-fold larger and more structurally diverse compound library than that used in our initial effort25 yielded compounds that were 10-fold and more potent than those reported in that previous study. Potencies in the mid-nanomolar range appear to be achieved through binding of the compounds via steric complementarity, hydrophobic, and hydrogen bonding interactions. While one substituent group (R1) is relatively unchangeable in terms of its size and physicochemical properties, the other group (R2) can vary greatly. The crystal structures of Eis-inhibitor complexes demonstrate that binding to variable structures is accompanied by conformational plasticity of the aminoglycoside-binding site of Eis, which adapts to different R2 groups, while the protein–R1 interactions are rigid body-like. Binding of the two inhibitors of Eis investigated structurally differs not only in protein conformation but also in the presence or absence of water-mediated contacts. Interestingly, the exquisite selectivity and potency of Gleevec toward its target Abl kinase have been recently elegantly shown to be a consequence of an induced-fit mechanism, where Abl undergoes unique conformational changes that stabilize Gleevec in the binding site of this kinase and result in a nanomolar affinity.31 Our Eis inhibitors also appear to be selective in that they do not inhibit other AAC enzymes. This selectivity must be due to the structurally different substrate binding pocket of Eis from that of regiospecific AAC enzymes. Indeed, we demonstrated previously that even though Mtb Eis shares main catalytic residues with Eis homologues from different bacteria and even AAC(2′)-Ic, their substrate binding pocket topographies and the surface charges differ dramatically.12 For example, the C-terminal domain of Eis contributing to the shape of the substrate binding pocket (and containing inhibitor interacting residue Glu401 and the C-terminal carboxyl group) is absent in the single-domain AAC(2′)-Ic and other AAC enzymes.4
Aminoglycosides are among the antibiotics of last resort for MDR and XDR-TB patients, and resistance to them strongly decreases the chances of a favorable treatment outcome. The strategy of using KAN in combination with an Eis inhibitor is meant not only to overcome KAN resistance due to Eis upregulation, but also to curb emergence of new resistant strains by mutagenesis of the eis promoter, as such mutation would not have a survival benefit in the face of combination therapy. This study not only validates AACs as drug targets, but it sets an important precedent for HTS-driven discovery for AACs that cause resistance in clinically useful non-TB pathogens, such as AAC(3) and AAC(6′) in Klebsiella pneumoniae.32–34 As with TB, such inhibitory agents could play an important role in both overcoming existing resistance and curbing the acquisition of resistance.
Conclusions
In conclusion, we identified potent inhibitors of Eis enzymatic activity with resulting sensitization of KAN-resistant Mtb cells, in which the resistance to KAN is caused by Eis upregulation. The inhibitors bind in the aminoglycoside binding pocket, blocking the access of aminoglycosides to the active site of the enzyme. The inhibitor binding is accompanied by conformational changes of the protein. These compounds have great potential for further development as KAN adjuvants in Mtb.
EXPERIMENTAL SECTION
Detailed versions of the abbreviated protocols below are provided in the Supporting Information.
Protein, Reagents, and Small-Molecule Libraries
The wild-type Eis from Mtb,4 EisC204A,10 AAC(6′)/APH(2″),35 AAC(3)-IV,35 and AAC(2′)-Ic4 were overexpressed and purified as previously reported, with minor modifications. All reagents were purchased from Sigma with the exception of albumin-dextrose-catalase, which was from BD Biosciences. Eis was screened at the Center for Chemical Genomics (CCG, University of Michigan) against 123 000 compounds from three libraries. Compounds were retested as fresh powder samples purchased from ChemDiv.
Eis Chemical Library Screening and Hit Validation
Screening of small-molecule libraries, hit validation, and dose response analysis (represented by Figure S3) were performed as previously described.25
Inhibitor Selectivity
Compounds 46b and 46c were tested against three additional AAC enzymes: AAC(6′)/APH(2″), AAC(3)-IV, and AAC(2′)-Ic at optimal reaction conditions for each enzyme (Table S2).
Mycobacterial MIC Determination by Alamar Blue Assay
The assay was done with Mtb strains H37Rv and K204 in 7H9 broth. Compounds were tested at either 100× IC50 (if known) or 20 µM for their inhibitory effect on Mtb growth and on KAN MIC in these two strains.
Crystallization, Diffraction Data Collection, and Structure Determination and Refinement of EisC204A-CoA-inhibitor Complexes
Crystals were grown by the hanging drop method and then underwent a complex harvesting process. The cryoprotectant solution contained 0.5 mM inhibitor. The diffraction data were collected at synchrotron beamline 22-ID of the Advanced Photon Source at the Argonne National Laboratory (Argonne, IL) at 100 K. The data were processed and the structures were built and refined according to previously described methods.10 The data collection and structure refinement statistics are given in Table S3.
Supplementary Material
Acknowledgments
Funding
This work was supported by a National Institutes of Health (NIH) Grant AI090048 (S.G.-T.), a grant from the Firland Foundation (S.G.-T.), a grant from the Center for Chemical Genomics (CCG) at the University of Michigan (S.G.-T.), as well as by startup funds from the College of Pharmacy at the University of Kentucky (S.G.-T. and O.V.T).
We thank S. Vander Roest, M. Larsen, and P. Kirchhoff from the CCG at the University of Michigan for help with HTS. We thank the staff of sector 22 (SER-CAT) of the Advanced Photon Source at the Argonne National Laboratories for their assistance with the remote X-ray diffraction data collection. The beamline use was supported, in part, by the Center for Structural Biology at the University of Kentucky.
ABBREVIATIONS
- AAC
aminoglycoside acetyltransferase
- CoA
coenzyme A
- Eis
enhanced intracellular survival
- HTS
high-throughput screening
- KAN
kanamycin
- MDR
multidrug-resistant
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- NEO
neomycin B
- TB
tuberculosis
- XDR
extensively drug-resistant
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Detailed experimental protocols for all experiments performed, including (1) proteins, reagents, and small molecule libraries; (2) Eis chemical library screening; (3) hit validation; (4) inhibition kinetics; (5) inhibitor selectivity; (6) mycobacterial MIC determination by alamar blue assay; (7) purification of EisC204A; and (8) crystallization, diffraction data collection, and structure determination and refinement of EisC204A-CoA-inhibitor complexes; Figures showing the structures of the compounds tested in this study (Figure S1), the comparison of the structure of a bound inhibitor, 13g, and of a bound tobramycin to EisC204A (Figure S2), and of representative IC50 plots for the inhibition of Eis using fresh powder (Figure S3); Tables summarizing the overall Eis activity of compounds tested against purified enzyme and in some cases against Mtb (Table S1), the inhibition of other AACs by two potent Eis inhibitors (Table S2), and X-ray diffraction data collection and refinement statistics of the EisC204A-CoA inhibitor 11c and EisC204A-CoA inhibitor 13g ternary complex structures (Table S3) (PDF)
Author Contributions
M.J.W. performed the determination of MIC values in Mtb. K.D.G. purified all enzymes, performed HTS assays, determined IC50 values, and confirmed the specificity of the inhibitors toward Eis. C.S.G. and C.H. crystallized and determined the two structures of EisC204A-CoA-inhibitor complexes. M.J.W., O.V.T, J.E.P, and S.G.-T. designed the experiments, analyzed data, and wrote the manuscript. K.D.G. and C.S.G. wrote the experimental sections of the manuscript. All authors provided feedback and corrections on the complete manuscript.
The authors declare no competing financial interest.
REFERENCES
- 1.World Health Organization. Global Tuberculosis Report, 2014. 2014 ISBM 978 992 974 156580 156589.
- 2.Green KD, Garneau-Tsodikova S. Front. Microbiol. 2013;4:208. doi: 10.3389/fmicb.2013.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell PJ, Morlock GP, Sikes RD, Dalton TL, Metchock B, Starks AM, Hooks DP, Cowan LS, Plikaytis BB, Posey JE. Molecular detection of mutations associated with first- and second-line drug resistance compared with conventional drug susceptibility testing of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2011;55:2032–2041. doi: 10.1128/AAC.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Biswas T, Porter VR, Tsodikov OV, Garneau-Tsodikova S. Unusual regioversatility of acetyltransferase Eis, a cause of drug resistance in XDR-TB. Proc. Natl. Acad. Sci. U. S. A. 2011;108:9804–9808. doi: 10.1073/pnas.1105379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W, Green KD, Tsodikov OV, Garneau-Tsodikova S. Aminoglycoside multiacetylating activity of the enhanced intracellular survival protein from Mycobacterium smegmatis and its inhibition. Biochemistry. 2012;51:4959–4967. doi: 10.1021/bi3004473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Green KD, Garneau-Tsodikova S. Cosubstrate tolerance of the aminoglycoside resistance enzyme Eis from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012;56:5831–5838. doi: 10.1128/AAC.00932-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Houghton JL, Green KD, Pricer RE, Mayhoub AS, Garneau-Tsodikova S. Unexpected N-acetylation of capreomycin by mycobacterial Eis enzymes. J. Antimicrob. Chemother. 2013;68:800–805. doi: 10.1093/jac/dks497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jennings BC, Labby KJ, Green KD, Garneau-Tsodikova S. Redesign of substrate specificity and identification of the aminoglycoside binding residues of Eis from Mycobacterium tuberculosis. Biochemistry. 2013;52:5125–5132. doi: 10.1021/bi4002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsodikov OV, Green KD, Garneau-Tsodikova S. A random sequential mechanism of aminoglycoside acetylation by Mycobacterium tuberculosis Eis protein. PLoS One. 2014;9:e92370. doi: 10.1371/journal.pone.0092370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houghton JL, Biswas T, Chen W, Tsodikov OV, Garneau-Tsodikova S. Chemical and structural insights into the regioversatility of the aminoglycoside acetyltransferase Eis. Chem Bio Chem. 2013;14:2127–2135. doi: 10.1002/cbic.201300359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pricer RE, Houghton JL, Green KD, Mayhoub AS, Garneau-Tsodikova S. Biochemical and structural analysis of aminoglycoside acetyltransferase Eis from Anabaena variabilis. Mol. BioSyst. 2012;8:3305–3313. doi: 10.1039/c2mb25341k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green KD, Biswas T, Chang C, Wu R, Chen W, Janes BK, Chalupska D, Gornicki P, Hanna PC, Tsodikov OV, Joachimiak A, Garneau-Tsodikova S. Biochemical and structural analysis of an Eis family aminoglycoside acetyltransferase from Bacillus anthracis. Biochemistry. 2015;54:3197–3206. doi: 10.1021/acs.biochem.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green KD, Pricer RE, Stewart MN, Garneau-Tsodikova S. Comparative study of Eis-like enzymes from pathogenic and non-pathogenic bacteria. ACS Infect. Dis. 2015;1:272–283. doi: 10.1021/acsinfecdis.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, 3rd, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Sun Y, Wang Y, Lu M, He J, Liu J, Chen Q, Zhang X, Zhou F, Wang G, Sun X. Non-antibiotic agent ginsenoside 20(S)-Rh2 enhanced the antibacterial effects of ciprofloxacin in vitro and in vivo as a potential NorA inhibitor. Eur. J. Pharmacol. 2014;740:277–284. doi: 10.1016/j.ejphar.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 16.Shlaes DM. New beta-lactam-beta-lactamase inhibitor combinations in clinical development. Ann. N. Y. Acad. Sci. 2013;1277:105–114. doi: 10.1111/nyas.12010. [DOI] [PubMed] [Google Scholar]
- 17.Zhanel GG, Lawson CD, Adam H, Schweizer F, Zelenitsky S, Lagace-Wiens PR, Denisuik A, Rubinstein E, Gin AS, Hoban DJ, Lynch JP, 3rd, Karlowsky JA. Ceftazidime-avibactam: a novel cephalosporin/beta-lactamase inhibitor combination. Drugs. 2013;73:159–177. doi: 10.1007/s40265-013-0013-7. [DOI] [PubMed] [Google Scholar]
- 18.Sader HS, Castanheira M, Flamm RK, Farrell DJ, Jones RN. Antimicrobial activity of ceftazidime-avibactam against Gram-negative organisms collected from U.S. medical centers in 2012. Antimicrob. Agents Chemother. 2014;58:1684–1692. doi: 10.1128/AAC.02429-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao F, Yan X, Auclair K. Synthesis of a phosphonate-linked aminoglycoside-coenzyme a bisubstrate and use in mechanistic studies of an enzyme involved in aminoglycoside resistance. Chem. - Eur. J. 2009;15:2064–2070. doi: 10.1002/chem.200802172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao F, Yan X, Baettig OM, Berghuis AM, Auclair K. Regio- and chemoselective 6′-N-derivatization of aminoglycosides: bisubstrate inhibitors as probes to study aminoglycoside 6′-N-acetyltransferases. Angew. Chem., Int. Ed. 2005;44:6859–6862. doi: 10.1002/anie.200501399. [DOI] [PubMed] [Google Scholar]
- 21.Gao F, Yan X, Shakya T, Baettig OM, Ait-Mohand-Brunet S, Berghuis AM, Wright GD, Auclair K. Synthesis and structure-activity relationships of truncated bisubstrate inhibitors of aminoglycoside 6′-N-acetyltransferases. J. Med. Chem. 2006;49:5273–5281. doi: 10.1021/jm060732n. [DOI] [PubMed] [Google Scholar]
- 22.Gao F, Yan X, Zahr O, Larsen A, Vong K, Auclair K. Synthesis and use of sulfonamide-, sulfoxide-, or sulfone-containing aminoglycoside-CoA bisubstrates as mechanistic probes for aminoglycoside N-6′-acetyltransferase. Bioorg. Med. Chem. Lett. 2008;18:5518–5522. doi: 10.1016/j.bmcl.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boehr DD, Draker KA, Koteva K, Bains M, Hancock RE, Wright GD. Broad-spectrum peptide inhibitors of aminoglycoside antibiotic resistance enzymes. Chem. Biol. 2003;10:189–196. doi: 10.1016/s1074-5521(03)00026-7. [DOI] [PubMed] [Google Scholar]
- 24.Suga T, Ishii T, Iwatsuki M, Yamamoto T, Nonaka K, Masuma R, Matsui H, Hanaki H, Omura S, Shiomi K. Aranorosin circumvents arbekacin-resistance in MRSA by inhibiting the bifunctional enzyme AAC(6′)/APH(2″) J. Antibiot. 2012;65:527–529. doi: 10.1038/ja.2012.53. [DOI] [PubMed] [Google Scholar]
- 25.Green KD, Chen W, Garneau-Tsodikova S. Identification and characterization of inhibitors of the aminoglycoside resistance acetyltransferase Eis from Mycobacterium tuberculosis. Chem Med Chem. 2012;7:73–77. doi: 10.1002/cmdc.201100332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boehr DD, Daigle DM, Wright GD. Domain-domain interactions in the aminoglycoside antibiotic resistance enzyme AAC(6′)-APH(2″) Biochemistry. 2004;43:9846–9855. doi: 10.1021/bi049135y. [DOI] [PubMed] [Google Scholar]
- 27.Caldwell SJ, Berghuis AM. Small-angle X-ray scattering analysis of the bifunctional antibiotic resistance enzyme aminoglycoside (6′) acetyltransferase-ie/aminoglycoside (2″) phosphotransferase-ia reveals a rigid solution structure. Antimicrob. Agents Chemother. 2012;56:1899–1906. doi: 10.1128/AAC.06378-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magalhaes ML, Blanchard JS. The kinetic mechanism of AAC3-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry. 2005;44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ainsa JA, Perez E, Pelicic V, Berthet FX, Gicquel B, Martin C. Aminoglycoside 2′-N-acetyltransferase genes are universally present in mycobacteria: characterization of the aac(2′)-Ic gene from Mycobacterium tuberculosis and the aac(2′)-Id gene from Mycobacterium smegmatis. Mol. Microbiol. 1997;24:431–441. doi: 10.1046/j.1365-2958.1997.3471717.x. [DOI] [PubMed] [Google Scholar]
- 30.Vetting MW, Hegde SS, Javid-Majd F, Blanchard JS, Roderick SL. Aminoglycoside 2′-N-acetyltransferase from Mycobacterium tuberculosis in complex with coenzyme A and aminoglycoside substrates. Nat. Struct. Biol. 2002;9:653–658. doi: 10.1038/nsb830. [DOI] [PubMed] [Google Scholar]
- 31.Agafonov RV, Wilson C, Otten R, Buosi V, Kern D. Energetic dissection of Gleevec’s selectivity toward human tyrosine kinases. Nat. Struct. Mol. Biol. 2014;21:848–853. doi: 10.1038/nsmb.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mainardi JL, Zhou XY, Goldstein F, Mohler J, Farinotti R, Gutmann L, Carbon C. Activity of isepamicin and selection of permeability mutants to beta-lactams during aminoglycoside therapy of experimental endocarditis due to Klebsiella-pneumoniae-Cf104 producing an aminoglycoside acetyltransferase 6′ modifying enzyme and a Tem-3 beta-lactamase. J. Infect. Dis. 1994;169:1318–1324. doi: 10.1093/infdis/169.6.1318. [DOI] [PubMed] [Google Scholar]
- 33.Stoesser N, Batty EM, Eyre DW, Morgan M, Wyllie DH, Del Ojo Elias C, Johnson JR, Walker AS, Peto TEA, Crook DW. Predicting antimicrobial susceptibilities for Escherichia coli and Klebsiella pneumoniae isolates using whole genomic sequence data. J. Antimicrob. Chemother. 2013;68:2234–2244. doi: 10.1093/jac/dkt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Filippa N, Carricajo A, Grattard F, Fascia P, El Sayed F, Defilippis JP, Berthelot P, Aubert G. Outbreak of multidrug-resistant Klebsiella pneumoniae carrying qnrB1 and bla-(CTX-M15) in a French intensive care unit. Ann. Intensive Care. 2013;3:18. doi: 10.1186/2110-5820-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Green KD, Chen W, Houghton JL, Fridman M, Garneau-Tsodikova S. Exploring the substrate promiscuity of drug-modifying enzymes for the chemoenzymatic generation of N-acylated aminoglycosides. Chem Bio Chem. 2010;11:119–126. doi: 10.1002/cbic.200900584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.