

Abstract
Aberrant protein folding beyond the capacity of endoplasmic reticulum (ER) quality control leads to stress response in the ER. The Lys-Asp-Glu-Leu (KDEL) receptor, a retrieval receptor for ER chaperones in the early secretory pathway, contributes to ER quality control. To elucidate the function of the KDEL receptor in vivo, we established transgenic mice expressing a mutant KDEL receptor. We found that the mutant KDEL receptor sensitized cells to ER stress and that the mutant mice developed dilated cardiomyopathy. Ultrastructural analyses revealed expanded sarcoplasmic reticulums and protein aggregates that obstructed the adjacent transverse tubules of the mutant cardiomyocytes. Cardiomyocytes from the mutant mice were sensitive to ER stress when treated with tunicamycin and showed a functional defect in the L-type Ca2+ current. We observed ubiquitinated protein aggregates, enhanced expression of CHOP (a death-related transcriptional factor expressed upon ER stress), and apoptosis in the mutant hearts. These findings suggest that impairment of the KDEL receptor disturbs ER quality control, resulting in accumulation of misfolded proteins in the ER in an in vivo system, and that the dilated cardiomyopathy found in the mutant KDEL receptor transgenic mice is associated with ER stress.
The endoplasmic reticulum (ER) provides a folding environment for newly synthesized secretory and membrane proteins (10). Aberrant protein folding due to extracellular stimuli, such as ischemia and oxidative stress, and genetic mutation lead to the accumulation of misfolded proteins in the ER, which in turn evokes the unfolded protein response (43), which reduces the amount of misfolded proteins by inducing the production of ER chaperones that promote protein folding, reducing general protein synthesis (16) and enhancing the degradation of misfolded proteins via a ubiquitin-proteasome system termed ER-associated degradation (7, 9, 60). The persistent accumulation of misfolded proteins beyond the capacity of ER quality control causes cellular dysfunction and cell death (24, 25, 46). This process is involved in diverse human disorders, including diabetes mellitus (14, 42) and neurodegenerative diseases such as Alzheimer's (23) and Parkinson's (20).
Misfolded proteins had been believed to remain in the ER, but recent genetic analyses in Saccharomyces cerevisiae have indicated that the unfolded protein response involves the whole secretory pathway (56) and that some misfolded proteins require transport between the ER and the Golgi complex for ER-associated degradation (17, 41, 53, 58). In addition, certain misfolded proteins in mammalian cells have also been reported to exit the ER and recycle between the ER and post-ER compartments, associating with ER chaperones. The KDEL receptor mediates this retrieval, suggesting that the secretion of misfolded proteins from the ER and their retrieval may contribute to ER quality control (12, 62).
The KDEL receptor has been identified as a retrieval receptor for luminal ER chaperones that have a carboxyl-terminal Lys-Asp-Glu-Leu (KDEL) sequence (28, 29, 36). These ER chaperones, when secreted from the ER, are recognized by the KDEL receptor in post-ER compartments, and then both the chaperones and the receptor are sorted into coat protein complex (COP) I vesicles for retrograde transport to the ER. Besides being a retriever, the KDEL receptor has been recognized as a regulator of membrane trafficking in the early secretory pathway. The deletion of ER retention-defective complementation group 2 (ERD2), the yeast homologue of the KDEL receptor, causes the accumulation of membrane structure and disturbs transport through the Golgi complex (49). The activation of the KDEL receptor by the ligand accelerates the formation of COPI-coated vesicles (2, 32).
The KDEL receptor has been studied extensively in yeast and mammalian cells; however, its function and the outcomes of its dysfunction in animals and humans in vivo are totally unknown. Impairment of the KDEL receptor is expected to perturb ER quality control, which may cause diseases associated with ER stress. We took advantage of previous studies on the KDEL receptor and made stable cell lines expressing a transport-mutant human KDEL receptor (55). We found that the mutant KDEL receptor disturbed the circulation of misfolded proteins between the ER and the Golgi complex, resulting in accumulation of misfolded proteins in the ER. As a result, these cells became sensitive to ER stress. This finding prompted us to make transgenic mice expressing a mutant KDEL receptor possibly sensitive to ER stress. The mutant mice developed dilated cardiomyopathy and heart failure. We discuss the possible role of ER stress on the pathogenesis of dilated cardiomyopathy.
MATERIALS AND METHODS
Cells and reagents.
The cell cultures were described previously (62). The following antibodies were used: mouse monoclonal antibody 9E10 against the Myc epitope (American Type Culture Collection, Manassas, Va.), a rabbit antiserum against TCRα (62), a mouse monoclonal antibody against Golgi p58, a mouse monoclonal antibody against γ-tubulin (Sigma Chemical, St. Louis, Mo.), mouse monoclonal antibody SPA-827 against immunoglobulin heavy chain-binding protein (BiP) (KDEL sequence), mouse monoclonal antibody SPA-810 against HSP72 (Stressgen, Victoria, Canada), mouse monoclonal antibody AF8 against calnexin (kindly provided by M. Brenner, Boston, Mass.) (18), a rabbit antiserum against C/EBP homologous protein (CHOP), a goat polyclonal antiserum against troponin T, a rabbit antiserum against X-box binding protein 1 (XBP-1), and a rabbit antiserum against ubiquitin (Santa Cruz Biotechnology, Santa Cruz, Calif.). The following reagents were used: endoglycosidase H (New England Biolabs, Beverly, Mass.), dithiothreitol (Sigma Chemical), and tunicamycin (Nacali Tasque, Kyoto, Japan).
Plasmids and transfection.
The Myc-tagged wild-type human KDEL receptor 1 and a transport mutant, D193N (55), were gifts from H. R. B. Pelham (Cambridge, United Kingdom). CD8E19 and CDE193S were kindly provided by M. R. Jackson (San Diego, Calif.). Transfection was performed by the calcium phosphate method (62). To generate a stable cell line, Myc-tagged D193N cDNA was cotransfected with a construct containing a neomycin resistance gene into HeLa cells by the calcium phosphate method. Cells were selected in complete medium containing 0.5 mg of geneticin (Invitrogen, Carlsbad, Calif.) ml−1. Stable transfectants were screened by immunofluorescence microscopy. The mutant KDEL receptor stable cell line expressed as much Myc-tagged protein as the wild-type KDEL receptor stable cell line (63).
Immunofluorescence microscopy and biochemistry.
Immunofluorescence microscopy, immunoprecipitation, RNA isolation, and RNA blot analysis were conducted as previously described (62, 63).
Metabolic labeling and chase experiment.
Cells were transiently transfected with CD8E19 or CD8E193S. Forty hours later, the cells were incubated in labeling medium (Dulbecco's modified Eagle's medium without methionine supplemented with 2% fetal bovine serum, 2 mM glutamine, 50 μg of streptomycin ml−1 and 50 U of penicillin G ml−1) for 20 min at 37°C, labeled with [35S]methionine at 250 μCi ml−1 for 10 min, washed, and then chased in complete medium for 0 to 60 min. The cells were collected, lysed, and subjected to immunoprecipitation with an anti-CD8 monoclonal antibody (OKT8). The immunoprecipitates were washed, boiled in sample buffer, and separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) under reducing conditions. The gels were then analyzed with BAS 2500 and Image Gauge software (Fuji Photo Film Co. Ltd., Tokyo, Japan).
Sucrose gradient experiment.
The cells were washed twice with ice-cold phosphate-buffered saline and once with homogenization buffer (10 mM triethanolamine, 1 mM EDTA, 250 mM sucrose), removed from the dishes, and pelleted by centrifugation for 10 min at 800 × g. The pellet was resuspended in 1 ml of homogenization buffer containing 10 μg of aprotinin, 10 μg of leupeptin, and 30 μg of N-acetyl-l-leucinal-l-lecinal-l-norleucinal (ALLN; Sigma Chemical Co.) ml−1 and homogenized by passing it four times through a ball-bearing homogenizer (EMBL, Heidelberg, Germany). A postnuclear supernatant was obtained by centrifugation for 10 min at 800 × g at 4°C. The postnuclear supernatant was loaded on a continuous sucrose gradient (20 to 50%) prepared with a Gradient Master (Biocomp Instruments, Inc., Fredericton, Canada). The postnuclear supernatant on the gradient was centrifuged for 2 h at 40,000 rpm with a Beckman SW41Ti rotor. Twelve fractions were obtained from each sample. One twentieth of each fraction was separated by SDS-PAGE under reducing conditions and then transferred onto polyvinylidene fluoride membranes. The distribution of TCRα, calnexin, and Golgi p58 was determined by Western blotting.
Generation of transgenic mice.
All animal experimental procedures were in accordance with a protocol approved by the Institutional Animal Care Committee of Chiba University, Chiba, Japan. We made an inducible expression vector of the wild-type and mutant KDEL receptors with the Cre-loxP-based recombination system (see Fig. 2A). The Myc-tagged wild-type and mutant (D193N) KDEL receptor cDNAs were cloned into the transgenic construct after the chicken beta-actin (CAG) promoter-loxP-CAT-poly(A)-loxP sites. The construct was linearized and injected into the male pronucleus of fertilized single-cell embryos of C57BL/6 mice to produce transgenic mice. The transgene-positive mice were mated with mice generating Cre, driven by a CAG promoter. The offspring's tail DNA was screened by PCR with oligonucleotides 3 (5′-CTGCTAACCATGTTCATGCC-3′) and 4 (5′-AGGAATCGGAAGAGATTCAT-3′), which were within the CAG promoter and the first exon of human KDEL receptor 1, respectively. Recombination occurred in all mice that had both the transgene and the Cre gene, and this was passed on to the offspring regardless of the Cre gene (see Fig. 2B and C).
FIG. 2.

Generation of wild-type and mutant human KDEL receptor transgenic mice. (A) Transgenic construction for the Myc-tagged wild-type and mutant human KDEL receptors. The PCR (arrows 1 and 2) detected a 2.0-kb DNA fragment before and a 0.4-kb DNA fragment after recombination by the Cre-loxP system. (B) Northern blotting probed with the human KDEL receptor cDNA detected endogenous and transgene products in C57BL/6 mutant (lines B and F) and wild-type (lines D and F) human KDEL receptor transgenic mice. All the transgenic mice were Cre gene negative. The relative ratio of expression of the transgene (a) to that of the endogenous KDEL receptor (b) was assessed by densitometry. (C) The Myc-tagged mutant KDEL receptor is expressed ubiquitously in transgenic mice (line B), as determined by Western blotting with an anti-Myc monoclonal antibody.
Heart histology and TUNEL staining.
The hearts were isolated and fixed in 4% paraformaldehyde for 24 h. After fixation, they were dehydrated in increasing concentrations of ethanol and embedded in paraffin wax. Sections with 8-μm thickness were stained with hematoxylin and eosin. Apoptotic cells were visualized by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay (Roche, Basel, Switzerland).
Ultrastructural analysis.
The mice were sacrificed by cervical dislocation under pentobarbital anesthesia, and the hearts were removed. The left ventricle was dissected, sliced, and immersion fixed in a solution of 2% paraformaldehyde and 2.5% glutaraldehyde in phosphate-buffered saline. Thin sections of the slice were osmicated, dehydrated, and embedded in epoxy resin. Ultrathin sections were prepared, stained with lead citrate and uranyl acetate, and observed under a Hitachi (Tokyo, Japan) H-600 transmission electron microscope.
Western blotting of heart lysates.
The hearts were removed from the mice and homogenized in a buffer containing 0.4% Nonidet P-40, 0.2% N-lauroylsarcosine, 30 mM Tris-HCl pH 8.0, 1 mM EDTA, 10 μg of aprotinin ml−1, 10 μg of leupeptin ml−1, and 30 μg of ALLN ml−1. The lysates were boiled in sample buffer and separated by SDS-PAGE under reducing conditions. For Western blotting, gels were transferred onto polyvinylidene fluoride membranes (Immobilon-P; Millipore Corp., Bedford, Mass.), blocked, incubated with primary antibody followed by peroxidase-conjugated donkey anti-goat, -mouse, or -rabbit immunoglobulin G antibody, and developed by chemiluminescence (ECL kit; Amersham Pharmacia Biotech, Buckinghamshire, United Kingdom). Imaging was obtained by LAS1000 and Image Gauge software (Fuji Photo Film Co. Ltd.).
Myocyte isolation and culture.
Primary cardiac myocyte cultures were prepared as previously described (64) with modifications. Each ventricle from 0-day-old mice was treated. The ventricles were first perfused with nominally Ca2+-free solution for 5 min; this was immediately followed by 40 min of recirculating perfusion with the same solution containing 0.9 mg of class II collagenase (Worthington Biochemicals, Lakewood, N.J.) ml−1. Purified yachts from each mouse were plated on gelatin-coated culture dishes in minimal essential medium (Sigma Chemical) with 5% iron-fortified bovine calf serum (JRH Biosciences, Lenexa, Kans.) and maintained for 48 h.
Electrophysiology.
Mice aged 19 to 24 weeks were used for the functional experiments. Single cells were isolated by conventional enzymatic digestion, and whole-cell membrane currents were recorded by the patch-clamp method, as previously described (52). Recordings of the L-type Ca2+ current were performed at 36.0°C with a glass patch pipette filled with 120 mM CsOH, 110 mM l-aspartame, 20 mM CsCl, 1 mM MgCl2, 5 K2-ATP, 1.42 mM CaCl2, 10 mM EGTA, and 5 mM HEPES (pH 7.4). The external solution contained 143 mM NaCl, 5.4 mM CsCl, 0.33 mM NaH2PO4, 0.5 mM MgCl2, 1.8 mM CaCl2, 5.5 mM glucose, and 5 mM HEPES (pH 7.4). Membrane currents were elicited by 300-ms depolarizing pulses from a holding potential of −40 mV, delivered at 0.1 Hz. Membrane capacitance was measured and used as an index of cell surface area.
Mechanical function study.
The heart was quickly removed from mice anesthetized with urethane (1.5 mg g−1, intraperitoneally), and the left ventricular pressure of the Langendorff-perfused hearts was continuously measured as previously described (52).
Reverse transcription-PCR.
The nucleotide sequences of the primers used in this study were follows: mouse atrial natriuretic peptide, 5′-CCGTGACAAGCTTTGCCGAA-3′ and 5′-GATCTGTGTTGGACACCGCA-3′; mouse brain natriuretic peptide, 5′-CAAGATGCAGGTGAGCACTG-3′ and 5′-GGGGCCTCTAAGGGTAGCAT-3′; and mouse glyceraldehyde-3-phosphate dehydrogenase, 5′-ATGGGGTGAGGCCGGTGCTG-3′ and 5′-CTTGATGTCATCATACTTGG-3′.
Aortic constriction of rats.
Sprague-Dawley male rats weighing 290 to 300 g were anesthetized with 2% isoflurane. The right carotid artery was cannulated with a polyethylene catheter connected to a pressure transducer, and the mean carotid arterial pressures were measured. Under sterile conditions, the abdominal aorta was exposed and constricted at the suprarenal level with a 25-gauge needle to establish the diameter of ligature as previously described (34). Thereafter, the abdominal incision was closed. A similar mock procedure was performed except for the constriction.
RESULTS
Misfolded proteins recycled between the ER and the Golgi complex, and the mutant KDEL receptor disturbed the recycling.
The mutant KDEL receptor (D193N) recognizes KDEL proteins; however, the receptor is not transported to the ER upon ligand recognition but stays in post-ER compartments (55). To assess the effects of the mutant KDEL receptor on transport between the ER and the Golgi complex, we used a chimeric protein, CD8E19, that has the extracellular region and transmembrane region of CD8 and the cytoplasmic region of adenovirus E19 protein with a dilysine retrieval motif that is recognized directly by the COPI complex (27) and aids in locating the protein in the ER (21). CD8E193S has a mutation in the dilysine motif; thus it reaches the cell surface. CD8 is a glycoprotein that takes both O-linked glycosylation in the cis-Golgi and N-linked glycosylation in the ER (21).
HeLa cells and cells stably expressing either wild-type or mutant Myc-tagged KDEL receptor (63) were transiently transfected with CD8E19 or CD8E193S and subjected to metabolic pulse-chase labeling. CD8E19 acquired N-glycosylation immediately (Fig. 1A, i) and O-glycosylation gradually (Fig. 1A, ii) (21). The fraction acquiring O-glycosylation may represent the circulating fraction of the CD8E19 that reaches the cis-Golgi. Interestingly, the O-glycosylation of CD8E19 in the cells stably expressing the mutant KDEL receptor was suppressed in comparison to that in HeLa cells and in the wild-type KDEL receptor stable cells (Fig. 1A, lower panel). On the other hand, CD8E193S was transported through the medial Golgi complex and acquired a complex N-linked glycosylation to a similar extent in all cell types (Fig. 1A, iii).
FIG. 1.

Transport mutant KDEL receptor perturbed recycling of misfolded proteins between the ER and the Golgi complex. (A) Cells were transiently transfected with CD8E19 or CD8E193S. Addition of ER-specific N-linked glycosylation (i), cis-Golgi-specific O-linked glycosylation (ii), or Golgi-specific N-linked glycosylation (iii) was assessed by pulse-chase experiments followed by immunoprecipitation with an anti-CD8 monoclonal antibody. The recycling of CD8E19 was assessed by calculating the fraction with O-linked glycosylation (ii/[i + ii]) by densitometry. The line graph represents the mean value ± standard error from three experiments. Statistical analyses were performed with analysis of variance and the Scheffe test among the three groups. The values in the mutant cells at 30 min (*, 0.306 ± 0.023) and 60 min (**, 0.417 ± 0.012) were significantly different from those in HeLa cells (0.429 ± 0.014 and 0.612 ± 0.010, respectively) and wild-type cells (0.459 ± 0.015 and 0.678 ± 0.015, respectively). *, P < 0.01; **, P < 0.0001. (B) The distribution of TCRα, KDEL receptors, Golgi p58, and calnexin in wild-type (upper panel) and mutant KDEL receptor (middle panel) stable cells on a continuous sucrose gradient (top, fraction 1; bottom, fraction 12) was determined by Western blotting. Aliquots of the ER (fraction 9 in the middle panel) and the post-ER (fraction 3 in both the upper and middle panels) fractions were digested with endoglycosidase H and analyzed by SDS-PAGE, followed by Western blotting with a rabbit anti-murine TCRα antiserum (lower panel). (C) Wild-type (white columns) and mutant KDEL receptor (black columns) stable cells were treated with dithiothreitol (5 mM) for 0 to 48 h. After collection of the cells, cell viability was determined by trypan blue staining. The graph represents the mean value ± standard deviation of the surviving fraction from three experiments.
These results suggest that the mutant KDEL receptor might selectively disturb the recycling of proteins between the ER and the Golgi complex but not the anterograde transport to the secretory pathway from the ER.
We previously showed that the α subunit of the T-lymphocyte antigen receptor complex (TCRα) was circulating as a misfolded protein between the ER and the Golgi complex when it expressed itself in a cell without other subunits (62). To evaluate the effects of the mutant KDEL receptor on the circulation of misfolded proteins, we transfected TCRα into stable cells and examined its subcellular localization by sucrose gradient experiments. Calnexin is an integral membrane protein and one of the ER chaperones located in the ER (18). Thus, we used it in the sucrose gradient experiment as a marker for the ER fraction. In the wild-type KDEL receptor stable cells, TCRα was distributed in both the ER and post-ER compartments (Fig. 1B, upper panel). Even the TCRα in the ER acquired a complex Golgi glycosylation that was resistant to endoglycosidase H digestion (Fig. 1B, lower panel, p43), suggesting that the TCRα was circulating between the ER and the Golgi complex. On the other hand, most of the TCRα in the mutant KDEL receptor stable cells was located in the ER as a core-glycosylated ER form and remained sensitive to digestion (Fig. 1B, lower panel, p38).
These findings show that the mutant KDEL receptor might disturb the circulation of misfolded proteins in the early secretory pathway. As a result, the mutant KDEL receptor stable cells became sensitive to ER stress when treated with an agent such as dithiothreitol that perturbed protein folding and caused the accumulation of misfolded proteins in the ER (Fig. 1C); this suggests that the secretion of misfolded proteins from the ER and their retrieval may contribute to ER quality control and that impairment of the KDEL receptor may sensitize cells to ER stress.
Generation of transgenic mice.
In order to investigate the function of the KDEL receptor in vivo, we created transgenic mice expressing the human wild-type and transport mutant KDEL receptors with the Cre-loxP-based recombination system (48). We established two lines of the wild-type and two lines of the mutant KDEL receptor transgenic mice after crossing them with CAG promoter-Cre transgenic mice (Fig. 2A). The recombinant transgene was passed on to the offspring and expressed its products without the Cre gene. The Northern blot revealed that the expression level of the mutant KDEL receptor was as high as that of the endogenous one (Fig. 2B). Western blotting confirmed that the mutant KDEL receptor was expressed ubiquitously (Fig. 2C).
Transgenic mice expressing the mutant KDEL receptor developed dilated cardiomyopathy.
The transgenic mice expressing the mutant KDEL receptor seemed to grow normally until early adulthood. In the course of the study, we found that the transport mutant KDEL receptor transgenic mice of both lines died sporadically after attaining the age of more than 14 weeks. They appeared dyspneic, lethargic, and motionless. These mice developed peripheral edema, ascites, and cardiomegaly and seemed to die due to heart failure (Fig. 3A).
FIG. 3.

Transgenic mice expressing the transport mutant KDEL receptor developed dilated cardiomyopathy. (A) Hearts were obtained from a wild-type KDEL receptor transgenic (TG) mouse with the Cre gene (20 weeks old), a mutant KDEL receptor transgenic mouse with the Cre gene, and its littermate C57BL/6 mouse (20 weeks old). The top panels show gross sections fixed with 4% paraformaldehyde and dehydrated. The bottom panels show coronal sections stained with hematoxylin and eosin. Each division on the scale represents 1 mm. (B) Heart weight to body weight ratios among the mutant KDEL receptor transgenic mice (B and F), the wild-type KDEL receptor transgenic mice, and the C57BL/6 mice. All the mice were Cre gene positive. Statistical analyses were performed with the Mann-Whitney U test between the two groups. *, P < 0.01; †, P < 0.05. (C) Histological findings from the hearts of the wild-type KDEL receptor transgenic mouse with the Cre gene (12 weeks old), the mutant KDEL receptor transgenic mouse with the Cre gene (line B, 12 weeks old), and the C57BL/6 mouse (12 weeks old) stained with hematoxylin and eosin. The top panels show low-power photomicrographs of longitudinal sections. The bottom panels show higher-power photomicrographs of axial sections. Arrows indicate vacuolization. Bar, 100 μm. (D) The diameters of individual myofilaments in the longitudinal section were examined for each group in panel C. A statistical analysis among the three groups was performed with analysis of variance and the Scheffe test. Mean values ± standard deviation are shown. *, P < 0.0001, n = 100. (E) Cardiomyocytes from wild-type transgenic mice (11 weeks old), mutant KDEL receptor transgenic mice (line F), and littermate C57BL/6 mice (12 weeks old) were evaluated for DNA fragmentation by the TUNEL assay. Bar, 100 μm. TUNEL-positive cells were scored, and values are means ± standard deviation for five experiments. A statistical analysis was performed with analysis of variance and the Scheffe test among the three groups. *, P < 0.0001, n = 100. All mice were Cre gene negative.
We observed marked four-chamber dilation without wall thickening. The average heart-to-body weight ratio of the mutant KDEL receptor transgenic mice was significantly higher than that of the wild-type KDEL receptor transgenic mice and the parental C57BL/6 mice (Fig. 3B). Histological examination revealed that the cardiomyocytes of the mutant transgenic mice were of various sizes but on average were significantly enlarged. There was also an expanded interstitial fibrosis and vacuolization. On the other hand, the histology of the wild-type transgenic mice appeared normal (Fig. 3C and D). With TUNEL staining, we detected significantly more apoptotic cells in the hearts of the mutant KDEL receptor transgenic mice than in those of the control C57BL/6 mice and the wild-type transgenic mice (Fig. 3E). We found no significant difference in systemic arterial blood pressure between the mutant KDEL receptor transgenic mice and the C57BL/6 mice, suggesting that the cardiomegaly in the mutant mice was not a secondary change due to systemic hypertension. Taken together, these results suggest that the mutant KDEL receptor transgenic mice developed primary dilated cardiomyopathy.
Ultrastructural analyses revealed the accumulation of protein aggregates in the expanded sarcoplasmic reticulum of the mutant cardiomyocytes.
In order to gain insight into the mechanism responsible for dilated cardiomyopathy in the mutant KDEL receptor transgenic mice, we examined the ultrastructure of the mutant cardiomyocytes. The contractile apparatus appeared intact, i.e., the arrangement of myofibril structures and the banding of myofilaments were little different in the myocardia of control C57BL/6 mice (Fig. 4A) and mutant transgenic mice (Fig. 4B). The arrangement of mitochondria was also intact in the transgenic mice. On the other hand, the proliferation of sarcoplasmic reticulum was prominent around the transverse tubule area, and the transverse tubule structure was narrowed significantly in mutant transgenic mice with (Fig. 4C) and without (Fig. 4D) apparent cardiomegaly, in comparison to the transverse tubules and surrounding the sarcoplasmic reticulum in the control (Fig. 4A and E). Furthermore, an aggregation of degenerate membrane structures was found among the proliferated sarcoplasmic reticulums (Fig. 4F), and lamellated or further fused membrane structures were found in the transverse tubule area in the mutant mice (Fig. 4D). We observed electron-dense materials, possibly protein aggregates, in the expanded sarcoplasmic reticulum (Fig. 4G and H). These aggregates were associated with polyribosomes, suggesting that they were part of the ER network. On the other hand, the ultrastructure of the cardiomyocytes from the wild-type KDEL receptor transgenic mice appeared normal.
FIG. 4.

Transmission electron microscopic analysis of cardiomyocytes. The pictures in panels A and E are from a control C57BL/6 mouse (15 weeks old). The pictures in panels B and D are from mutant transgenic mouse line F without apparent cardiomegaly (13 weeks old), and the pictures in panels C, F, G, and H are from mutant transgenic mouse line B with apparent cardiomegaly (15 weeks old). All the mice were Cre gene positive. (A) Myocardium from the control. (B) Myocardium from mutant transgenic mouse F. Though the arrangement of myofibrils and the banding patterns of myofilaments are intact, the structures around the transverse tubules are aberrant (arrows). (C) Proliferation of the sarcoplasmic reticulum is evident, and the transverse tubule lumen is significantly narrowed (asterisk) in the mutant B. mt, mitochondrion. (D) Lamellated membrane structures (arrows) are found to be apposed to the narrowed T-tubule (asterisk) in the mutant F. (E) Higher magnification of a transverse tubule (large arrows) and surrounding structures, such as the sarcoplasmic reticulum (small arrows) and mitochondria, in the control. (F) The aggregation of degenerate membrane structures (large arrows) is found to be apposed to the proliferated sarcoplasmic reticulum (small arrows) in mutant B. (G and H) Electron-dense structures accumulated in the mutant cardiomyocytes were associated with polyribosomes. g, Golgi complex. Bars: panels A to C, G, and H, 1 μm; panels D to F, 200 nm.
The morphological changes found in the cardiomyocytes of the mutant KDEL receptor transgenic mice suggest that the mutant KDEL receptor might have impaired ER quality control, which led to the accumulation of misfolded proteins in the sarcoplasmic reticulum.
Mutant KDEL receptor transgenic mice revealed defective L-type Ca2+ current in the ventricular myocytes.
We measured the L-type Ca2+ current (ICa) in the ventricular cells with patch-clamp techniques to evaluate functional changes in the mutant hearts, since L-type Ca2+ channels reside on the transverse tubules. The density of basal ICa in the mutant transgenic cells was significantly lower than in the control cells (C57BL/6). The β-adrenoceptor agonist isoproterenol increased ICa in both control and transgenic cells. The percent increase in ICa after isoproterenol treatment in the transgenic cells was not significantly different from that in the control cells; the increase in ICa at −10 mV in the control cells (n = 8) and the transgenic cells (n = 12) was 168 ± 25% and 130 ± 13%, respectively, after 100 nM isoproterenol. However, the densities of ICa after the application of 10 or 100 nM isoproterenol in the transgenic cells were significantly lower than those in the control cells (Fig. 5A and B).
FIG. 5.

L-type Ca2+ current in ventricular cells and cardiac function in isolated hearts of control and mutant transgenic mice. (A) Representative current traces at baseline after administration of 10 and 100 nM isoproterenol (ISO). The traces were obtained by delivering 300-ms depolarizing pulses from a holding potential of −40 to −10 mV. (B) Summarized data for peak ICa measured at −10 mV. The data represent the mean ± standard error of 8 to 12 cells. *, P < 0.05 versus the control. (C and D) Summarized data for changes in heart rate (C) and left ventricular developed pressure (D) after 100 nM isoproterenol in the isolated hearts of control (C57BL/6) and mutant transgenic (TG) mice. Solid squares and open circles, control and transgenic mouse hearts, respectively. The data are means ± standard error for control (n = 6) and transgenic (n = 5) mouse hearts. *, P < 0.05 versus the control; #, P < 0.05 versus the baseline value. (E) Expression of markers for congestive heart failure in hearts from mutant transgenic mice with the Cre gene and a C57BL/6 mouse was evaluated by semiquantitative reverse transcription-PCR. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a standard to verify equal amounts of cDNA. ANP, mouse atrial natriuretic peptide. BNP, mouse brain natriuretic peptide.
We then measured the left ventricular pressure of the mutant transgenic hearts before and after the application of isoproterenol. Although the basal heart rate in the Langendorff-perfused hearts of the transgenic mice was significantly lower than that in the control (C57BL/6) hearts (Fig. 5C), there was no significant difference between the two groups in the basal values of the left ventricular developed pressure (Fig. 5D). While treatment with isoproterenol (100 nM) produced significant increases in heart rate, i.e., positive chronotropic responses, in both the control (n = 6) and transgenic (n = 5) hearts, it produced a significant increase in left ventricular developed pressure only in the control hearts.
These results suggest that the transduction system of β-adrenoceptors remains intact in mutant transgenic ventricular cells, at least for the changes in ICa and chronotropic response. Although the mutant transgenic mice used in these experiments did not show overt signs of heart failure yet, their cardiac function may have been latently impaired due to a decreased density of functional L-type Ca2+ channels in the sarcolemma of the heart cells. In fact, reverse transcription-PCR analyses showed enhanced expressions of atrial and brain natriuretic peptides in the mutant hearts (Fig. 5E), which indicated that the mutant transgenic mice suffered from chronic heart failure.
Hearts in the mutant KDEL receptor transgenic mice might have suffered from ER stress.
We took neonatal cardiomyocytes as a primary cell culture. When treated with tunicamycin, which disturbs protein glycosylation in the ER, neonatal cardiomyocytes from the control mice showed diffuse expression of BiP, apparently in the ER distribution. To the contrary, we observed cell shrinkage with uneven distribution of BiP in the cardiomyocytes of the mutant KDEL receptor transgenic mice, suggesting that those cardiomyocytes were sensitive to ER stress (Fig. 6A). Accumulation of misfolded proteins in the ER leads to protein degradation in a ubiquitin-proteasome system. Western blotting revealed marked accumulation of ubiquitinated protein aggregates in the hearts of the mutant KDEL receptor transgenic mice but not the wild-type KDEL receptor transgenic mice, suggesting that misfolded proteins might have saturated the ubiquitin-proteasome system during a circumstance in which the mutant KDEL receptor might have impaired the capacity for ER quality control (Fig. 6B). We found slightly increased expression of BiP and significant accumulations of CHOP in the adult mutant hearts (Fig. 6C). Since CHOP is induced by ER stress and has been acknowledged to cause apoptosis during the unfolded protein response (67), this might account for the enhanced apoptosis in the mutant hearts (Fig. 2E). These results suggest that the mutant hearts might have suffered from ER stress, which might have contributed to the pathogenesis of the cardiomyopathy found in the mutant mice.
FIG. 6.
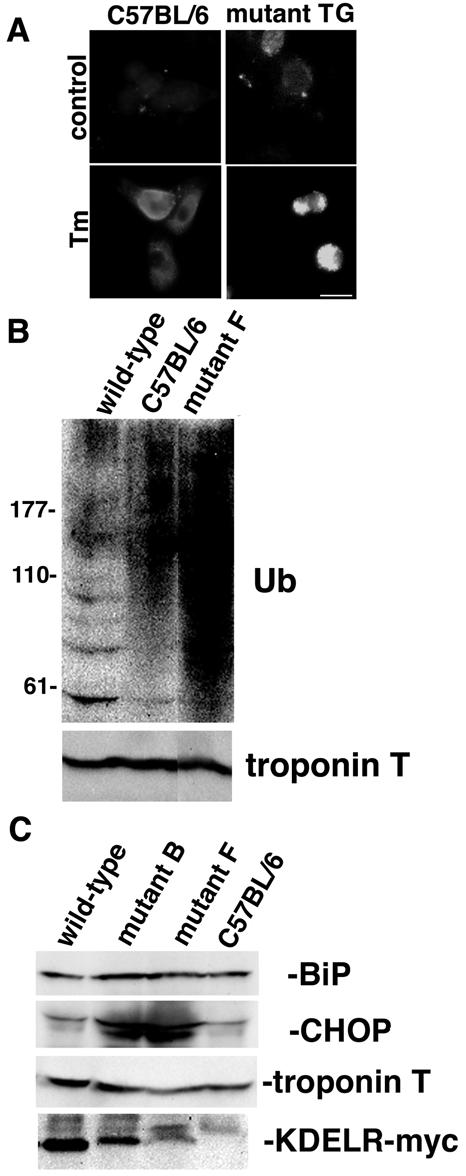
Hearts from mutant transgenic mice might have suffered from ER stress. (A) Neonatal myocytes from a mutant KDEL receptor transgenic (TG) mouse and its littermate C57BL/6 mouse were cultured. Forty-eight hours later, the cells were fixed with or without treatment with tunicamycin (Tm, 3 μg ml−1) at 37°C for 12 h. The myocytes were evaluated by fluorescence microscopy with a mouse anti-BiP monoclonal antibody. Bar, 10 μm. (B) Accumulation of ubiquitinated proteins in the hearts of mutant KDEL receptor transgenic mice. Western blotting of heart lysates with an anti-ubiquitin antiserum (Ub) and an antitroponin T antiserum. Lane 1, wild-type KDEL receptor transgenic mouse (56 weeks old); lane 2, C57BL/6 mouse (34 weeks old); lane 3, mutant KDEL receptor transgenic mouse (line F, 34 weeks old). Both transgenic mice were Cre gene positive. (C) Expression of BiP, CHOP, troponin T, and Myc-tagged KDEL receptors in hearts from wild-type, mutant KDEL receptor transgenic (lines B and F), and C57BL/6 mice (8 weeks old) was evaluated by Western blotting. All the mice were Cre gene negative.
Pressure load might contribute to ER stress on the heart.
We investigated whether the mutant KDEL receptor affected any tissues, specifically heart tissue. One possible assumption is that mechanical stress to the heart may disturb the environment for protein folding in cardiomyocytes. We performed supportive experiments with rats (Fig. 7A). Increased pressure load to the hearts due to abdominal aortic banding in normal rats induced the expression of heat shock protein 70, a chaperone in the cytosol, and BiP, a chaperone in the ER (Fig. 7B). Since both chaperones play crucial roles in protein folding and are induced upon stress, pressure overload may elicit a stress response not only in the cytosol but also in the ER of cardiomyocytes. Furthermore, we found expression of CHOP and XBP-1, transcriptional factors induced upon ER stress, in the rats' hearts, suggesting that the heart might suffer from ER stress by pressure load.
FIG. 7.

Pressure overload disturbed protein folding in normal hearts. (A) A typical recording of the right carotid artery (A.) blood pressure (BP) of a rat before and after abdominal aortic banding. (B) Expression of HSP70, BiP, CHOP, XBP-1 (s, spliced form; u, unspliced form), and troponin T in hearts from rats 6 to 72 h after banding or from a rat 72 h after a sham operation was evaluated by Western blotting.
DISCUSSION
Dilated cardiomyopathy is characterized by increased ventricular chamber size and reduced contractility of the heart, typically accompanied by loss of cardiomyocytes (38). The mutant KDEL receptor transgenic mice demonstrated cardiac dilation with congestive heart failure, interstitial fibrosis, myocyte heterogeneity, vacuolization, and apoptosis, which closely resembles the pathological and clinical features of human dilated cardiomyopathy. Clinically and experimentally, dilated cardiomyopathy is caused by a variety of factors (45, 54), including abnormalities in force transmission and other things that cause the failure of compensatory hypertrophy. Apoptosis of the cardiomyocytes may play a role in the development of dilated cardiomyopathy. Although a recent report showed that the mitochondrial cell death pathway (66) was involved in such cell death, the molecular mechanism that leads to apoptosis of the cardiomyocytes in dilated cardiomyopathy has not been well understood (22).
ER stress brings on human disorders such as neurodegenerative diseases (25); however, it is uncertain whether it also causes cardiac diseases. Cardiomyocytes are exposed to mechanical stress throughout their lives. While mechanical stress may be sensed by cardiomyocytes specifically through integrins that transduce signals from the extracellular matrix to the cytoskeletal proteins and cytosolic signal transduction molecules (8, 47), it may also cause deformation of the sarcolemma, which may lead to conformational changes in the proteins (30, 47). This effect seems to be nonspecific. Since newly synthesized proteins have not folded correctly and are unstable, their folding process might also be affected. In fact, mechanical stress induces in hearts the transcription of heat shock protein genes such as HSP70 (4). Heat shock proteins are molecular chaperones that assist in the folding of newly synthesized proteins. A perturbation of protein folding causes the accumulation of misfolded proteins in the cytosol, which dissociates HSP70 from the heat shock transcriptional factor and induces stress response (4). Similarly, the accumulation of misfolded proteins in the ER dissociates BiP, an ER-resident homologue of HSP70, from ER kinases such as IRE1, PERK, and activating transcription factor 6; this dissociation initiates the unfolded protein response, the stress response in the ER (6, 50). We observed an accumulation of HSP70 as well as BiP upon pressure overload in normal rat hearts. Thus, mechanical stress may disturb the folding of newly synthesized proteins, which in turn induces the stress response in the ER as well as in the cytosol.
We observed accumulation of CHOP and ubiquitinated proteins in the hearts of the mutant KDEL receptor transgenic mice. Hyperubiquitination of proteins has been observed in human dilated cardiomyopathy (59). Accumulations of both cytosolic and membranous misfolded proteins impair the ubiquitin-proteasome protein degradation system, which sensitizes cells to ER stress (3). Aggregates of cytosolic polyglutamine proteins and membranous Pael receptors have been thought to cause Huntington's disease and Parkinson's disease, respectively, via ER stress (20, 39). Similarly, misfolded proteins might saturate the ubiquitin-proteasome system and cause ER stress, especially in mutant KDEL receptor transgenic mice, in which the ER quality control system might be impaired. In fact, the presence of Cre seemed to enhance the phenotype of the mutant mice, although all the transgenic mice had the recombinant transgene and expressed its products even without the Cre gene. As foreign protein, Cre might also impair the ubiquitin-proteasome system. In support of this assumption, a large overexpression of cytosolic green fluorescence protein (19) or β2-adrenoreceptor (31) in transgenic mice has been reported to cause dilated cardiomyopathy. As we showed that the increased pressure load to the hearts caused upregulation of BiP, CHOP, and XBP-1 as well as HSP70, a marker for general cellular stress, the phenotype of the mutant transgenic mice might be caused by the combined effects of general cellular stress and ER stress.
While the KDEL receptor has been considered a retrieval receptor for the KDEL proteins, we observed that the transport mutant KDEL receptor suppressed the recycling of CD8E19 with a dilysine retrieval motif. The dilysine motif is recognized directly by the COPI complex (27). Thus, our results support the idea that the KDEL receptor is not merely a retrieval receptor for the KDEL proteins but also a regulator for COPI transport (2, 32). Since bidirectional transport between the Golgi complex and the ER may be coupled to each other, we suppose that the mutant KDEL receptor suppresses not only COPI retrograde transport from the Golgi to the ER but also anterograde transport from the ER to the Golgi. This suppressive effect of the mutant KDEL receptor on anterograde transport from the ER to the Golgi seems to be selective, because it reduces the recycling of CD8E19 but not the secretion of CD8E193S, which does not have the dilysine retrieval motif.
Similar selective effects on transport have been reported in a study with S. cerevisiae showing that a COPI mutation allows certain proteins to be transported unhindered to the secretory pathway, while other secretions from the ER are disturbed (11). We propose that misfolded proteins to be degraded or refolded are recycling between the Golgi and the ER to some extent and that the mutant KDEL receptor may disturb this recycling, resulting in the accumulation of misfolded proteins in the ER and causing ER stress. Interestingly, a large overexpression of β2-adrenoreceptor in transgenic mice (31) accompanied the upregulation of Rab GTPases (61), which regulate transport in the secretory pathway. Rab1 specifically plays a role in the recruitment of COPI proteins to the Golgi complex (1). Transgenic mice expressing Rab1a developed cardiac hypertrophy and failure (61), suggesting the critical role of transport between the ER and the Golgi complex in the pathogenesis of cardiomyopathy (37).
Although the precise molecular mechanism that leads to dilated cardiomyopathy in the mutant mice is still open, we speculate that there is a kind of vicious cycle. Pressure load may induce the accumulation of misfolded proteins in the sarcoplasmic reticulum, especially in the mutant cardiomyocytes, where the capacity for ER quality control is limited. Expanded sarcoplasmic reticulums and obstructed transverse tubules may disturb Ca2+ homeostasis, which may impair cardiac contractility and in turn increase the pressure overload. Persistent pressure overload causes further accumulation of misfolded proteins in the sarcoplasmic reticulum, which elicits apoptosis of the cardiomyocytes through an intense unfolded protein response and ultimately results in dilated cardiomyopathy and heart failure (Fig. 8).
FIG. 8.

Possible model for dilated cardiomyopathy (DCM) in mutant KDEL receptor transgenic mice.
ER stress initiates the unfolded protein response by activation of the ER kinases ATF6, PERK, and IRE1; this subsequently produces XBP-1, a transcriptional factor for proteins related to the unfolded protein response, such as ER chaperones (26, 65). Several attempts have been made to assess the in vivo effects of ER stress with animal models. PERK−/− mice developed progressive diabetes mellitus (15). PERK plays a major role in cells adapting to ER stress by the suppression of protein synthesis through phosphorylating eukaryotic translation initiation factor 2α. Although this phenotype might represent the common phenotype of ER stress on mice, it might also reflect the distribution of PERK, which is expressed at high levels in the pancreas (51). Since activation of PERK leads to the induction of CHOP through ATF4 (13), the apoptosis of β cells in PERK−/− mice may not be dependent on CHOP. We found that CHOP accumulated in the mutant transgenic hearts. If CHOP plays an important role in apoptosis in cardiomyopathy, it may be possible that the hearts of PERK−/− mice are less sensitive to ER stress because CHOP induction is impaired in PERK−/− mice.
Another animal model, IRE1β−/− mice, revealed an increased sensitivity to gastrointestinal insults, resulting in the development of colitis, which might reflect the restricted expression of IRE1β in the epithelial cells of the gastrointestinal tract (5). IRE1α is ubiquitously expressed, and IRE1α−/− mice died as embryos (57). Information about ATF6−/− mice is not available at this time. XBP-1−/− (also called TREB5−/−) mice died as embryos between days 10.5 and 14.5 and displayed cellular necrosis of cardiac myocytes (33) as well as hypoplastic fetal livers and anemia (44). BiP is an ER chaperone that plays an important role in the unfolded protein response. Although information about BiP−/− mice is not available, they might also die as embryos because yeast cells deleted of Kar2 (a yeast homologue of BiP) are not viable (40). Calreticulin is another ER chaperone that plays a major role in ER quality control, and calreticulin−/− mice died as embryos between days 12.5 and 18.5 due to the failure of cardiac development (35).
Collectively, based on the above studies, we are not sure whether there are any established features of ER stress in adult mice in vivo at this time. Each animal seems to have a specific phenotype derived from the specific function and expression pattern of the molecule responsible. At least XBP-1−/−(TREB5−/−) mice and calreticulin−/− mice have a cardiac phenotype during their embyogenesis.
The mutant KDEL receptor transgene is driven by the CAG promoter and is believed to be ubiquitously expressed. The expression level is moderate, as high as the endogenous level. Although all tissues of the mutant KDEL receptor transgenic mice might be sensitive to ER stress, one of the most important factors that affect whether ER stress may cause a disease is the regenerative ability of the tissue concerned. Neurodegenerative diseases and diabetes mellitus are usually regarded as typical ER stress-associated diseases or conformational diseases (14, 25, 42) because a neuron and a β cell live as long as the individual's life span and are therefore susceptible to the accumulation of misfolded proteins. In this regard, cardiomyocytes have the same life span and are exposed to harsh conditions, such as mechanical and oxidative stress. This study suggests that a cardiomyocyte may also be a target for ER stress. Whether a failing human heart shares this novel molecular mechanism caused by ER stress will be an interesting issue to explore.
Acknowledgments
We thank H. R. B. Pelham and M. R. Jackson for providing reagents. We also thank H. Gin and T. Toyozaki for technical support.
This work was supported by Grants-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan to T.A.
REFERENCES
- 1.Alvarez, C., R. Garcia-Mata, E. Brandon, and E. Sztul. 2003. COPI recruitment is modulated by a Rab1b-dependent mechanism. Mol. Biol. Cell 14:2116-2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoe, T., A. J. Lee, E. van Donselaar, P. J. Peters, and V. W. Hsu. 1998. Modulation of intracellular transport by transported proteins: insight from regulation of COPI-mediated transport. Proc. Natl. Acad. Sci. USA 95:1624-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bence, N. F., R. M. Sampat, and R. R. Kopito. 2001. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292:1552-1555. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin, I. J., and D. R. McMillan. 1998. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ. Res. 83:117-132. [DOI] [PubMed] [Google Scholar]
- 5.Bertolotti, A., X. Wang, I. Novoa, R. Jungreis, K. Schlessinger, J. H. Cho, A. B. West, and D. Ron. 2001. Increased sensitivity to dextran sodium sulfate colitis in IRE1beta- deficient mice. J. Clin. Investig. 107:585-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertolotti, A., Y. Zhang, L. M. Hendershot, H. P. Harding, and D. Ron. 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded- protein response. Nat. Cell Biol. 2:326-332. [DOI] [PubMed] [Google Scholar]
- 7.Bonifacino, J. S., and A. M. Weissman. 1998. Ubiquitin and the control of protein fate in the secretory and endocytic pathways. Annu. Rev. Cell Dev. Biol. 14:19-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brancaccio, M., L. Fratta, A. Notte, E. Hirsch, R. Poulet, S. Guazzone, M. De Acetis, C. Vecchione, G. Marino, F. Altruda, L. Silengo, G. Tarone, and G. Lembo. 2003. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 9:68-75. [DOI] [PubMed] [Google Scholar]
- 9.Brodsky, J. L., and A. A. McCracken. 1999. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 10:507-513. [DOI] [PubMed] [Google Scholar]
- 10.Ellgaard, L., and A. Helenius. 2003. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell. Biol. 4:181-191. [DOI] [PubMed] [Google Scholar]
- 11.Gaynor, E. C., and S. D. Emr. 1997. COPI-independent anterograde transport: cargo-selective ER to Golgi protein transport in yeast COPI mutants. J. Cell Biol. 136:789-802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammond, C., and A. Helenius. 1994. Quality control in the secretory pathway: retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 126:41-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harding, H. P., I. I. Novoa, Y. Zhang, H. Zeng, R. Wek, M. Schapira, and D. Ron. 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6:1099-1108. [DOI] [PubMed] [Google Scholar]
- 14.Harding, H. P., and D. Ron. 2002. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51(Suppl. 3):S455-S461. [DOI] [PubMed] [Google Scholar]
- 15.Harding, H. P., H. Zeng, Y. Zhang, R. Jungries, P. Chung, H. Plesken, D. D. Sabatini, and D. Ron. 2001. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell 7:1153-1163. [DOI] [PubMed] [Google Scholar]
- 16.Harding, H. P., Y. Zhang, and D. Ron. 1999. Protein translation and folding are coupled by an endoplasmic-reticulum- resident kinase. Nature 397:271-274. [DOI] [PubMed] [Google Scholar]
- 17.Haynes, C. M., S. Caldwell, and A. A. Cooper. 2002. An HRD/DER-independent ER quality control mechanism involves Rsp5p-dependent ubiquitination and ER-Golgi transport. J. Cell Biol. 158:91-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hochstenbach, F., V. David, S. Watkins, and M. B. Brenner. 1992. Endoplasmic reticulum resident protein of 90 kilodaltons associates with the T- and B-cell antigen receptors and major histocompatibility complex antigens during their assembly. Proc. Natl. Acad. Sci. USA 89:4734-4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang, W. Y., J. Aramburu, P. S. Douglas, and S. Izumo. 2000. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat. Med. 6:482-483. [DOI] [PubMed] [Google Scholar]
- 20.Imai, Y., M. Soda, H. Inoue, N. Hattori, Y. Mizuno, and R. Takahashi. 2001. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105:891-902. [DOI] [PubMed] [Google Scholar]
- 21.Jackson, M. R., T. Nilsson, and P. A. Peterson. 1993. Retrieval of transmembrane proteins to the endoplasmic reticulum. J. Cell Biol. 121:317-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang, P. M., and S. Izumo. 2003. Apoptosis in heart: basic mechanisms and implications in cardiovascular diseases. Trends Mol. Med. 9:177-182. [DOI] [PubMed] [Google Scholar]
- 23.Katayama, T., K. Imaizumi, N. Sato, K. Miyoshi, T. Kudo, J. Hitomi, T. Morihara, T. Yoneda, F. Gomi, Y. Mori, Y. Nakano, J. Takeda, T. Tsuda, Y. Itoyama, O. Murayama, A. Takashima, P. St George-Hyslop, M. Takeda, and M. Tohyama. 1999. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1:479-485. [DOI] [PubMed] [Google Scholar]
- 24.Kaufman, R. J. 2002. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 110:1389-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopito, R. R., and D. Ron. 2000. Conformational disease. Nat. Cell Biol. 2:E207-E209. [DOI] [PubMed] [Google Scholar]
- 26.Lee, K., W. Tirasophon, X. Shen, M. Michalak, R. Prywes, T. Okada, H. Yoshida, K. Mori, and R. J. Kaufman. 2002. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 16:452-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Letourneur, F., E. C. Gaynor, S. Hennecke, C. Demolliere, R. Duden, S. D. Emr, H. Riezman, and P. Cosson. 1994. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell 79:1199-1207. [DOI] [PubMed] [Google Scholar]
- 28.Lewis, M. J., and H. R. Pelham. 1990. A human homologue of the yeast HDEL receptor. Nature 348:162-163. [DOI] [PubMed] [Google Scholar]
- 29.Lewis, M. J., D. J. Sweet, and H. R. Pelham. 1990. The ERD2 gene determines the specificity of the luminal ER protein retention system. Cell 61:1359-1363. [DOI] [PubMed] [Google Scholar]
- 30.Li, C., and Q. Xu. 2000. Mechanical stress-initiated signal transductions in vascular smooth muscle cells. Cell Signal. 12:435-445. [DOI] [PubMed] [Google Scholar]
- 31.Liggett, S. B., N. M. Tepe, J. N. Lorenz, A. M. Canning, T. D. Jantz, S. Mitarai, A. Yatani, and G. W. Dorn 2nd. 2000. Early and delayed consequences of beta(2)-adrenergic receptor overexpression in mouse hearts: critical role for expression level. Circulation 101:1707-1714. [DOI] [PubMed] [Google Scholar]
- 32.Majoul, I., M. Straub, S. W. Hell, R. Duden, and H. D. Soling. 2001. KDEL-cargo regulates interactions between proteins involved in COPI vesicle traffic: measurements in living cells using FRET. Dev. Cell 1:139-153. [DOI] [PubMed] [Google Scholar]
- 33.Masaki, T., M. Yoshida, and S. Noguchi. 1999. Targeted disruption of CRE-binding factor TREB5 gene leads to cellular necrosis in cardiac myocytes at the embryonic stage. Biochem. Biophys. Res. Commun. 261:350-356. [DOI] [PubMed] [Google Scholar]
- 34.Massart, P. E., J. Donckier, J. Kyselovic, T. Godfraind, G. R. Heyndrickx, and M. Wibo. 1999. Carvedilol and lacidipine prevent cardiac hypertrophy and endothelin-1 gene overexpression after aortic banding. Hypertension 34:1197-1201. [DOI] [PubMed] [Google Scholar]
- 35.Mesaeli, N., K. Nakamura, E. Zvaritch, P. Dickie, E. Dziak, K. H. Krause, M. Opas, D. H. MacLennan, and M. Michalak. 1999. Calreticulin is essential for cardiac development. J. Cell Biol. 144:857-868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munro, S., and H. R. Pelham. 1987. A C-terminal signal prevents secretion of luminal ER proteins. Cell 48:899-907. [DOI] [PubMed] [Google Scholar]
- 37.Muslin, A. J. 2001. Road rage: cardiac Rab1 and ER-to-Golgi traffic. Circ. Res. 89:1087-1088. [PubMed] [Google Scholar]
- 38.Narula, J., N. Haider, R. Virmani, T. G. DiSalvo, F. D. Kolodgie, R. J. Hajjar, U. Schmidt, M. J. Semigran, G. W. Dec, and B. A. Khaw. 1996. Apoptosis in myocytes in end-stage heart failure. N. Engl. J. Med. 335:1182-1189. [DOI] [PubMed] [Google Scholar]
- 39.Nishitoh, H., A. Matsuzawa, K. Tobiume, K. Saegusa, K. Takeda, K. Inoue, S. Hori, A. Kakizuka, and H. Ichijo. 2002. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 16:1345-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Normington, K., K. Kohno, Y. Kozutsumi, M. J. Gething, and J. Sambrook. 1989. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell 57:1223-1236. [DOI] [PubMed] [Google Scholar]
- 41.Oda, Y., N. Hosokawa, I. Wada, and K. Nagata. 2003. EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299:1394-1397. [DOI] [PubMed] [Google Scholar]
- 42.Oyadomari, S., A. Koizumi, K. Takeda, T. Gotoh, S. Akira, E. Araki, and M. Mori. 2002. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 109:525-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patil, C., and P. Walter. 2001. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 13:349-355. [DOI] [PubMed] [Google Scholar]
- 44.Reimold, A. M., A. Etkin, I. Clauss, A. Perkins, D. S. Friend, J. Zhang, H. F. Horton, A. Scott, S. H. Orkin, M. C. Byrne, M. J. Grusby, and L. H. Glimcher. 2000. An essential role in liver development for transcription factor XBP-1. Genes Dev. 14:152-157. [PMC free article] [PubMed] [Google Scholar]
- 45.Ross, J., Jr. 2002. Dilated cardiomyopathy: concepts derived from gene deficient and transgenic animal models. Circ. J. 66:219-224. [DOI] [PubMed] [Google Scholar]
- 46.Rutkowski, D. T., and R. J. Kaufman. 2004. A trip to the ER: coping with stress. Trends Cell Biol. 14:20-28. [DOI] [PubMed] [Google Scholar]
- 47.Ruwhof, C., and A. van der Laarse. 2000. Mechanical stress-induced cardiac hypertrophy: mechanisms and signal transduction pathways. Cardiovasc. Res. 47:23-37. [DOI] [PubMed] [Google Scholar]
- 48.Sakai, K., K. Mitani, and J. Miyazaki. 1995. Efficient regulation of gene expression by adenovirus vector-mediated delivery of the CRE recombinase. Biochem. Biophys. Res. Commun. 217:393-401. [DOI] [PubMed] [Google Scholar]
- 49.Semenza, J. C., K. G. Hardwick, N. Dean, and H. R. Pelham. 1990. ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 61:1349-1357. [DOI] [PubMed] [Google Scholar]
- 50.Shen, J., X. Chen, L. Hendershot, and R. Prywes. 2002. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 3:99-111. [DOI] [PubMed] [Google Scholar]
- 51.Shi, Y., K. M. Vattem, R. Sood, J. An, J. Liang, L. Stramm, and R. C. Wek. 1998. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 18:7499-7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki, M., R. A. Li, T. Miki, H. Uemura, N. Sakamoto, Y. Ohmoto-Sekine, M. Tamagawa, T. Ogura, S. Seino, E. Marban, and H. Nakaya. 2001. Functional roles of cardiac and vascular ATP-sensitive potassium channels clarified by Kir6.2-knockout mice. Circ. Res. 88:570-577. [DOI] [PubMed] [Google Scholar]
- 53.Taxis, C., F. Vogel, and D. H. Wolf. 2002. ER-golgi traffic is a prerequisite for efficient ER degradation. Mol. Biol. Cell 13:1806-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Towbin, J. A., and N. E. Bowles. 2002. The failing heart. Nature 415:227-233. [DOI] [PubMed] [Google Scholar]
- 55.Townsley, F. M., D. W. Wilson, and H. R. Pelham. 1993. Mutational analysis of the human KDEL receptor: distinct structural requirements for Golgi retention, ligand binding and retrograde transport. EMBO J. 12:2821-2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Travers, K. J., C. K. Patil, L. Wodicka, D. J. Lockhart, J. S. Weissman, and P. Walter. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249-258. [DOI] [PubMed] [Google Scholar]
- 57.Urano, F., X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H. P. Harding, and D. Ron. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287:664-666. [DOI] [PubMed] [Google Scholar]
- 58.Vashist, S., W. Kim, W. J. Belden, E. D. Spear, C. Barlowe, and D. T. Ng. 2001. Distinct retrieval and retention mechanisms are required for the quality control of endoplasmic reticulum protein folding. J. Cell Biol. 155:355-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weekes, J., K. Morrison, A. Mullen, R. Wait, P. Barton, and M. J. Dunn. 2003. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics 3:208-216. [DOI] [PubMed] [Google Scholar]
- 60.Wiertz, E. J., D. Tortorella, M. Bogyo, J. Yu, W. Mothes, T. R. Jones, T. A. Rapoport, and H. L. Ploegh. 1996. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384:432-438. [DOI] [PubMed] [Google Scholar]
- 61.Wu, G., M. G. Yussman, T. J. Barrett, H. S. Hahn, H. Osinska, G. M. Hilliard, X. Wang, T. Toyokawa, A. Yatani, R. A. Lynch, J. Robbins, and B. S. Cain. 2001. Increased myocardial Rab GTPase expression: a consequence and cause of cardiomyopathy. Circ. Res. 89:1130-1137. [DOI] [PubMed] [Google Scholar]
- 62.Yamamoto, K., R. Fujii, Y. Toyofuku, T. Saito, H. Koseki, V. W. Hsu, and T. Aoe. 2001. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 20:3082-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yamamoto, K., H. Hamada, H. Shinkai, Y. Kohno, H. Koseki, and T. Aoe. 2003. The KDEL receptor modulates the endoplasmic reticulum stress response through mitogen-activated protein kinase signaling cascades. J. Biol. Chem. 278:34525-34532. [DOI] [PubMed] [Google Scholar]
- 64.Yao, A., O. Kohmoto, T. Oyama, Y. Sugishita, T. Shimizu, K. Harada, H. Matsui, I. Komuro, R. Nagai, H. Matsuo, T. Serizawa, T. Maruyama, and T. Takahashi. 2003. Characteristic effects of alpha1-beta1,2-adrenergic blocking agent, carvedilol, on [Ca2+]i in ventricular myocytes compared with those of timolol and atenolol. Circ. J. 67:83-90. [DOI] [PubMed] [Google Scholar]
- 65.Yoshida, H., T. Matsui, A. Yamamoto, T. Okada, and K. Mori. 2001. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107:881-891. [DOI] [PubMed] [Google Scholar]
- 66.Yussman, M. G., T. Toyokawa, A. Odley, R. A. Lynch, G. Wu, M. C. Colbert, B. J. Aronow, J. N. Lorenz, and G. W. Dorn 2nd. 2002. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat. Med. 8:725-730. [DOI] [PubMed] [Google Scholar]
- 67.Zinszner, H., M. Kuroda, X. Wang, N. Batchvarova, R. T. Lightfoot, H. Remotti, J. L. Stevens, and D. Ron. 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12:982-995. [DOI] [PMC free article] [PubMed] [Google Scholar]