

Significance
The increasing prevalence of multidrug-resistant strains of tuberculosis has created an urgent need for novel therapies to treat tuberculosis infections. Here we have demonstrated the successful utilization of the DNA-encoded X-Chem technology for the discovery inhibitors of Mycobacterium tuberculosis enoyl–acyl-carrier protein (ACP) reductase, InhA, a validated target for the treatment of tuberculosis. The identified inhibitors are cofactor specific and have activity in multiple cellular assays. Crystal structures of representative compounds from five chemical series revealed that the compounds bind adjacent to the NADH cofactor and adopt a variety of conformations, including two previously unreported binding modes. The compounds identified may serve as useful leads in the development of new antibacterial drugs with efficacy against multidrug-resistant tuberculosis.
Keywords: Mycobacterium tuberculosis, DNA-encoded X-Chem technology, DNA-encoded libraries, multidrug resistance, InhA
Abstract
Millions of individuals are infected with and die from tuberculosis (TB) each year, and multidrug-resistant (MDR) strains of TB are increasingly prevalent. As such, there is an urgent need to identify novel drugs to treat TB infections. Current frontline therapies include the drug isoniazid, which inhibits the essential NADH-dependent enoyl–acyl-carrier protein (ACP) reductase, InhA. To inhibit InhA, isoniazid must be activated by the catalase-peroxidase KatG. Isoniazid resistance is linked primarily to mutations in the katG gene. Discovery of InhA inhibitors that do not require KatG activation is crucial to combat MDR TB. Multiple discovery efforts have been made against InhA in recent years. Until recently, despite achieving high potency against the enzyme, these efforts have been thwarted by lack of cellular activity. We describe here the use of DNA-encoded X-Chem (DEX) screening, combined with selection of appropriate physical properties, to identify multiple classes of InhA inhibitors with cell-based activity. The utilization of DEX screening allowed the interrogation of very large compound libraries (1011 unique small molecules) against multiple forms of the InhA enzyme in a multiplexed format. Comparison of the enriched library members across various screening conditions allowed the identification of cofactor-specific inhibitors of InhA that do not require activation by KatG, many of which had bactericidal activity in cell-based assays.
Tuberculosis (TB) infects millions of people per year and contributes to the deaths of over 1.5 million annually. It is the second leading cause of death from infectious disease worldwide. In 2012, 8.6 million people fell ill with TB, and 1.3 million died from TB. More than 95% of TB deaths occur in developing countries, and it is among the top three causes of death for women aged 15–44 y. TB is a leading killer of people living with HIV, causing one quarter of all deaths in this population. The causative agent of TB, Mycobacterium tuberculosis (Mtb), has been increasingly observed to possess resistance to the frontline therapies rifampicin, and isoniazid commonly used to treat TB. For this reason, new therapeutic modalities to fight Mtb infection are desperately needed.
The enoyl-acyl-carrier protein (ACP) reductase, InhA, thought to be the primary target of the anti-Mtb drug isoniazid, catalyzes the NADH-dependent reduction of the 2-trans double bond of the lipid-modified ACP via an enoyl intermediate forming part of the fatty acid biosynthetic pathway essential for the formation of the outer membrane of Mtb (1, 2). Isoniazid is used as part of a combination therapy for the treatment of Mtb but is a prodrug that requires activation by KatG. Upon activation by KatG, isoniazid forms a covalent adduct with the cofactor NADH (Fig. 1). The isoniazid–NADH adduct acts an inhibitor of InhA by competing with NADH (Table 1) (3, 4). Many multidrug-resistant (MDR) TB strains exhibit resistance to isoniazid associated with mutations in at least five genes linked to isoniazid prodrug conversion, and the majority of those mutations are linked to defects in the katG gene and its upstream promoter (5–7). Direct inhibitors of InhA would provide TB drugs for the isoniazid-resistance strains without cross-resistance to isoniazid; however, until recently, discovery of InhA inhibitors with cellular activity has been challenging. The lack of bioactive compounds with cellular activity has thwarted efforts to develop InhA lead compounds with appropriate in vivo properties.
Fig. 1.

InhA inhibitors showing cellular activity in Mtb previously described in the literature. (1) Isoniazid adduct (23). (2) PT70 (24, 25). (3) Pyridomycin (26). (4) Methyl thiazole (15). (5) Pyrazole ELT hit (13). (6) Pyridine dione (27).
Table 1.
Biochemical and cellular activity of InhA inhibitors described in the literature (Fig. 1) that show cellular activity in Mycobacterium tuberculosis
| Compound | Reported affinity, nM | Cellular activity: MIC50, µM (H37Rv) | cLogP | LLE | Predominant InhA binding |
| 1 | 0.79* | 0.158 | −0.7† | 9.8 | Apo |
| 2 | 0.02‡ | 10.000 | 7.0 | 3.7 | NAD+ |
| 3 | 6309.57§ | 0.398 | 3.1 | 2.1 | Apo |
| 4 | 3.16§ | 0.199 | 2.3 | 6.2 | NADH |
| 5 | 3.98¶ | 0.501# | 1.4 | 7.0 | ND |
| 6 | 630.96¶ | 0.079 | 5.3 | 0.9 | NADH‖ |
cLogP, the calculated logarithms of water-octanol partition coefficients; LLE, log10 of reported affinity − cLogP; ND, not determined.
Ki measurement.
cLogP calculated for prodrug isoniazid rather than active drug INH-NAD adduct.
K1 measurement.
Kd measurement.
IC50.
MIC90.
A close analog, NITD-529, showed binding only to NADH form of InhA.
The successful use of DNA-encoded library technologies to discover novel chemical matter against a variety of target classes has been reported, including the discovery of novel inhibitors of InhA (8–14). DNA-encoded library technologies have provided the ability to interrogate large compound libraries (109–1011 unique small molecules) against a protein target in solution very rapidly and requires only microgram quantities of protein. Targets can be interrogated under multiple experimental conditions in parallel. These conditions could include different target concentrations, target bound to different cofactors/inhibitors, and off-targets, among others. Multiple conditions are included with the goal of identifying small molecules with desired mechanisms of action. Recently, we reported on inhibitors with activity in cellular assays that bind tightly to the InhA:NADH complex (Fig. 1 and Table 1) (15). Here we describe the use of the DNA-encoded X-Chem technology (DEX) for the discovery of both NADH- and NAD+-specific InhA inhibitors with activity in multiple cellular assays.
Results
InhA Selections.
A pool of 11 DNA-encoded libraries comprising more than 66 billion on-DNA compounds was used for selections against various forms of InhA including apo InhA, InhA:NAD+, and InhA:NADH. The chemical libraries were constructed using standard split-and-pool methodology with concomitant DNA encoding for synthetic steps (8, 16–18). InhA containing an N-terminal 6xHis tag was mixed with the library pool in the presence or absence of NADH or NAD+. Complexes of the target and bound small molecules then were captured on His-select nickel affinity resin via the protein affinity tag. The complexes then were washed to remove nonbinders, and the bound molecules were eluted by heat denaturation of the protein. This selection process was repeated using the eluted library members and fresh protein to refine the population of small molecule binders further. The final eluted fraction then was amplified and sequenced to determine the identities of the small molecule binders.
Eight individual selection conditions were run in parallel to identify library compounds with the desired mechanisms of action, in this case, binding selectively to the apo, NADH, or NAD+ complexes of InhA. The isoniazid-resistant mutant of InhA, S94A, was also included to aid in the identification of small molecules with activity against isoniazid-resistant TB. The selection conditions were as follows: no protein; 10 µM apo InhA; or 10 µM InhA plus saturating amounts of NADH, NAD+, or NADH in combination with a known NADH-dependent, tight binding InhA inhibitor (IC50 = 0.4 µM). A second tight-binding inhibitor with a known binding mode (IC50 = 0.8 µM) was conjugated to DNA and included in the library pool at a low concentration. Inclusion of the on-DNA positive control allowed validation of the selection conditions by assessment of enrichment by sequencing.
Analysis of the selection output resulted in four general profiles of enriched library members: (i) enriched only in the presence of apo InhA, (ii) enriched only in the presence of the InhA:NAD+ complex, (iii) enriched only in the presence of the InhA:NADH complex but not in the presence of the included tight-binding small molecule, and (iv) enriched in the presence of InhA:NAD+ and InhA:NADH but not in the presence of the included tight-binding small molecule. The on-DNA control compound was significantly enriched in the selections against InhA:NADH, including the S94A mutant of InhA, but was not enriched in the selections containing apo protein or InhA:NAD+. In the selection containing the WT InhA:NADH complex, the on-DNA control compound was enriched by a factor of 1,100-fold over two cycles of selection when normalized to a corresponding unfunctionalized oligonucleotide. As expected, the on-DNA control compound was competed away by the NADH-dependent small molecule which was included in selections. The majority (70%) of the enriched families (groups of structurally related compounds with the same profile) and, correspondingly, off-DNA compounds we chose to synthesize, fell into the third profile. In each of the profile classes, the majority of the families showed equivalent enrichment against both the WT and S94A mutant of InhA. Compounds that showed a significant reduction in enrichment against the S94A mutant relative to the WT protein were not synthesized. Representative compounds from highly enriched families were resynthesized without the DNA tag and tested for their activity in in vitro and cell-based assays. Approximately 50 representative small molecules across each of the four profiles were chosen for off-DNA synthesis based on potential mechanism of action, enrichment level, and their physicochemical properties.
Assay Results.
Off-DNA compounds were tested in in vitro assays which monitored the WT InhA-dependent conversion of NADH to NAD+ by reading the fluorescence at 420 nm. Two forms of the assay were used, one in which the protein was incubated in the presence of NADH (NADH-dependent) and another in which the protein was incubated with a mixture of NADH and NAD+ in which NAD+ was in excess (NAD+-dependent). Of the roughly 50 compounds synthesized, 25 had detectable activity with four having an IC50 in the 10- to 30-µM range, seven having an IC50 in the 1- to 10-µM range, and 14 having an IC50 less than 1 µM in one of the two assays. These compounds represented families observed in multiple DNA-encoded libraries.
Compounds with IC50 values less than 20 µM in either of the in vitro assays were sent for testing in the TB panel provided by Infectious Disease Research Institute (IDRI), which is part of the Microbiology and Infectious Disease Resources supported by the National Institute of Allergy and Infectious Disease (NIAID). This panel of assays includes determinations of minimum inhibitory concentration (MIC); IC50; the concentration that results in 90% inhibition of growth (IC90) against Mtb H37Rv under both aerobic and anaerobic conditions; Mtb H37Rv minimum bacterial concentration (MBC); IC50 and IC90 in an intracellular activity assay measuring the ability of compounds to kill Mtb-infected human cells; and MIC against a set of five drug-resistant strains of Mtb. Multiple chemical series were identified with an MIC less than 50 µM in the aerobic Mtb H37Rv assay. Although multiple representatives of the NADH- and NAD+-dependent profiles had cellular activity, no compound that was uniquely enriched against apo InhA and was active in one of the in vitro assays showed activity in the MIC panel.
Compounds with an MIC value less than 100 µM in the aerobic Mtb H37Rv assay were submitted for affinity determination in surface plasmon resonance (SPR) experiments under three conditions: binding against apo InhA, binding to InhA in the presence of NAD+, and binding to InhA in the presence of NADH. Notably, the NADH and NAD+ cofactor-dependent profiles observed in selection corresponded to the behavior observed in in vitro and biophysical assays. Compounds that were enriched only in the presence of NAD+ were significantly more potent in the NAD+-dependent assay (Table 2). Likewise, compounds with a preference for NADH- or NAD+-bound forms of InhA in selection showed increased affinities for their respective complexes in SPR experiments (Table 2). Structure–activity relationships and crystal structures of the most potent of the series with cell-based activity are described subsequently.
Table 2.
Comparison of selection profiles, in vitro assay potencies, and SPR-binding constants for InhA inhibitors
| Compound | Series | Profile class | InhA NAD+ assay IC50, µM | InhA NADH assay IC50, µM | InhA NAD+ SPR Kd, µM | InhA NADH SPR Kd, µM | InhA Apo SPR Kd, µM |
| 11 | 11 | 1 | 0.389 ± 0.007 | 0.168 (n = 1) | NT | NT | NT |
| 10a | 10 | 2 | 0.038 ± 0.006 | 0.198 ± 0.009 | 0.26 ± 0.12 | 5 ± 1.20 | 12.4 ± 1.41 |
| 12 | 12 | 2 | 0.682 ± 0.207 | 6.838 ± 0.175 | NT | NT | NT |
| 2a* | 1–3 | 3 | 0.060 ± 0.004 | 0.057 ± 0.006 | 46.7 ± 11.6 | 0.094 ± 0.06 | >100 |
| 13 | 13 | 3 | 0.791 ± 0.008 | 0.609 ± 0.114 | NT | NT | NT |
| 8a* | 8 and 9 | 3 | NT | 0.130 ± 0.006 | 49 ± 2.90 | 0.055 ± 0.03 | >100 |
| 6a* | 6 and 7 | 3 | NT | 5.917 ± 1.22 | >100 | 36.8 ± 3.25 | >100 |
| 4a* | 4 and 5 | 3 | NT | 0.297 ± 0.053 | >100 | 0.25 ± 0.11 | >100 |
| 1a* | 1–3 | 3 | NT | 0.065 ± 0.008 | 13.4 ± 4.30 | 0.34 ± 0.22 | >100 |
| 14 | 14 | 3 | NT | 5.568 ± 0.777 | >100 | 6.3 | >100 |
Profile classes: 1, enriched only in the presence of apo InhA; 2, enriched only in the presence of the InhA:NAD+ complex; 3, enriched only in the presence of the InhA:NADH complex but not in presence of small molecule. NT, not tested. Chemical structures for compounds 11–14 are given in Fig. S1.
WT InhA cocrystal structures reported herein.
Fig. S1.

Chemical structures of biochemically active compounds identified from InhA selections and referenced in Table 2.
Series 1–3.
Series 1–3 were identified from a capped dipeptide library containing 225 million on-DNA compounds. These series are defined by a conserved cyclohexyl core, a variety of benzyl or heterocyclic substituents at the position proximal to the methyl amide, which serves as the chemical handle for DNA attachment, and most often a benzyl triazole or methylbenzothiophene at the position distal to the methyl amide (Table 3). Crystal structures of the two parent compounds, compound 1a (IC50 = 0.065 µM) and compound 2a (IC50 = 0.057 µM), in complex with InhA:NADH were obtained (Fig. 2). These structures revealed that the amide linking the substituent at the hydrophobic pocket, known as “site II,” to the cyclohexyl core formed the warhead of the molecule binding into the catalytic site, site I, adjacent to the nicotinamide ring of NADH. In the substrate-bound state, site II is occupied by the lipophilic chain of ACP. The protein active site adopts a Tyr158-in conformation in which the carboxylic oxygen of the warhead forms a pair of bridging hydrogen bonds between the cofactor and side-chain oxygen atom of Tyr158. The cyclohexyl core forms packing interactions with the main-chain carbon atoms near Phe97 (site III). In both cocrystal structures, the methyl amide and the proximal benzyl substituents were disordered, and the solvent was exposed. Those substituents were modeled as occupying site II. The active site loop of InhA was also disordered in these structures. Multiple analogs were synthesized in an attempt to improve cell-based activity. The parent compounds along with their analogs were evaluated in the NADH-dependent in vitro assay, and compounds with an IC50 <1 µM were submitted for testing in the TB panel (Table 3). Compound 1a had an MIC of 13 µM under aerobic conditions and an IC50 of 4 µM in the intracellular activity assay. The intracellular activity did not appear to be caused by nonspecific cytotoxicity, as demonstrated by an MIC >50 µM in the cytotoxicity assay (Table S1). Compound 1c, a closely related analog of compound 3, demonstrated cellular activity comparable to that observed for the parent compound, and both compounds had MIC’s in the 12- to 31-µM range against a panel of isoniazid and rifampicin resistant strains (Table S2).
Table 3.
Series 1–3 structure–activity relationships
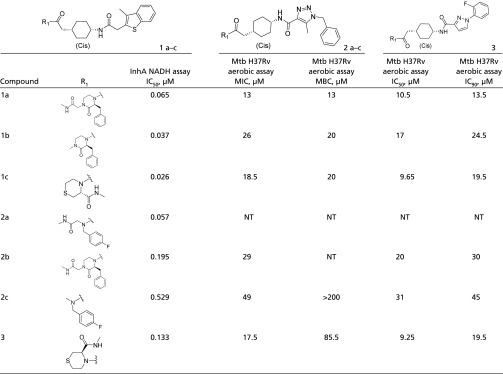 |
NT, not tested.
Fig. 2.

(A) Crystal structure of compound 1a (green carbon atoms) bound to InhA (yellow carbon atoms) (PDB ID code 5G0S). The inhibitor occupies three distinct subsites, which are depicted by colored ovals; red oval: catalytic site (site I); yellow oval: hydrophobic site (site II); green oval: hydrophilic site (site III). Selected residues are displayed as sticks. NADH is displayed with pink carbon atoms. The active site loop of InhA is displayed in orange. (B) Overlay of all five series [compounds 1a (green carbon atoms) (PDB ID code 5G0S); 2a (yellow) (PDB ID code 5G0T); 4a (orange) (PDB ID code 5G0U); 6a (cyan) (PDB ID code 5G0V); and 8a (red) (PDB ID code 5G0W)]. The proteins from the complexes were superimposed, and only the bound compounds and NADH from the complex of InhA with compound 1a are shown for clarity. The compounds occupy the three sites marked in A, and compounds 6a and 8a occupy an additional subsite above the adenine of NADH marked in cyan. A and B are shown in same orientation.
Table S1.
Compound 1a activity in THP-1 intracellular activity and cytotoxicity assays
| Compound | Series | THP-1 cell cytotoxicity IC50, µM | THP-1 intracellular activity IC50, µM | THP-1 intracellular activity IC90, µM |
| Compound 1a | 1–3 | >50 | 4.4 | 6.9 |
Table S2.
Compound 1c activity in THP-1 intracellular activity and cytotoxicity assays
| Compound | Series | INH-R1 IC50, µM | INH-R1 IC90, µM | INH-R1 MIC, µM | INH-R2 IC50, µM | INH-R2 IC90, µM | INH-R2 MIC, µM | RIF-R2 IC50, µM | RIF-R2 IC90, µM | RIF-R2 MIC, µM |
| Compound 1c | 1–3 | 11.9 | 27 | 24.5 | 12 | 22 | 24 | 12 | 22 | 24 |
Series 4 and 5.
Series 4 and 5 were identified from a pyridine core library with two points of diversity containing 1 million on-DNA compounds. The off-DNA parent compound, compound 4a, contains fluorophenoxybenzyl and piperidinyl pyridine substituents. The crystal structure of InhA:NADH in complex with compound 4a (IC50 = 0.310 µM; MIC = 25 µM) was obtained (Fig. 3) and revealed that the piperidinyl pyridine substituent formed the warhead of the molecule binding into site I and that the hydrophobic pocket at site II was occupied by the fluorophenoxybenzyl substituent, which forms hydrophobic packing interactions with residues in the active site loop including Ile202. The pyridine core formed the linker portion of the molecule, packing against the side chain of Phe97 in site III. The active site loop is ordered, and Tyr158 adopts the “out” conformation, rotated away from the nicotinamide ring of NADH and toward the side chains of Ile215 and Leu218. Most members of the series have bicyclic or biaryl substituents at both positions. Although most modifications of the parent compound led to a loss in potency, the replacement of the ether linkage of the fluorophenoxybenzyl group with an amine in compound 4b did lead to a 10-fold improvement in potency in the in vitro assay (IC50 = 0.026 µM), possibly because of the formation of an additional hydrogen-bonding interaction with the main-chain carbonyl oxygen atom of Ala198, and a threefold improvement in bactericidal activity (MIC = 7.9 µM) (Table 4).
Fig. 3.

X-ray crystal structure of InhA:NADH in complex with compound 4a (PDB ID code 5G0U) from series 4. Carbon atoms for compounds are shown in green. The protein backbone cartoon is represented in yellow. Selected atoms for the InhA side chains including F97, M98, and Ile202 in the active site loop are shown as sticks. NADH is shown as sticks with magenta carbon atoms. Refined (2fo-fc) electron density contoured at 1.0 σ is represented as a wire mesh. Some atoms of the active-site covering loop represented in orange have been removed for clarity.
Table 4.
Series 4 and 5 structure–activity relationships
 |
NA, not applicable; NT, not tested.
Series 6 and 7.
Series 6 and 7 were identified from a two-cycle library containing 3.8 million on-DNA compounds. The off-DNA compounds that comprise these series contain a tertiary amine core decorated with three substituents, typically the N-methylacetamide and two bicyclic groups. The parent compound of series 6 and 7, compound 6a (IC50 = 6 µM), has a piperidinyl pyridine moiety that, in the cocrystal structure with InhA:NADH, forms the warhead of the molecule binding adjacent to the nicotinamide ring of NADH (Fig. 4) in a fashion similar to that of the piperidinyl pyridine of compound 4a from series 4 and 5. The phenyl pyrazole of compound 6a appears to bypass the typical stacking interaction with Phe97 and instead reaches into a previously unobserved pocket defined by the side chains of Phe41 and Arg43. The active site loop is disordered, and the nearby hydrophobic pocket is unoccupied. In this crystal structure Tyr158 adopts the out conformation. Compound 6a has an MIC of 50 µM under aerobic conditions. Multiple analogs were synthesized in an attempt to improve cell-based activity; however, the majority of the changes made to the molecule reduced or eliminated activity. One analog, compound 7, had improved activity in the in vitro assay (IC50 = 0.5 µM) but was less active in the cell-based assay (MIC = 57 µM), potentially because of an increased cLogP (the calculated logarithms of water-octanol partition coefficients) (Table 5).
Fig. 4.

X-ray crystal structure of InhA:NADH in complex with compound 6a (PDB ID code 5G0V) from series 6. Carbon atoms for compounds are shown in green. The protein backbone cartoon is represented in yellow. Selected atoms for the InhA side chains including the catalytic residue Y158, F97, and M98 are shown as sticks. NADH is shown as sticks with magenta carbon atoms. Refined (2fo-fc) electron density contoured at 1.0 σ is represented as a wire mesh. Some atoms of the active-site covering loop represented in orange have been removed for clarity.
Table 5.
Series 6 and 7 structure–activity relationships
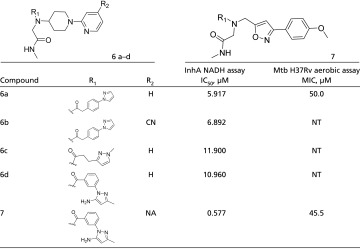 |
NA, not applicable; NT, not tested.
Series 8 and 9.
Series 8 and 9 were identified from the same library as series 1–3 described previously. These series are exemplified by compound 8a (IC50 = 0.130 µM), which has a conserved pyrrolidine core and three substituents: benzoyl and 1-t-Butoxy ethyl groups that are directly connected to the pyrrolidine core and a pyrazole benzaldehyde connected via the t-Butoxy ethyl. The compound 8a cocrystal structure showed the pyrazole group forming the warhead by binding into site I adjacent to the nicotinamide ring of NADH (Fig. 5). The t-Butoxy ethyl group forms packing interactions with the active site loop, which is ordered. Tyr158 adopts the out conformation. The benzoyl forms a previously unobserved π–π stacking interaction with Phe97. The methyl amide forms a pair of hydrogen bonds with the side chain of Arg43, also a previously unobserved interaction. When tested in the TB panel, compound 8a had an MIC of 19 µM (Table 6).
Fig. 5.

X-ray crystal structure of InhA:NADH in complex with compound 8a (PDB ID code 5G0W) from series 8. Carbon atoms for compounds are shown in green. The protein backbone cartoon is represented in yellow. Selected atoms for the InhA side chains including Phe41, Arg43, F97, and M98 are shown as sticks. NADH is shown as sticks with magenta carbon atoms. Refined (2fo-fc) electron density contoured at 1.0 σ is represented as a wire mesh. Some atoms of the active-site covering loop represented in orange have been removed for clarity.
Table 6.
Series 8 and 9 structure–activity relationships
 |
Me, methyl; NA, not applicable; NT, not tested.
Series 10.
Series 10 was identified from the same library as series 1–3 described previously. This series of compounds was based on compound 10a, which was identified from a family of on-DNA compounds uniquely enriched in the presence of NAD+. Compound 10a had an IC50 of 0.198 µM in the NADH-dependent assay, but the potency improved fivefold to 0.038 µM when tested in the NAD+-dependent assay, confirming the preference of the compound for the InhA:NAD+ complex (Table 2). SPR experiments further confirmed this preference. The Kd of compound 10a for the InhA:NADH complex is 5 µM, whereas the Kd for the InhA:NAD+ complex is 260 nM. The series 10 compounds contain a 4-phenylpiperidine-4-carboxylate core. Compound 10a has a pyrazole connected to the nitrogen atom of the piperidine ring and a trifluorobenzamide at the 4 position (Table 7). Repeated attempts to cocrystallize InhA:NADH and InhA:NAD+ in complex with compound 10a were unsuccessful. The NAD+-dependent compound 10a had an MIC of 12 µM under aerobic conditions.
Table 7.
Series 10 structure–activity relationships
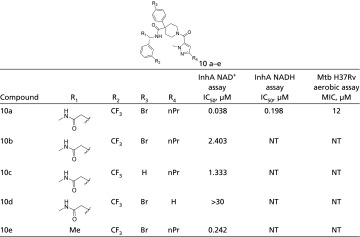 |
CF3, trifluoromethyl; Br, bromine; nPr, n-propyl; NT, not tested.
Discussion
A pool of 11 DNA-encoded libraries comprising more than 66 billion unique small molecules was interrogated against InhA in the apo-, NAD+-, NADH-, and NADH-inhibitor–bound forms. Comparison of the profiles of enriched library members across selections allowed the discovery of small molecules that inhibited the enzyme in a cofactor-specific manner as demonstrated in the in vitro and biophysical assays. Fourteen of the nearly 50 library members synthesized off-DNA had IC50 values of less than 1 µM in the in vitro assays (Table 2). Seven of the off-DNA compounds initially evaluated had MIC values less than 50 µM in the aerobic Mtb assay. Of those seven, three compounds—compound 1a from series 1–3, compound 8a from series 8 and 9, and compound 10a from series 10—had MICs of 12–13 µM.
The crystal structures obtained revealed a diversity of binding modes in which site I, adjacent to the nicotinamide ring of NADH, could be occupied by a variety of substituents: a triazole (series 1–3), a piperidinyl pyridine (series 4, 5, and 10), or a pyrazole (series 8 and 9). Compounds from series 1–3 and from series 4 and 5 formed the previously observed packing interactions with main chain atoms near Phe97. In three of the four crystal structures, Tyr158 adopted the out conformation, rotated away from the nicotinamide ring of NADH and toward the side chains of Ile215 and Leu218, under the active site loop. The parent compound of series 8 and 9 forms a unique π–π stacking interaction with the side chain of Phe97, whereas the series 6 and 7 compound bypassed Phe97 altogether, adopting an unusual binding mode in which the phenyl pyrazole binds into a previously unobserved pocket defined by the side chains of Phe41 and Arg43.
InhA was discovered to be the molecular target of isoniazid in 1996 (19). Since then ongoing efforts to develop novel InhA inhibitors as drugs for the treatment of TB have met with no success. Here, we have further demonstrated the successful application of DEX technology in identifying drug leads against a difficult protein target. Using the DEX technology, we have identified five unique, cofactor-specific chemical series with demonstrated activity in cell-based assays. Structure-guided drug design was combined with structure–affinity relationships revealed from selection in the design and synthesis of analogs from multiple chemical series that showed cell-based activity. The most broadly explored series were series 4 and 5 with 32 analogs made. These compounds showed a range of activities in the TB panel as described in Table 4. Analogs from series 4 and 5 made in an attempt to lower the molecular weight of the molecule (removal of methyl amide, cyclization, and replacement of phenyl rings with smaller isosteres) did not improve activity. Replacement of the ether linkage of the fluorophenoxybenzyl group with an amine did lead to a 10-fold improvement in potency in the in vitro assay and to a threefold improvement in the MIC. Although 12 analogs were made in series 6 and 7, only one resulted in improvement in the IC50, and that compound was less potent in the TB panel. The parent compound of the series 8 and 9 had an MIC of 12 µM. Five analogs in this series were synthesized, all of which were less potent than the parent in the in vitro assay, and none was evaluated in the TB panel. Series 10 was the only NAD+-dependent series explored. In contrast to previous reports of cell-based activity for NADH-dependent compounds (13), the parent compound of the series 10, compound 10a, had an MIC of 12 µM, equivalent to the two most potent compounds from NADH-dependent series reported herein, demonstrating that bactericidal compounds can bind to either reduced or oxidized cofactor-bound forms of InhA. This result suggests that further efforts to identify InhA inhibitors that bind specifically to the NAD+-bound form of the enzyme could yield bactericidal compounds.
Utilization of the DEX technology allowed the identification of multiple classes of Mtb InhA inhibitors, many of which had cell-based activity, directly from the primary affinity-based screen. Compounds were identified as cofactor-specific binders of Mtb InhA with direct target engagement further demonstrated by both in vitro activity assays and SPR experiments. Of the 50 initial compounds synthesized, 25 inhibited in vitro enzyme activity. Seven compounds with in vitro activity inhibited Mtb bacterial growth under aerobic conditions. Series 1–3 both inhibited bacterial growth in Mtb MIC assays and killed Mtb-infected THP-1 cells. Further efforts are needed to understand how these compounds enter bacteria and whether they are subject to efflux. This understanding would allow the development of a next generation of lead compounds that are effective therapeutics for the treatment of MDR Mtb infections.
Experimental Methods
Protein Expression/Purification.
Mtb InhA WT was cloned in the pET15b vector and expressed in the Escherichia coli BL21 Star (DE3) strain. The cell pellet was suspended in 50 mL lysis buffer [50 mM Tris⋅HCl (pH 8.0), 150 mM NaCl, 10 mM imidazole, 1 mM PMSF, 50 µg/mL lysozyme, and one tablet of EDTA-free protease inhibitor mixture], incubated on ice for 30 min, and sonicated. The cell lysate was centrifuged at 100,000 × g at 4 °C for 1 h, and the supernatant was collected. Qiagen Ni-NTA resin was equilibrated with five column volumes of lysis buffer, and the clarified lysate was passed through the column. The column was washed with five column volumes of wash buffer [50 mM Tris⋅HCl (pH 8.0), 150 mM NaCl, 50 mM imidazole] to 12.5% (vol/vol) of buffer B [50 mM Tris⋅HCl (pH 8.0), 150 mM NaCl, 400 mM imidazole]. Ni-NTA–bound proteins were eluted using buffer B [12.5 to 100% (vol/vol) B] over seven column volumes. Peak fractions were pooled and dialyzed into 30 mM Pipes (pH 6.8), 150 mM NaCl, 1 mM EDTA, and 10% (vol/vol) glycerol. Precipitation was seen in both pools after dialysis. Samples were centrifuged to remove precipitated protein, and supernatant was loaded onto the gel-filtration column. Peak fractions were pooled, concentrated, and evaluated for activity.
Expression and purification of Mtb inhA (S94A) were identical to that of the WT InhA with the exception of the lysis buffer, which was comprised of 50 mM Tris HCl (pH 8.0), 10 mM imidazole, 1 mM PMSF, 50 µg/mL lysozyme, and one tablet of EDTA-free protease inhibitor mixture.
Affinity-Mediated Selection of DNA-Encoded Libraries for InhA-Binding Small Molecules.
All reagents were acquired from Sigma-Aldrich unless otherwise noted. Eleven different DNA-encoded chemical libraries comprising in total ∼100 billion different encoded building-block combinations were combined in solution, and affinity-mediated selection for InhA binders was initiated by multiple incubations in 60 µL of a model cytosol incubation buffer containing Hepes (20 mM), potassium acetate (134 mM), sodium acetate (8 mM), sodium chloride (4 mM), magnesium acetate (0.8 mM), sheared salmon sperm DNA (1 mg/mL; Invitrogen), imidazole (5 mM), and Tween 20 [0.02% (vol/vol)] at pH 7.2. Different incubation samples contained different combinations of InhA (10 µM), NAD+ (5 mM), NADH (500 µM), and a known NADH-dependent InhA inhibitor (50 µM). Individual libraries were included in the library mixture at a concentration of 50 nM. The on-DNA positive control compound was included in each 60-µL selection at a final concentration of 1.67 pM. An unsubstituted headpiece was included as a negative control (subsequently ligated) at a concentration of 167 nM (10 pmol). After 1 h of incubation the mixture was flowed over a 5-µL bed of nickel affinity matrix (His-Select High-Flow Nickel Affinity Gel; Phynexus) with 20 passages followed by eight washes with 200-µL incubation buffer aliquots. Retained library members were eluted by incubation with 60 µL of incubation buffer at 85 °C for 5 min followed by a further incubation with a second 5-µL resin bed of His-Select High-Flow Nickel Affinity Gel to remove any eluted protein. This entire selection protocol was repeated with a fresh addition of InhA and cofactor where appropriate to half of the round-one eluate to regenerate a 10-µM InhA concentration. Encoding oligonucleotides present in the output of the second selection round were amplified using Platinum PCR Supermix (Invitrogen) with denaturation at 94 °C, annealing at 55 °C, and extension at 72 °C for 24 cycles using 5′- and 3′-oligonucleotides (each at 0.5 µM) that each incorporate sequences complementary to the tailpiece or headpiece along with Illumina READ1 or READ2 sequences required to support clustering and subsequent single-read 100-bp sequencing on an Illumina HiSeq 2500 system. Sequencing also was performed for PCR-amplified samples of the naive (unselected) library and the output of a no-target selection performed in the absence of InhA. From the combined samples, 398 million sequence reads were determined. Sequence data were converted back into encoded chemical information computationally, and demographic and statistical information was calculated for individual building-block combinations.
In Vitro Assays.
In the NADH-dependent assay, the 2-trans-dodecenoyl-CoA (DDCoA) substrate and NADH cofactor were prepared in the assay buffer [30 mM Pipes (pH 6.8), 50 mM NaCl, 0.005% (vol/vol) Brij detergent, 2 mM DTT, 0.1 mM EDTA]. InhA was diluted in a buffer comprised of 30 mM Pipes (pH 6.8), 150 mM NaCl, and 1 mM EDTA. Compounds were prepared at 90× the final concentration in 100% (vol/vol) DMSO and then were diluted to 3× in assay buffer. A 3× solution of InhA:NADH was prepared by further dilution of the enzyme in 3× NADH. Positive control wells contained DMSO in lieu of the compound. Negative control wells did not contain enzyme. The InhA:NADH was preincubated with the compound for 1 h at room temperature followed by the addition of 3× DDCoA. The final concentrations of the components were 10 nM InhA, 50 µM NADH, and 150 µM DDCoA. Upon the addition of DDCoA, the reaction was run for 30 min at 30 °C. End-point fluorescence (F) was read at excitation λ = 340 nM and emission λ = 420 nM. Compounds were tested at a top concentration of 30 µM serially titrating down threefold for a total of 11 doses. The percent inhibition of enzyme was calculated as 100*(1−(Fcompound well − Fneg control)/(Fpos control − Fneg control))
In the NAD+-dependent assay, the DDCoA substrate and stock solution of InhA were prepared as described for the NADH-dependent assay. A mixture of NADH and NAD+ cofactors were prepared in the assay buffer [30 mM Pipes (pH 6.8), 50 mM NaCl, 0.005% Brij, 2 mM DTT, and 0.1 mM EDTA]. Compounds were prepared as described for the NADH-dependent assay. A 3× solution of InhA was prepared by further dilution of the enzyme in 3× NADH:NAD+. Positive control wells contained DMSO in lieu of the compound. Negative control wells did not contain enzyme. The InhA:NADH:NAD+ mixture was preincubated with the compound for 1 h at room temperature followed by the addition of 3× DDCoA. Upon the addition of DDCoA, the reaction was run for 30 min at 30 °C. The final concentrations of the components were 10 nM InhA, 50 µM NADH, 2 mM NAD+, and 150 µM DDCoA. End-point fluorescence (F) was read at excitation λ = 340 nM and emission λ = 420 nM. The percent inhibition of enzyme was calculated as described for the NADH-dependent assay.
SPR Assay.
A BIAcore 4000 instrument (GE Healthcare) was used to monitor binding interactions using a direct binding assay format. Before activation, the research-grade CM5 chip surface was preconditioned using two 50-µL injections each of 10 mM HCl, 50 mM NaOH, 0.1% SDS, and 0.085% H3PO4, at a flow rate of 100 µL/min. Full-length InhA was immobilized on the sensor surface using standard amine coupling. Amine coupling was achieved by activating the sensor surface using 7-min injections of a mixture of 11.5 mg/mL N-hydroxysuccinimide with 75 mg/mL 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride. Protein immobilization was accomplished using a 10-min injection of full-length InhA (100 µg/mL) in 100 mM sodium acetate (pH 5.5) buffer. Remaining reactive esters were blocked using a 7-min injection of 100 mM Tris⋅HCl (pH 8.5) at a flow rate of 10 μL/min. Immobilization levels typically were around 5,000 resonance units. Reference flow cells were prepared without the protein. All binding measurements were performed in 50 mM Hepes (pH 7.5), 150 mM NaCl, 0.005% (vol/vol) T20, and 1% DMSO at a flow rate of 50 μL/min. Compound concentrations were injected over the active protein and reference surface with at least 90-s association and 600-s dissociation times. Solvent calibration and double-referencing subtractions were made to eliminate changes in the refractive index and injection noise using Biacore Evaluation software (GE Healthcare). Surface regeneration was achieved using dissociation for a time period, allowing the response to return to baseline. Control injections of a fixed, saturating compound concentration of 100 nM and buffer were interspersed with injections of compound to allow monitoring of the functionality of the immobilized protein surface. Compounds were tested at a top concentration of 100-µM threefold dilution series at nine concentrations. Compounds were injected either alone, in the presence of 2 mM NAD+, or in the presence of 100 µM NADH + 1 mM Tris(2-carboxyethyl)phosphine (TCEP). Excess cofactor was included in the compound injections to ensure that the protein remained saturated with cofactor. NADH injections (50 µM) were interspersed with compound injections to ensure that the chip surface had not degraded. SPR equilibrium binding data, consisting of Req values from eight-point concentration series, were analyzed by fitting a simple 1:1 binding model to yield Rmax and Kd values using Grafit (Erithacus software).
Crystallography.
Crystals of InhA with compounds were grown using the hanging-drop method at 293 K. The reservoir solution contained 12% (wt/vol) PEG 4000, 0.1 M N-(2-acetamido)iminodiacetic acid (ADA) (pH 6.8), 6 mM DMSO, 0.1 M ammonium acetate, 1% glycerol, and 4.5 mM NAD+. Drops were set up with 1.5 µL protein and 1 µL reservoir. Trays were incubated at 20 °C, and crystals appeared after 3–6 d. Crystals were transferred to a solution containing 5 mM compound and 12% (wt/vol) PEG 4000, 0.1 M ADA (pH 7.2), 6 mM DMSO, 0.1 M ammonium acetate, 1% glycerol, and 4.5 mM NADH and were incubated for 24 h. Crystals then were cryo-protected after increasing the concentrations of PEG 4000 to 15% (wt/vol) and glycerol to 20% (vol/vol). Crystals were frozen directly into a cryo-stream at 100 K.
Diffraction data were collected at Diamond beam-line I04-1 equipped with a Dectris Pilatus 6M X-ray detector, using a Si111 monochromatic wavelength of 0.92 Å. Data were processed using MOSFLM and AIMLESS and were reduced using CCP4 software (20). The structures were solved by molecular replacement using coordinates Mtb InhA in complex [Protein Data Bank (PDB) ID code 4D0R] as a trial model using CCP4 software. Protein and inhibitor were modeled into the electron density using COOT (21). The model was refined using BUSTER (22). Crystallographic statistics are reported in Table S3.
Table S3.
Crystallographic statistics
| PDB ID | 5G0S | 5G0T | 5G0U | 5G0V | 5G0W |
| Compound | 1a | 2a | 4a | 6a | 8a |
| Space group | P1211 | P1211 | P1211 | P1211 | P1211 |
| Cell constants: a, b, c, Å b | 65.3, 112.6, 68.1, 98.2 | 65.2, 112.1, 67.8, 98.4 | 65.5, 114.1, 68.5, 97.6 | 65.2, 111.7, 67.8, 98.3 | 65.4, 112.7, 68.0, 97.7 |
| Resolution range, Å | 57.9–1.74 (2.00–1.74) | 67.1–1.54 (1.68–1.54) | 67.9–1.73 (2.00–1.73) | 57.5–1.79 (2.07–1.79) | 57.8–1.79 (2.07–1.79) |
| Rmerge* | 0.053 (0.375) | 0.072 (0.560) | 0.061 (0.485) | 0.056 (0.338) | 0.077 (0.453) |
| Total observations | 338,423 (118,407) | 488,016 (116,316) | 348,798 (121,092) | 308,853 (112,305) | 318,898 (113,999) |
| Reflections, unique | 97,477 (34,069) | 128,781 (30,560) | 97,637 (33,822) | 88,619 (31264) | 90,585 (31,791) |
| I/SigI† | 15.4 (3.5) | 12.0 (2.6) | 13.9 (2.4) | 13.3 (3.4) | 11.5 (3.2) |
| CC1/2 | 0.99 (0.92) | 0.99 (0.78) | 0.99 (0.89) | 0.99 (0.90) | 0.98 (0.85) |
| Completeness, % | 98.4 (98.4) | 90.2 (89.8) | 94.4 (93.6) | 98.0 (99.0) | 98.6 (99.1) |
| Multiplicity | 3.5 (3.5) | 3.8 (3.8) | 3.6 (3.6) | 3.5 (3.6) | 3.5 (3.6) |
| Rvalue overall‡, % | 16.3 | 16 | 18.2 | 15.9 | 16.5 |
| Rvalue free§, %) | 18.6 | 18.2 | 19.7 | 18.8 | 19.5 |
| Nonhydrogen protein atoms | 7,623 | 7,473 | 7,660 | 7,501 | 7,643 |
| Nonhydrogen ligand atoms | 84 | 156 | 78 | 32 | 86 |
| Solvent molecules | 910 | 1072 | 897 | 836 | 974 |
| Rmsd from ideal values | |||||
| Bond lengths, Å | 0.01 | 0.01 | 0.01 | 0.01 | 0.01 |
| Bond angles, degrees | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Average B values, Å2 | |||||
| Protein main chain atoms | 26.3 | 17.9 | 30.1 | 24.4 | 25.3 |
| Protein all atoms | 27.8 | 19.3 | 32.0 | 26.2 | 26.8 |
| Ligand | 44.4 | 34.5 | 30.9 | 35.1 | 41.5 |
| Solvent | 40.3 | 34.8 | 39.8 | 40.0 | 39.1 |
| Φ, Ψ angle distribution for residues¶ | |||||
| In most favored regions,% | 91 | 91.2 | 91.8 | 91.2 | 92.2 |
| In additional allowed regions, % | 7.7 | 7.5 | 7 | 7.6 | 6.5 |
| In generously regions, % | 0 | 0 | 0.1 | 0.2 | 0 |
| In disallowed regions, % | 1.3 | 1.3 | 1.1 | 0.9 | 1.3 |
Rmerge = Σhkl [(Σi |Ii - ‹I›|)/ Σi Ii].
I/sigI avg is the mean I/sig for the unique reflections in the output file.
Rvalue = Σhkl ||Fobs| - |Fcalc||/Σhkl |Fobs|.
Rfree is the cross-validation R factor computed for the test set of 5% of unique reflections.
Ramachandran statistics as defined by PROCHECK.
IDRI TB Panel.
MIC under aerobic conditions.
The MICs of compounds were determined by measuring bacterial growth after 5 d in the presence of test compounds. Compounds were prepared as 10-point twofold serial dilutions in DMSO and were diluted into 7H9-Tw-OADC medium in 96-well plates with a final DMSO concentration of 2% (vol/vol). The highest concentration of compound was 200 µM where compounds were soluble in DMSO at 10 mM. For potent compounds, assays were repeated at lower starting concentrations. Each plate included assay controls for background (medium/DMSO only, no bacterial cells), zero growth (100 µM rifampicin), and maximum growth (DMSO only) as well as a rifampicin dose–response curve. Plates were inoculated with Mtb and incubated for 5 d; growth was measured by OD590 and fluorescence (excitation 560/emission 590) using a BioTek Synergy 4 plate reader. Growth was calculated separately for OD590 and relative fluorescence units (RFU). To calculate the MIC, the 10-point dose–response curve was plotted as percent of growth and was fitted to the Gompertz model using GraphPad Prism 5. The MIC was defined as the minimum concentration at which growth was completely inhibited and was calculated from the inflection point of the fitted curve to the lower asymptote (zero growth). In addition dose–response curves were generated using the Levenberg–Marquardt algorithm, and the IC50 and IC90 were determined.
MBC.
Mtb was grown aerobically to logarithmic phase and inoculated into liquid medium containing four different compound concentrations with a final maximum concentration of 2% (vol/vol) DMSO. For compounds with an MIC <20 µM (from task group 1 assay), the concentrations selected were 10× MIC, 5× MIC, 1× MIC, and 0.25× MIC. For compounds with MIC >20 µM, the highest concentration possible was tested (200, 100, 20, and 5 µM). Cultures were exposed to compounds for 21 d, and cell viability was measured by enumerating colony-forming units on agar plates on days 0, 7, 14, and 21. The MBC was defined as the minimum concentration required to achieve a 2-log kill in 21 d. For compounds with >1-log kill, an assessment of time- and/or concentration-dependence was determined from the kill kinetics. DMSO was used as a positive control for growth.
SI Experimental Methods
THP-1 Intracellular Activity Assay.
The activity of compounds against intracellular bacteria was determined by measuring viability in infected THP-1 cells after 3 d in the presence of test compounds. Compounds were prepared as 10-point threefold serial dilutions in DMSO. The highest concentration of compound tested was 50 µM for compounds that were soluble in DMSO at 10 mM.
THP-1 cells were cultured in complete RPMI and were differentiated into macrophage-like cells using 80 nM phorbol12-myristate13-acetate (PMA) overnight at 37 °C, 5% CO2. THP-1 cells were infected with a luminescent strain of H37Rv (which constitutively expresses luxABCDE) at a multiplicity of infection of 1 and were incubated overnight at 37 °C, 5% CO2. Infected cells were recovered using Accutase/EDTA solution, washed twice with PBS to remove extracellular bacteria, and seeded into assay pates. Compound dilutions were added to a final DMSO concentration of 0.5%. Assay plates were incubated for 72 h at 37 °C, 5% CO2. Each run included isoniazid as a control. RLU were measured using a BioTek Synergy 2 plate reader. The dose–response curve was fitted using the Levenberg–Marquardt algorithm. The IC50 and IC90 were defined as the compound concentrations that produced 50% and 90% inhibition of growth, respectively.
Two isoniazid-resistant strains (INH-R1 and INH-R2) and two rifampicin-resistant strains (RIF-R1 and RIF-R2) were tested. INH-R1 was derived from H37Rv and is a katG mutant (Y155* = truncation). INH-R2 is strain ATCC35822. RIF-R1 was derived from H37Rv and is an rpoB mutant (S522L). RIF-R2 is strain ATCC35828.
Cytotoxicity Assay.
The cytotoxicity of compounds was determined by measuring THP-1 cell viability after 3 d in the presence of test compounds. Compounds were prepared as 10-point threefold serial dilutions in DMSO. The highest concentration of compound tested was 50 µM for compounds that were soluble in DMSO at 10 mM. THP-1 cells were cultured in complete RPMI and were differentiated into macrophage-like cells using 80 nM PMA overnight at 37 °C, 5% CO2. Cells were inoculated into assay plates and were cultured for 24 h before compound dilutions were added to a final DMSO concentration of 0.5%. Each run included staurosporine as a control. Assay plates were incubated for 3 d at 37 °C, 5% CO2; growth was measured using the CellTiter-Glo Luminescent Cell Viability Assay (Promega), which uses ATP as an indicator of cell viability. RLU were measured using a BioTek Synergy 4 plate reader. The dose–response curve was fitted using the Levenberg–Marquardt algorithm. The IC50 was defined as the compound concentration that produced 50% inhibition of growth.
Acknowledgments
This work was supported by NIH National Institute of Allergy and Infectious Diseases Contract HHSN272201100009I.
Footnotes
Conflict of interest statement: H.H.S., P.C., M.A.C., J.W.C., M.-A.G., S.H., A.D.K., K.M.K., E.A.S., and Y.Z. are employees of X-Chem Pharmaceuticals. DNA-encoded X-Chem technology (DEX) is a proprietary drug discovery platform discovered and developed by employees of X-Chem. A.D.F., G.D., E.R.S., J.B., P.M., and J.A.R. are employees of AstraZeneca.
This article is a PNAS Direct Submission.
Data deposition: Crystallography, atomic coordinates, and structure factors reported in this paper have been deposited in the Protein Data Bank database (ID codes 5G0S, 5G0T, 5G0U, 5G0V, and 5G0W).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1610978113/-/DCSupplemental.
References
- 1.Banerjee A, et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263(5144):227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 2.Dessen A, Quémard A, Blanchard JS, Jacobs WR, Jr, Sacchettini JC. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science. 1995;267(5204):1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 3.Rozwarski DA, Grant GA, Barton DHR, Jacobs WR, Jr, Sacchettini JC. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279(5347):98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 4.Chollet A, et al. Crystal structure of the enoyl-ACP reductase of Mycobacterium tuberculosis (InhA) in the apo-form and in complex with the active metabolite of isoniazid pre-formed by a biomimetic approach. J Struct Biol. 2015;190(3):328–337. doi: 10.1016/j.jsb.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder EK, de Souza N, Santos DS, Blanchard JS, Basso LA. Drugs that inhibit mycolic acid biosynthesis in Mycobacterium tuberculosis. Curr Pharm Biotechnol. 2002;3(3):197–225. doi: 10.2174/1389201023378328. [DOI] [PubMed] [Google Scholar]
- 6.Basso LA, Santos DS. Drugs that inhibit mycolic acid biosynthesis in Mycobacterium tuberculosis an update. Med Chem Res. 2005;2:393–413. doi: 10.2174/1389201023378328. [DOI] [PubMed] [Google Scholar]
- 7.Hazbón MH, et al. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2006;50(8):2640–2649. doi: 10.1128/AAC.00112-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark MA, et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat Chem Biol. 2009;5(9):647–654. doi: 10.1038/nchembio.211. [DOI] [PubMed] [Google Scholar]
- 9.Gilmartin AG, et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat Chem Biol. 2014;10(3):181–187. doi: 10.1038/nchembio.1427. [DOI] [PubMed] [Google Scholar]
- 10.Deng H, et al. Discovery of highly potent and selective small molecule ADAMTS-5 inhibitors that inhibit human cartilage degradation via encoded library technology (ELT) J Med Chem. 2012;55(16):7061–7079. doi: 10.1021/jm300449x. [DOI] [PubMed] [Google Scholar]
- 11.Ding Y, et al. Discovery of Potent and Selective Inhibitors for ADAMTS-4 through DNA-Encoded Library Technology (ELT) ACS Med Chem Lett. 2015;6(8):888–893. doi: 10.1021/acsmedchemlett.5b00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kollmann CS, et al. Application of encoded library technology (ELT) to a protein-protein interaction target: Discovery of a potent class of integrin lymphocyte function-associated antigen 1 (LFA-1) antagonists. Bioorg Med Chem. 2014;22(7):2353–2365. doi: 10.1016/j.bmc.2014.01.050. [DOI] [PubMed] [Google Scholar]
- 13.Encinas L, et al. Encoded library technology as a source of hits for the discovery and lead optimization of a potent and selective class of bactericidal direct inhibitors of Mycobacterium tuberculosis InhA. J Med Chem. 2014;57(4):1276–1288. doi: 10.1021/jm401326j. [DOI] [PubMed] [Google Scholar]
- 14.Yang H, et al. Discovery of a Potent Class of PI3Kα Inhibitors with Unique Binding Mode via Encoded Library Technology (ELT) ACS Med Chem Lett. 2015;6(5):531–536. doi: 10.1021/acsmedchemlett.5b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shirude PS, et al. Methyl-thiazoles: A novel mode of inhibition with the potential to develop novel inhibitors targeting InhA in Mycobacterium tuberculosis. J Med Chem. 2013;56(21):8533–8542. doi: 10.1021/jm4012033. [DOI] [PubMed] [Google Scholar]
- 16.Litovchick A, et al. encoded library synthesis using chemical ligation and the discovery of sEH inhibitors from a 334-million member library. Sci Rep. 2015;5:10916. doi: 10.1038/srep10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keefe AD, Clark MA, Hupp CD, Litovchick A, Zhang Y. Chemical ligation methods for the tagging of DNA-encoded chemical libraries. Curr Opin Chem Biol. 2015;26:80–88. doi: 10.1016/j.cbpa.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 18.Goodnow RA., Jr . A Handbook for DNA-Encoded Chemistry: Theory and Applications for Exploring Chemical Space and Drug Discovery. John Wiley & Sons; New York: 2014. [Google Scholar]
- 19.Wheeler PR, Anderson PM. Determination of the primary target for isoniazid in mycobacterial mycolic acid biosynthesis with Mycobacterium aurum A+ Biochem J. 1996;318(Pt 2):451–457. doi: 10.1042/bj3180451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Bricogne G, et al. BUSTER version 2.11.5. Global Phasing Ltd.; Cambridge, UK: 2011. [Google Scholar]
- 23.Rawat R, Whitty A, Tonge PJ. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: Adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100(24):13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luckner SR, Liu N, am Ende CW, Tonge PJ, Kisker C. A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J Biol Chem. 2010;285(19):14330–14337. doi: 10.1074/jbc.M109.090373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan P, Tonge PJ. Targeting InhA, the FASII enoyl-ACP reductase: SAR studies on novel inhibitor scaffolds. Curr Top Med Chem. 2012;12(7):672–693. doi: 10.2174/156802612799984535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartkoorn RC, et al. Pyridomycin bridges the NADH- and substrate-binding pockets of the enoyl reductase InhA. Nat Chem Biol. 2014;10(2):96–98. doi: 10.1038/nchembio.1405. [DOI] [PubMed] [Google Scholar]
- 27.Manjunatha UH, et al. Direct inhibitors of InhA are active against Mycobacterium tuberculosis. Sci Transl Med. 2015;7:269ra3. doi: 10.1126/scitranslmed.3010597. [DOI] [PMC free article] [PubMed] [Google Scholar]