

Abstract
The aberrant activation of B-cells has been implicated in several types of cancers and hematological disorders. BTK and PI3Kδ are kinases responsible for B-cell signal transduction, and inhibitors of these enzymes have demonstrated clinical benefit in certain types of lymphoma. Simultaneous inhibition of these pathways could result in more robust responses or overcome resistance as observed in single agent use. We report a series of novel compounds that have low nanomolar potency against both BTK and PI3Kδ as well as acceptable PK properties that could be useful in the development of treatments against B-cell related diseases.
Keywords: BTK, PI3K, p110δ, inhibitor, B-cell, BCR, pyrazolopyrimidine
B-cell receptor (BCR) signaling is essential for normal B-cell development, proliferation, and survival.1 Constitutive activation of the signaling pathway downstream of BCR is a hallmark of B-cell malignancies such as chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL), follicular B-cell non-Hodgkin lymphoma (FL), small lymphocytic lymphoma (SLL), and diffuse large B-cell lymphoma (DLBCL).2,3 Inhibition of BCR signaling is used clinically to treat B-cell malignancies, and there are several intervention points in the downstream signaling pathway that are clinically relevant including LYN, SYK, BTK, and PI3Kδ.3,4
Bruton’s tyrosine kinase (BTK) is a member of the TEC family of nonreceptor tyrosine kinases and is an upstream activator of multiple antiapoptotic signaling molecules and pathways, which include phosphoinositide 3-kinase (PI3K). PI3K constitutes a family of lipid kinases, which, like BTK, are involved in cellular signaling and control a broad number of cellular processes. The PI3K family comprises four isoforms, α, β, γ, and δ that catalyze the phosphorylation of phosphatidylinositol-4,5-biphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), a key signaling molecule that impacts cellular growth, proliferation, chemotaxis, differentiation, and survival.
The approval of ibrutinib5 (formerly PCI-32765) and idelalisib6 (formerly GS-1101) by the US FDA between 2013 and 2014 demonstrated the therapeutic benefit of inhibiting BTK (ibrutinib) and PI3Kδ (idelalisib) for the treatment of certain B-cell malignancies such as CLL and MCL. However, de Rooij et al. reported that roughly one-third of patients receiving ibrutinib for CLL and MCL show primary resistance.7−9 Moreover, it is expected that a percentage of responsive patients who receive prolonged treatment with ibrutinib will develop resistance due to mutations of BTK and phospholipase Cγ2 (PLCγ2), the kinase immediately downstream of BTK.10,11 In contrast to ibrutinib, mechanisms for resistance to idelalisib are not as well understood.12 Although both kinases regulate BCR signaling, they do so via parallel pathways,13 making combination therapy, either by codosing or with a dual agent, an attractive strategy for potentially achieving deeper durable responses as well as preventing resistance. Herein we report the discovery of reversible dual inhibitors of BTK and PI3Kδ for the potential treatment of B-cell malignancies.
In 2010, the Shokat group at UCSF reported a series of PI3Kδ/γ dual inhibitors, the most potent being SW13 (PI3Kδ IC50 = 0.7 nM; PI3Kγ IC50 = 33 nM).14 Recognizing the structural similarities between PCI-29732,15 a noncovalent analogue of ibrutinib (1, BTK IC50 = 8.2 nM), and SW13 (2), Figure 1, we speculated that appropriately designed molecules could maintain reasonable affinity for PI3Kδ while also demonstrating affinity for BTK.
Figure 1.
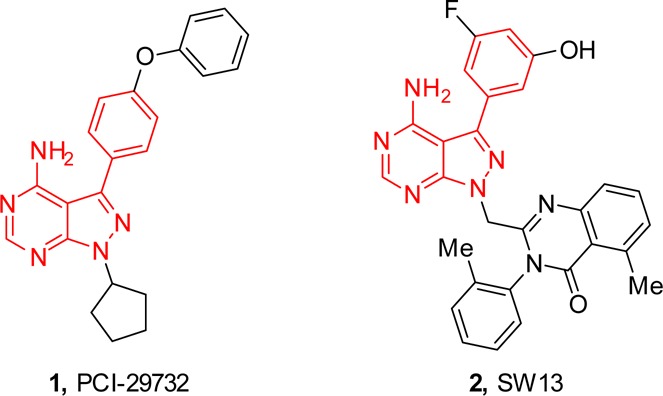
Structural similarities between PCI-29732 and SW13.
Toward that end, PCI-29732 was docked (Figure 2A) and SW13 was overlaid (Figure 2B) onto the BTK binding pocket of structure 3GEN,16 which has a pyrrolopyrimidine-containing ligand that is otherwise identical to PCI-29732. As expected, the docked pose of PCI-29732 makes the same important interactions as the cocrystallized ligand, namely, two hydrogen bonds with each hinge residue, Glu475 and Met477, as well as a pi–pi interaction with Phe540. The pose for SW13 was achieved by superimposing the pyrazolopyrimidine of SW13 on to the pyrrolopyrimidine of the 3GEN ligand The cocrystallized ligand was then removed for ease of viewing. This perspective view allowed us to quickly assess that the large tolyl-quinazolinone of SW13 would need to be modified to something smaller that would fit in this part of the BTK pocket. Although the propeller-like conformation is favorable for selective binding to PI3Kδ,17 we decided to address selectivity within the PI3K family at a later time. With this in mind, our initial analogues were designed with the methylene group removed to obviate the propeller-like bend, and the bicyclic-group attached to N-1 of the pyrazolopyrimidine was simplified.
Figure 2.
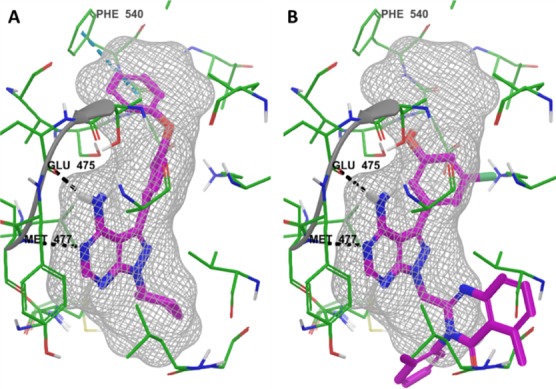
Docking of PCI-29732 (A) and an overlay of SW13 (B) to BTK (3GEN) using Glide. The bound ligand is removed for ease of viewing.
The analogues were prepared following the route in Scheme 1. Starting from iodide 3,18 a Suzuki coupling19 with boronic acid 4 afforded pyrazolopyrimidine 5, which was subsequently modified with several mesylates. Treatment of compounds 6–8 with BBr3 in CH2Cl2 cleaved the methyl-groups to afford the desired compounds 9–11.
Scheme 1.

Reagents and conditions: (a) 3-fluoro-5-methoxyphenylboronic acid (4), Na2CO3, Pd(PPh3)4, DMF, H2O, 75 °C, 2 h, 32%; (b) i. NaH, DMF, 0 °C-RT, ii. R-OMs, 80 °C, 2 h, 5–50%; (c) 1 M BBr3 in CH2Cl2, 0 °C, 18 h, 3–50%.
To our satisfaction, 9–11 not only retained PI3Kδ activity but also gained activity against BTK, Table 1. In our assay, SW13 (2) demonstrated subnanomolar inhibition of PI3Kδ, which was in agreement with the value reported by Shokat, and no inhibition of BTK. Replacing the methylene-linked quinazolinone group of SW13 with a 2-indanyl in 9 increased BTK activity to 67 nM while maintaining single-digit nanomolar activity against PI3Kδ. Although not as dramatic, similar results were observed for 10 and 11. Interestingly, the corresponding methoxy-containing analogues 6–8 had significantly attenuated activity against both kinases compared to 9–11, signifying that either the phenolic group was making important interactions with both targets or the methoxy group is sterically encumbered.
Table 1. N-1 Pyrazolopyrimidine Modifications to Give Compounds 6–11a.
| No. | R | BTK (IC50, nM) | PI3Kδ (IC50, nM) |
|---|---|---|---|
| 2 | NA | >10000 | <0.5 |
| 6 | 2-indanyl | 1970 | 2790 |
| 7 | 1-indanyl | 584 | 1830 |
| 8 | 2-tetralinyl | 891 | 1340 |
| 9 | 2-indanyl | 67 | 8 |
| 10 | 1-indanyl | 217 | 13 |
| 11 | 2-tetralinyl | 86 | 9 |
Compounds 7, 8, 10, and 11 were tested as racemates.
Having identified a scaffold that provided dual activity against both BTK and PI3Kδ, we next turned our attention toward evaluating the C-3 aryl group. As outlined in Scheme 2, C-3 modifications were prepared by alkylating pyrazolopyrimidine 3 with indan-2-yl methanesulfonate (12). Suzuki coupling of the resulting 3-iodopyrazolopyrimidine, 13, with various boronic acids provided compounds 14–21.
Scheme 2.

Reagents and conditions: (a) i. NaH, DMF, 0 °C-RT, ii. 12, 80 °C, 2 h, 51%; (b) Ar–B(OR)2, Na2CO3, Pd(PPh3)4, DMF, H2O, 75 °C, 2–3 h, 30–70%.
Interestingly, removal of the fluoro-substituent had little effect on BTK affinity but attenuated PI3Kδ activity as demonstrated by comparing 9 and 14, Table 2. Replacing the fluoro-substituent with a chloro-substituent (15) maintained moderate BTK activity while low nanomolar activity against PI3Kδ was restored, suggesting that substitution at the meta-position may be important for proper binding to PI3Kδ. This was further demonstrated when the halo-substituent was moved to the para-position as in 16–17. In these examples, the PI3Kδ IC50 increased roughly 8-fold, compared to their corresponding meta-substituted variants. To our surprise, the BTK IC50 decreased nearly 10-fold in the same examples. These were our first analogues where the potency against BTK was greater than PI3Kδ. Increasing the size of the para-substituent to bromo (18) maintained BTK activity while enhancing PI3Kδ activity.
Table 2. C-3 Pyrazolopyrimidine Modifications to Give Compounds 14–21.
| No. | Ar | BTK (IC50, nM) | PI3Kδ (IC50, nM) |
|---|---|---|---|
| 14 | 3-OH-Ph | NDa | NDb |
| 15 | 3-Cl-5-OH-Ph | 142 | 4 |
| 16 | 4-F-5-OH-Ph | 7 | 61 |
| 17 | 4-Cl-5-OH-Ph | 14 | 33 |
| 18 | 4-Br-5-OH-Ph | 12 | 8 |
| 19 | 3-OH-4-Me-Ph | 43 | 172 |
| 20 | 3-OH-4-OMe-Ph | 84 | 32 |
| 21 | 3,4-diF-5-OH-Ph | 27 | 3 |
IC50 not determined; inhibition of BTK was 66% at 100 nM 14.
IC50 not determined; inhibition of PI3Kδ was 41% at 100 nM 14.
At this point, compound 18 was the most potent dual BTK, PI3Kδ inhibitor we had made thus far. Replacing the para-bromo group with a methyl or methoxy group resulted in attenuated activity against both targets as observed in 19 and 20, respectively. This was somewhat surprising for BTK as we predicted that a pocket that could accommodate the phenyl ether of PCI-29732 and the bromo group of 18 would be more than large enough to accept moderately sized substituents such as methyl or methoxy. This suggests that electronic effects on the aryl-ring may also contribute given that electron donating groups such as the methoxy group in 20 appear to be weaker BTK binders than the corresponding electron withdrawing groups. Furthermore, in the case of 20, the disruption of an internal hydrogen bond between the hydroxy and methoxy groups would also decrease binding affinity. Substituting both the meta- and para-positions as in 21 affords a compound with good BTK activity and excellent activity against PI3Kδ. Inspired by the in vitro potency, we quickly sought to evaluate our compounds in vivo. Selected compounds from our initial hits were administered to mice and their pharmacokinetic (PK) properties are summarized in Table 3.
Table 3. In Vivo Mouse Pharmacokinetic Propertiesa of Initial Hits.
| No. | CL (L/h/kg) | Vz (L/kg) | AUCPO (μM·h) | PO t1/2 (h) | F (%) |
|---|---|---|---|---|---|
| 9b | 5.10 | 6.01 | 0.177 | 3.1 | 2.2 |
| 15b | 9.36 | 11.5 | 0.033 | 0.60 | 0.8 |
| 16b | 7.29 | 5.45 | 0.211 | 0.80 | 3.7 |
| 18c | 3.82 | 5.90 | 0.213 | 2.3 | 6.9 |
| 20b | 1.87 | 6.56 | 2.37 | 6.3 | 11 |
| 21b | 9.25 | 12.1 | 0.073 | 0.95 | 1.7 |
WinNonlin noncompartmental analysis.
Dosed 5 mg/kg IV and 15 mg/kg PO.
Dosed 1 mg/kg IV and 5 mg/kg PO.
Unfortunately, all of our initial compounds performed poorly in mouse PK studies. The results suggested that the compounds generally suffered from high clearance and poor bioavailability. Interestingly, 20 had markedly lower clearance and a longer oral half-life compared to the other examples. Appreciating that phenolic groups could have the propensity to form O-linked conjugates in vivo, we speculated whether the hydroxyl was a primary contributor to the high clearance. The results for 18 and 20 would appear to corroborate this as the relative size of the adjacent group could decrease the relative rate of bioconjugation. In an effort to elucidate the factors that contributed to our in vivo PK results, we tested the compounds in a series of in vitro metabolism studies for oxidation and glucuronidation stability, Table 4.
Table 4. In Vitro Metabolism Results of Initial Hitsa.
| No. | mLM (%R) | mUGT (%R) | hLM (%R) | hUGT (%R) |
|---|---|---|---|---|
| 9 | 25 | <1 | 67 | 1.3 |
| 18 | 53 | 13 | 66 | 5.2 |
| 20 | 14 | 12 | 52 | 7.0 |
| 21 | 78 | 5.1 | 75 | 3.7 |
Percent parent remaining (%R) after 30 min.
The in vitro metabolism assays were performed with either mouse or human liver microsomes, which were supplemented with either NADPH to assess oxidative stability (mLM or hLM) or UDP-glucuronic acid to assess glucuronidation transferase stability (mUGT or hUGT). The results demonstrate high turnover of substrate and identify glucuronidation as a clearance pathway in vitro for compounds 9, 18, 20, and 21. Moreover, in mice, glucuronidation appeared to be the primary clearance pathway for compounds 9, 18, and 21 given that the levels of parent remaining were significantly higher under NADPH conditions compared to when UDP-glucuronic acid was added. In human liver microsomes, phase II metabolism appeared to be more significant than oxidative metabolism for all compounds tested.
Armed with this information we set out to improve on our initial hits with a directed effort to replace the problematic phenolic group. Following Scheme 2, we prepared a series of analogues in which the hydroxy-group was replaced with either a fluoro- or amino-group, Table 5. In comparison to 21, trifluorophenyl-containing analogue, 22, had significantly decreased inhibition of both enzymes; however, stability toward bioconjugation was significantly increased. This reaffirms the importance of the phenol with respect to BTK and PI3Kδ affinity, while demonstrating its liability to PK properties. Compounds 23–25 had substituted anilines in place of the phenol. These compounds showed improvement in BTK inhibition but continued to lack significant activity against PI3Kδ. When reviewing our initial SAR, we noticed that 20 had over a 5-fold improvement in PI3Kδ IC50 compared to 19. This suggested to us that the methoxy-group was able to adopt a favorable conformation when binding to PI3Kδ. We prepared cyclic amino-ether 26 in an effort to tie-back the methylene group and preclude bond rotation. Gratifyingly, we observed nearly a 3-fold improvement in PI3Kδ inhibition with a moderate decrease in BTK inhibition and excellent stability toward UGT activity. Adding a meta-fluoro-substituent afforded 27, which further enhanced the PI3Kδ inhibition while maintaining BTK inhibition and UGT stability. Compound 27 was our first nonphenol containing analogue that achieved dual-inhibition at or below IC50 = 100 nM but more importantly demonstrated low levels of glucuronidation in our in vitro mouse UGT assay. The indole-containing analogue 28 had reduced PI3Kδ affinity while the benzimidazole containing analogue 29 lost nearly all activity against BTK compared to 27.
Table 5. C-3 Pyrazolopyrimidine Optimizations.

Kinase percent inhibition at 100 nM test article.
Percent (%R) parent remaining after 30 min.
Percent (%R) not determined.
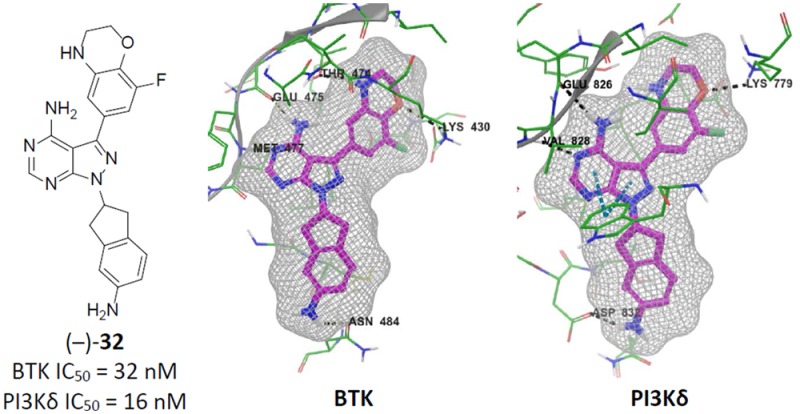
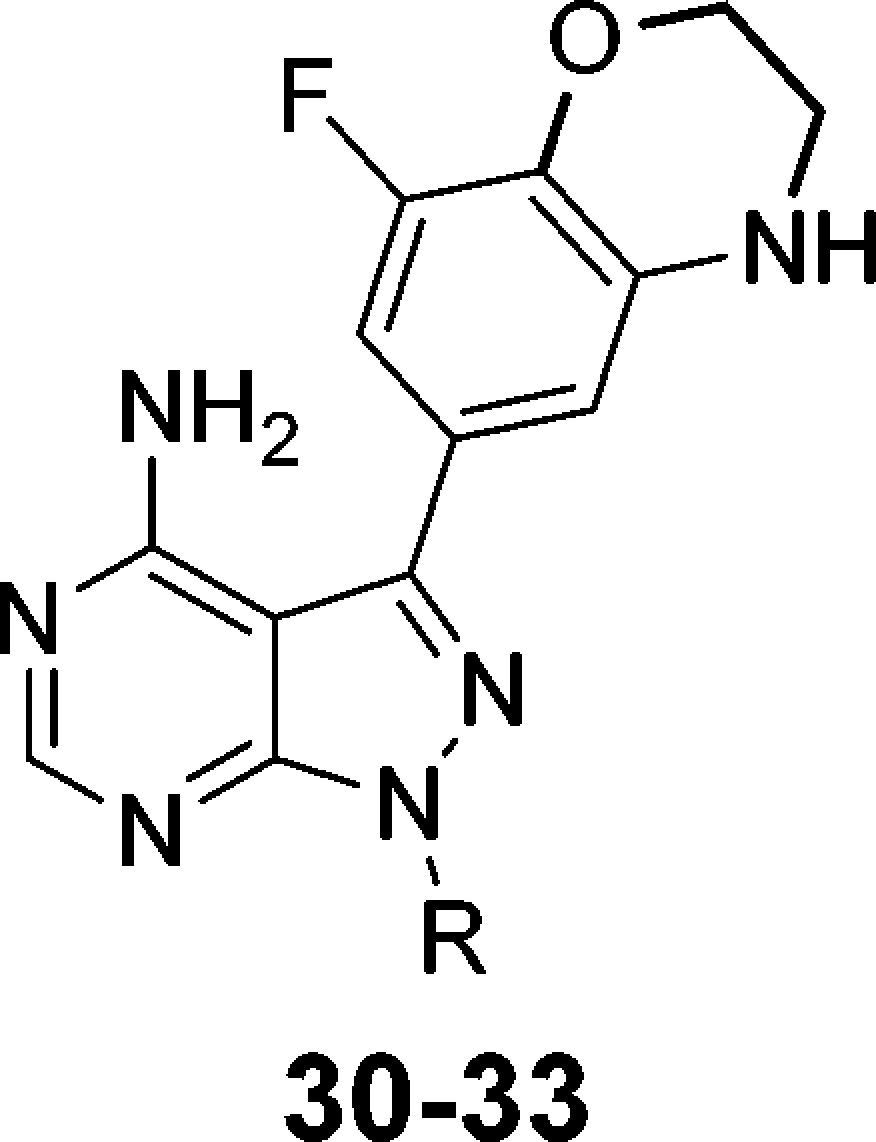
Having identified a potential substitute for the phenol, we next turned our attention toward improving the biochemical affinity against both BTK and PI3Kδ. In a similar manner to Scheme 1, we prepared optimized compounds 30–33, Table 6. Substituting the indane of 27 with a tetralin affords (±)-30. Compared to 27, the larger ring had little effect on PI3Kδ binding but diminished affinity for BTK. Adding a fluoro-substituent on the aryl portion of the indane reduced affinity for both enzymes even further as observed in compound (±)-31. Amino-substitutions such as in compounds 32 and 33 also had little effect on PI3Kδ binding but now provided improved BTK results compared to the unsubstituted analogues 27 and 30, respectively. Comparing docked poses of 27 and 32 suggested that the improvements in BTK binding were the result of an additional H-bond interaction with Asn484, Figure 3. Interestingly, both enantiomers of 32 exhibited excellent dual-inhibition of BTK and PI3Kδ, whereas (+)-33 had a 9-fold increase in BTK IC50 compared to (−)-33.
Table 6. N-1 Pyrazolopyrimidine Optimizations.
| No. | R | BTK (IC50, nM) | PI3Kδ (IC50, nM) |
|---|---|---|---|
| 27 | 2-indanyl | 104 | 35 |
| (±)-30 | 2-tetralinyl | NDa | NDb |
| (±)-31 | 2-(5-F-indanyl) | NDc | NDd |
| (+)-32 | 2-(5-NH2-indanyl) | 24 | 47 |
| (−)-32 | 2-(5-NH2-indanyl) | 32 | 16 |
| (+)-33 | 2-(7-NH2-tetralinyl) | 176 | 55 |
| (−)-33 | 2-(7-NH2-tetralinyl) | 19 | 72 |
IC50 not determined; inhibition of BTK was 34% at 100 nM (±)-30.
IC50 not determined; inhibition of PI3Kδ was 52% at 100 nM (±)-30.
IC50 not determined; inhibition of BTK was 6.6% at 100 nM (±)-31.
IC50 not determined; inhibition of PI3Kδ was 26% at 100 nM (±)-31.
Figure 3.
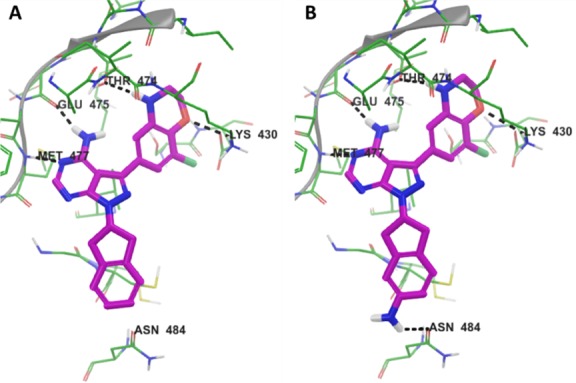
Docked poses of 27 (A) and 32 (B) with BTK indicate that 32 makes an additional H-bond interaction with Asn484.
We selected both enantiomers of 32 for additional biochemical profiling in an effort to distinguish between the two molecules, Table 7. In a lipid kinase panel consisting of the various PI3K isoforms, (−)-32 was more selective than (+)-32 when compared against PI3Kδ. The opposite trend was observed for the two nonreceptor tyrosine kinases. When comparing against BTK, (+)-32 was slightly more selective against BMX, nonreceptor tyrosine kinase, which is closely related to BTK, and TEC versus (−)-32. In general, both compounds performed similarly in our full metabolism panel, which included rat (rLM) and dog (dLM) liver microsomes. The only clear distinction was that (+)-32 appeared to be more stable than (−)-32 to oxidative metabolism in mouse and dog liver microsomes. More importantly, these analogues showed good stability in the low affinity albeit high capacity glucuronidation assay. Lastly, both enantiomers were equal in regards to P-gp efflux potential on Caco-2 cells and mouse plasma protein binding.
Table 7. Biochemical and in Vitro ADME Results for (+)-32 and (−)-32.
| kinase selectivity
(ratio) |
in vitro metabolismd (%R) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | PI3Kαa | PI3Kβa | PI3Kγa | BMXb | TECc | hLM | mLM | rLM | dLM | hUGT | mUGT | Caco-2e | PPBf (%) |
| (+)-32 | 6 | 34 | 2 | 4 | 20 | 71 | 28 | 45 | 65 | 91 | 93 | 1.3 | 98 |
| (−)-32 | 17 | 44 | 7 | 1 | 7 | 63 | 11 | 46 | 32 | 91 | 99 | 1.7 | 98 |
Ratio of PI3K IC50/PI3Kδ IC50.
Ratio of BMX IC50/BTK IC50.
Ratio of TEC IC50/BTK IC50.
Percent parent remaining (%R) after 30 min.
Efflux ratio, PappB → A/PappA → B.
Percent mouse plasma protein binding (PPB).
Although both compounds appear to be more stable in rats, as well as dogs, we chose to continue our studies in mice. First, we wanted to determine the extent of our optimizations in comparison to our previous analogues tested for mouse PK. Second, we were anticipating that mice would be the primary species for our preclinical in vivo efficacy models. Table 8 summarizes the mouse PK properties for (+)-32 and (−)-32. Compared to our initial leads, both compounds exhibited lower systemic clearance, which may largely be due to their improved stability against glucuronidation. Moreover, oral exposures increased over 125-fold for both compounds compared to 21, resulting in oral bioavailabilities of 57% and 40% for (+)-32 and (−)-32, respectively.
Table 8. In Vivo Mouse Pharmacokinetic Propertiesa of 32b.
| No. | CL (L/h/kg) | Vz (L/kg) | AUCPO (μM·h) | PO t1/2 (h) | F (%) |
|---|---|---|---|---|---|
| (+)-32 | 1.46 | 0.58 | 9.28 | 1.2 | 57 |
| (−)-32 | 0.59 | 3.44 | 15.2 | 1.3 | 40 |
WinNonlin noncompartmental analysis.
Dosed 2 mg/kg IV and 10 mg/kg PO.
We were pleased to observe that the systematic optimizations starting from SW13 and our initial hits resulted in a set of compounds that are (1) potent dual-inhibitors of BTK and PI3Kδ and (2) have improved mouse PK properties. These compounds will serve as the starting point for a subsequent round of optimizations, which will include cellular and in vivo proof-of-concept (PoC) studies.
Acknowledgments
The authors would like to thank Anup Barde, Vijay Sharma, Shailender Chauhan, Abhinandan Danodia, Kailas Thube, and Ankesh Sharma for their help with the scale-up of key intermediates; Diksha Sharma, Vishal Budha, and Rahul Takalkar for their assistance in the purification of intermediates and final compounds; and Shivam Dixit and Meena Bisht for their assistance in acquiring NMR data. We also thank Christopher Higgs for his analysis and comments regarding the preparation of this manuscript. This research was funded solely by Medivation, Inc.
Glossary
ABBREVIATIONS
- AUCpo
area under the concentration–time curve from time zero to the last quantifiable concentration after oral (PO) administration
- BCR
B-cell receptor
- BMX
bone marrow kinase on chromosome X
- CL
total body clearance after intravenous (IV) administration
- F
absolute oral bioavailability determined from AUCpo/AUCiv × doseiv/dosepo
- HPβCD
2-hydroxypropyl-β-cyclodextrin
- LM
liver microsomes
- LYN
Lck/Yes novel tyrosine kinase
- ND
not determined
- PEG
polyethylene glycol
- PO t1/2
terminal half-life after oral administration
- SYK
spleen tyrosine kinase
- TEC
tyrosine-protein kinase Tec
- UGT
uridine 5′-diphospho-glucuronosyltransferase
- Vz
total volume of distribution after IV administration
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00356.
Experimental procedures and analytical data for 3, 5–33; descriptions for in vitro biochemical and metabolic stability assays; descriptions for in vivo PK experiments; description for molecular modeling (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Niiro H.; Clark E. A. Regulation of B-Cell Fate by Antigen-Receptor Signals. Nat. Rev. Immunol. 2002, 2, 945–956. 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- Seda V.; Mraz M. B-Cell Receptor Signalling and Its Crosstalk with Other Pathways in Normal and Malignant Cells. Eur. J. Haematol. 2015, 94, 193–205. 10.1111/ejh.12427. [DOI] [PubMed] [Google Scholar]
- Rickert R. C. New Insights into Pre-BCR and BCR Signalling with Relevance to B Cell Malignancies. Nat. Rev. Immunol. 2013, 13, 578–591. 10.1038/nri3487. [DOI] [PubMed] [Google Scholar]
- Mato A.; Jauhari S.; Schuster S. J. Management of Chronic Lymphocytic Leukemia (CLL) in the Era of B-Cell Receptor Signal Transduction Inhibitors. Am. J. Hematol. 2015, 90, 657–664. 10.1002/ajh.24021. [DOI] [PubMed] [Google Scholar]
- Cameron F.; Sanford M. Ibrutinib: First Global Approval. Drugs 2014, 74, 263–271. 10.1007/s40265-014-0178-8. [DOI] [PubMed] [Google Scholar]
- Miller B. W.; Przepiorka D.; de Claro R. A.; Lee K.; Nie L.; Simpson N.; Gudi R.; Saber H.; Shord S.; Bullock J.; Marathe D.; Mehrotra N.; Hsieh L. S.; Ghosh D.; Brown J.; Kane R. C.; Justice R.; Kaminskas E.; Farrell A. T.; Pazdur R. FDA Approval: Idelalisib Monotherapy for the Treatment of Patients with Follicular Lymphoma and Small Lymphocytic Lymphoma. Clin. Cancer Res. 2015, 21, 1525–1529. 10.1158/1078-0432.CCR-14-2522. [DOI] [PubMed] [Google Scholar]
- De Rooij M. F. M.; Kuil A.; Kater A. P.; Kersten M. J.; Pals S. T.; Spaargaren M. Ibrutinib and Idelalisib Synergistically Target BCR-Controlled Adhesion in MCL and CLL: A Rationale for Combination Therapy. Blood 2015, 125, 2306–2309. 10.1182/blood-2014-12-619163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. L.; Rule S.; Martin P.; Goy A.; Auer R.; Kahl B. S.; Jurczak W.; Advani R. H.; Romaguera J. E.; Williams M. E.; Barrientos J. C.; Chmielowska E.; Radford J.; Stilgenbauer S.; Dreyling M.; Jedrzejczak W. W.; Johnson P.; Spurgeon S. E.; Li L.; Zhang L.; Newberry K.; Ou Z.; Cheng N.; Fang B.; McGreivy J.; Clow F.; Buggy J. J.; Chang B. Y.; Beaupre D. M.; Kunkel L. A.; Blum K. A. Targeting BTK with Ibrutinib in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2013, 369, 507–516. 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd J. C.; Furman R. R.; Coutre S. E.; Flinn I. W.; Burger J. A.; Blum K. A.; Grant B.; Sharman J. P.; Coleman M.; Wierda W. G.; Jones J. A.; Zhao W.; Heerema N. A.; Johnson A. J.; Sukbuntherng J.; Chang B. Y.; Clow F.; Hedrick E.; Buggy J. J.; James D. F.; O’Brien S. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyach J. A.; Furman R. R.; Liu T.-M.; Ozer H. G.; Zapatka M.; Ruppert A. S.; Xue L.; Li D. H.-H.; Steggerda S. M.; Versele M.; Dave S. S.; Zhang J.; Yilmaz A. S.; Jaglowski S. M.; Blum K. A.; Lozanski A.; Lozanski G.; James D. F.; Barrientos J. C.; Lichter P.; Stilgenbauer S.; Buggy J. J.; Chang B. Y.; Johnson A. J.; Byrd J. C. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. 10.1056/NEJMoa1400029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova N. L.; Burger J. A.; Wodarz D. Evolution of Ibrutinib Resistance in Chronic Lymphocytic Leukemia (CLL). Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 13906–13911. 10.1073/pnas.1409362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiestner A. The Role of B-Cell Receptor Inhibitors in the Treatment of Patients with Chronic Lymphocytic Leukemia. Haematologica 2015, 100, 1495–1507. 10.3324/haematol.2014.119123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H.; Matsuda S.; Terauchi Y.; Fujiwara M.; Ohteki T.; Asano T.; Behrens T. W.; Kouro T.; Takatsu K.; Kadowaki T.; Koyasu S. PI3K and Btk Differentially Regulate B Cell Antigen Receptor-Mediated Signal Transduction. Nat. Immunol. 2003, 4, 280–286. 10.1038/ni890. [DOI] [PubMed] [Google Scholar]
- Williams O.; Houseman B. T.; Kunkel E. J.; Aizenstein B.; Hoffman R.; Knight Z. A.; Shokat K. M. Discovery of Dual Inhibitors of the Immune Cell PI3Ks p110delta and p110gamma: A Prototype for New Anti-Inflammatory Drugs. Chem. Biol. 2010, 17, 123–134. 10.1016/j.chembiol.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z.; Scheerens H.; Li S.-J.; Schultz B. E.; Sprengeler P. A.; Burrill L. C.; Mendonca R. V.; Sweeney M. D.; Scott K. C. K.; Grothaus P. G.; Jeffery D. A.; Spoerke J. M.; Honigberg L. A.; Young P. R.; Dalrymple S. A.; Palmer J. T. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Marcotte D. J.; Liu Y.-T.; Arduini R. M.; Hession C. A.; Miatkowski K.; Wildes C. P.; Cullen P. F.; Hong V.; Hopkins B. T.; Mertsching E.; Jenkins T. J.; Romanowski M. J.; Baker D. P.; Silvian L. F. Structures of Human Bruton’s Tyrosine Kinase in Active and Inactive Conformations Suggest a Mechanism of Activation for TEC Family Kinases. Protein Sci. 2010, 19, 429–439. 10.1002/pro.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt A.; Miller S.; Williams O.; Le D. D.; Houseman B. T.; Pacold J. I.; Gorrec F.; Hon W.-C.; Liu Y.; Rommel C.; Gaillard P.; Ruckle T.; Schwarz M. K.; Shokat K. M.; Shaw J. P.; Williams R. L. The p110 Delta Structure: Mechanisms for Selectivity and Potency of New PI(3)K Inhibitors. Nat. Chem. Biol. 2010, 6, 117–124. 10.1038/nchembio.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai R.; Chakravarty S.; Green M. J.; Pham S. M.; Pujala B.; Agarwal A. K.; Nayak A. K.; Khare S.; Guguloth R.; Randive N. A.. Preparation of pyrazolo[3,4-D]pyrimidines and Other Heterocyclic Compounds That Are Inhibitors of Both Btk and PI3Kδ for Therapy of Cancer and Autoimmune Disorders. P.C.T. Int. Appl. No. WO2015058084A1, 2015.
- Suzuki A. Organoboranes in Organic Syntheses Including Suzuki Coupling Reaction. Heterocycles 2010, 80, 15–43. 10.3987/COM-09-S(S)Summary. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.