

Abstract
Scaffold-hopping of bioactive natural product aurones has been studied for the first time. 2-Arylideneimidazo[1,2-a]pyridinones as potential topoisomerase IIα (hTopoIIα)-targeting anticancer compounds were considered. A multifunctional activator, polyphosphoric acid, enabled to realize a cascade reaction of 2-aminopyridine with 2,3-epoxyesters toward synthesis of 2-arylideneimidazo[1,2-a]pyridinones. Most of the compounds exhibited hTopoIIα-selective poison activity with efficiency more than etoposide and DNA-binding property, while not interacting with hTopo I. The compounds showed pronounced antiproliferative activities in nanomolar range with relatively poor toxicity to normal cells, inhibition of invasiveness, and apoptotic effect. The activities for inhibition of tubulin assembly, CDK1 and pCDK1, were also observed. Interestingly, the hTopoIIα inhibitory (in vitro and ex vivo studies) and antiproliferative activities of representative potent compounds were found to be manifold higher compared to corresponding parent aurones bearing alike substitutions, indicating the importance of such scaffold-hopping strategy in medicinal chemistry research.
Keywords: Scaffold-hopping; human topoisomerase IIα; imidazo[1,2-a]pyridine; anticancer agents; aurones
Natural products are not only used as drugs in the treatment of various diseases but also have been considered as leading templates for the design, which have successfully led to development of a number of approved drugs and clinical trial agents.1,2 They introduce molecular diversity and structural novelty and have been valuable in recognition of novel targets. In particular, natural products have played critical role in anticancer drug discovery and development.3,4 According to the latest report by Newman and Cragg,5 approximately 83% (113 of 136) anticancer small molecules approved by FDA were either natural products, or based thereon, or mimicked natural products in some form. In order to have desired biological activity as well as ADMET properties, the lead optimization is required, which is usually done by modification/introduction of side chains or substituents. In this direction, interestingly, a complete change in molecular framework can be achieved by scaffold-hopping.
The term “scaffold-hopping” has been introduced by Schneider et al.,6 as the identification of iso-functional molecular structures with significantly different molecular backbones. The new molecule with changed core becomes patentable.7 There are number of examples of successful drug discovery and development employing scaffold-hopping.8,9 However, a few reports of scaffold-hopping of natural products are available.10 Recently, scaffold hopping of a natural product proteasome inhibitor Belactosin A has been reported, which led to development of highly potent nonpeptidic inhibitors.11
Toward the aim of topoisomerase II-targeting anticancer drug discovery and development,12−14 we considered scaffold-hopping of bioactive flavonoids class of compounds. We have already discovered scaffold-hopped analogues of flavones and isoflavones, 2/3-arylpyridopyrimidinones, as Topo II-targeting anticancer agents.10,15,16 In continuation, we were interested in the scaffold-hopping of aurone class of compounds. Aurones have been reported as promising anticancer agents, which show interference with various targets17 such as CDK1 inhibition,18 adenosine receptor inhibition,19 DNA scission, and telomerase inhibition.20 We focused on amalgamation of structural features of aurones and the imidazopyridine class of compounds, which we previously discovered as potent topoisomerase II-targeting anticancer agents.11,13 In addition, several other related bicyclic compounds21−23 have been reported as topoisomerase II inhibitors. We envisaged scaffold-hopped analogues of aurones, 2-arylideneimidazo[1,2-a]pyridinones, as potential topoisomerase II-targeting anticancer agents (Figure 1a).
Figure 1.

Design and synthesis of the target compounds.
Chemistry
Recently, we have developed a new cascade reaction of 2-aminopyridine with 2,3-epoxyesters to produce 2-arylideneimidazo[1,2-a]pyridinones.24 The reaction involves a set of sequential transformations: epoxide ring opening, aziridination, nucleophilic opening of aziridine, elimination to form enamine, and intramolecular transamidation. These cascade transformations required a distinct catalysis/activation, which was achieved by polyphosphoric acid as multifunctional activator. Several relevantly substituted 2-arylideneimidazo[1,2-a]pyridinones were synthesized bearing structural features of anticancer aurones (Figures 1b,c). 2-(4-Hydroxybenzylidene)imidazo[1,2-a]pyridinone was synthesized via BBr3-mediated ether cleavage.25
To evaluate the effectiveness of scaffold-hopping concept on the topoisomerase II inhibitory activities of aurones, two aurones possessing alike substitutions, completely mimicking the structures of investigated potent topo II-inhibiting 2-arylideneimidazo[1,2-a] pyridinones were prepared, such that a comparison in their activities can be made. Two aurones C1 and C2 bearing the same substitutions as that of compounds 3c and 3k were prepared following literature reported procedure (Figure 1d).27,28
Biological Studies
Inhibition of hTopoIIα-Mediated kDNA Decatenation
The synthesized 2-arylideneimidazo[1,2-a]pyridinones (3a–r) and parent aurones (C1–2) were tested for inhibition of the hTopoIIα-mediated decatenation of kinetoplast DNA (kDNA) (Figure 2).12 Etoposide, a known topoisomerase II-inhibiting anticancer drug, was used as a reference. In the presence of compounds 3b–e, 3i, 3k, 3m, 3n, 3p, and 3q, relatively higher inhibition of hTopoIIα-mediated kDNA decatenation compared to etoposide was observed. Interestingly, scaffold-hopping based structural modifications were found to result in increased hTopoIIα-inhibitory activities. For example, compared to parent aurones (C1 and C2), compounds 3c and 3k, which possess alike substitutions, were found to be more potent hTopoIIα-inhibitors.
Figure 2.
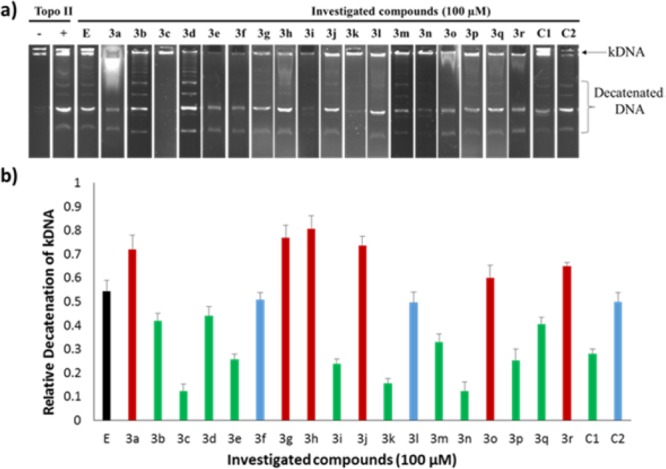
(a) Inhibition of hTopoIIα-mediated kDNA decatenation: kDNA was treated with hTopoIIα in the absence or presence of 100 μM etoposide (E) or investigated compounds. (b) Quantification of decatenation products.
hTopoIIα-DNA Cleavage Complex Formation
A hTopoIIα poison leads to stabilization of a TopoII-cleaved DNA complex (called as a cleavage complex), which appears as a linear band (Lin).12 In the presence of the active compounds 3c and 3k (100 μM), characteristic linear band was observed (Figure S1). It indicates that the compounds 3c and 3k act as hTopoIIα poisons at tested concentration.
Inhibition of hTopoI-Mediated DNA Relaxation
In order to evaluate the selectivity of inhibition for hTopoIIα, selected compounds 3c and 3k were screened for inhibition of hTopoI-mediated relaxation of negatively supercoiled DNA.12 In the presence of tested compounds, relatively less inhibition of hTopoI-mediated DNA-relaxation occurred as compared to camptothecin (Figure S2). Hence, tested compounds are not hTopoI inhibitors.
DNA Interaction Study
In order to check the DNA binding efficacy, UV-based study for in vitro DNA-drug binding was carried out. The exact spectrum of 260 nm was found for DNA without any agent. In the case of DNA treated with increasing concentrations of compounds 3c and 3k, a hypochromism in absorption was found to occur, indicating possible binding of the compounds to DNA (Figure S6). Kd was found to be 2 × 106 and 2.5 × 106 M–1 for compounds 3c and 3k, respectively.
Cytotoxic Potential of Selected Compounds in Cancer Cells
Cytotoxic potential of the potent hTopoIIα inhibitors (3a–d, 3i, 3k, 3n, 3o, 3r) and control compounds (C1 and C2) was measured by a well-known colorimetric-based MTT assay.12 The representative cancer cells, kidney cancer cell line (HEK-293T), its corresponding normal cell line (VERO), and breast cancer cell line (MCF-7) were considered for testing cytotoxic activities. The IC50 (concentration of compound to cause 50% cell growth inhibition) of compounds 3a–d, 3i, 3k, 3n, 3o, 3r were found remarkably low (10, 150, 60, 80, 30, 45, 45, 80, 40 nM, respectively) in HEK 293T cells compared to IC50 values obtained in VERO cells (200, 1500, 1500, 1500, 200, 200,1500, 1500, and 1500 nM, respectively) (Table S1a). The IC50 for etoposide was found to be 22 μM in HEK-293T cells and 50 μM in VERO cells. It is interesting to note that parent compounds C1 and C2 compared to their investigated scaffold-hopped analogues did not show significant cytotoxicity. The IC50 values of compounds C1 and C2 were found to be 0.7 and 0.9 μM in HEK-293T cells and 2.5 and 2.4 μM in VERO cells, respectively (Table S1a). In MCF-7 cells, the cytotoxicities of the investigated scaffold-hopped analogues were also found to be multifold higher compared to etoposide and parent aurones C1 and C2 (Table S1a). The cell viability was also checked in other breast cancer cell lines, i.e., MDA-MB-231 and MDA-MB-468. The IC50 for compound 3c were found to be 64 and 60 nM, and the IC50 for compound 3k were found to be 76 and 70 nM, for MDA-MB-231 and MDA-MB-468, respectively.
However, the IC50 for control compounds C1 and C2 in MDA-MB-231 and MDA-MB-468 were found to be much higher, 840–600 nM (Table S2). To further confirm the cytotoxicity a long-term cell survival assay (clonogenic assay) was performed.24 For compounds 3a–d, 3i, 3k, 3n, 3o, and 3r the LC50 values (50% cell death in culture) in HEK 293T cells were found to be 55, 60, 60, 75, 40, 20, 40, 40, and 50 nM, respectively, and in VERO cells 1000, 1500, 1500, 1500, 1500, 1500, 1500, 1500, and 1500 nM, respectively (Table S1b). In MCF-7 cells, these compounds exhibited also pronounced anticlonogenic properties (LC50 in nM ranges). In both HEK-293T and MCF-7 cells, the investigated imidazo-pyridinones were found to be prominently more potent (IC50: nanomolar vs micromolar) than etoposide and the parent aurones C1 and C2. In both MTT and clonogenic assays, concentration-dependent cytotoxicities of these imidazo-pyridinones were observed (Figure S3).
Studies of Apoptosis by Compound 3c and Inhibition of CDK1
From above cytotoxicity experiments it was observed that tested compounds exhibited appreciable toxicity in cancer cells with minimal effect to normal cells. To check whether these agents caused apoptosis in cancer cells, we considered a representative compound 3c and measured the apoptosis after treatment in MCF-7 cells using DAPI nuclear staining.28 The cells were treated with increasing concentrations of compound 3c for 48 h prior to addition of DAPI dye. Significantly higher chromatin condensation and nuclear fragmentation in treated cells were noticed compared to untreated cells (Figure S4). More than 6-fold increase in apoptotic nuclei was observed at 75 nM compared to untreated control.
To confirm that the investigated compounds caused apoptosis we did Western blot analysis29 of various pro- and antiapoptotic proteins in MCF-7 cellular lysate treated with compound 3c. An increase in the expressions of pro-apoptotic pBAX and decrease in the expression of antiapoptotic protein BCL-XL confirmed that compound 3c causes apoptosis in MCF-7 cells. Next we intended to check the possible involvement of other cell cycle regulatory proteins, i.e., CDK1 and pCDK1 (Thr 161). A significant decreased expression of CDK1 as well as pCDK1 (Thr 161) with increasing concentration of compound 3c (Figure 3a) was observed. Inhibition of these cell cycle regulatory proteins leads to arresting of cells in G2/M transition phase.
Figure 3.

(a) Expression pattern of BAX, BCL XL, p21, p53, CDK1, pCDK1 (Thr 161), and CASPASE 3 after treatment with compound 3c. GAPDH served as a loading control to check the equal loading of protein in each lane. (b) Immunocytochemistry of CASPASE 3 in MCF-7 cells treated with compound 3c. The numerical value above each panel shows the relative fold change in comparison with untreated control measured by densitometric analysis. All the experiments are carried out at least thrice.
Induction and degradation of mitochondrial enzyme CASPASE 3 is the hallmark of apoptosis. An increased expression of cleaved CASPASE 3 (17 kDa) was observed in compound 3c treated MCF-7 cellular lysate (Figure 3a). To further validate this result we have checked the expression of CASPASE 3 by immunocytochemistry after treating MCF-7 cells with increasing concentrations of compound 3c. Increase in the expression of CASPASE 3 was noticed, which further validates that compound 3c induced apoptosis in the MCF-7 cells (Figure 3b).
Compound 3c Inhibited the Invasiveness of MCF 7 Cells
Invasion into basement membrane is one of the important properties of cancer cell. The effect of compound 3c in the invasive property of MCF-7 cell was measured by a well-established matrigel cell invasion assay.30 It was observed that with increase in concentration of compound 3c the numbers of invaded colonies were significantly decreased with respect to untreated control. This indicates that compound 3c inhibited the in vitro cell invasion (Figure S5).
Inhibition of the Topoisomerase Activity in MCF-7 Cells; Comparison of Activities of Scaffold-Hopped Compounds with Parent Compounds
To check the ex vivo topoisomerase-inhibiting property of compound 3c and compound 3k, a plasmid based topoisomerase inhibition assay using nuclear lysate of MCF-7 and a plasmid NFkB was carried out according to protocol described in the experimental section. Etoposide was used as a positive control. According to the principle if any agent inhibits the topoisomerase activity, then more supercoiled DNA forms and run faster in the agarose gel compared to relaxed/linear one. Figure 4a,b demonstrates the formation of supercoiled DNA and migration into the gel after treatment with compounds 3c and 3k, respectively. In nuclear lysate without treatment of compound 3c/3k, the plasmid was relaxed or linear and thus did not enter into 0.8% agarose gel. However, with increase in the concentration of the agents (3c or 3k) the formation of linear/relaxed plasmid decreased, as a result more supercoiled DNA entered into the gel. The effect of topoisomerase inhibitory activity is directly proportional to the migration of DNA into the gel. Interestingly, compounds (3c and 3k) in comparison with parent compounds (C1/C2) exhibited significantly higher migration of bands into the gel. This indicates the superior activities of scaffold-hopped analogues than that of parent aurones C1 and C2 in ex vivo topoisomerase-inhibition.
Figure 4.

(a) Inhibition of topoisomerase activity by compound 3c with respect to control C1 and positive control etoposide in MCF-7 cell nuclear lysate. (b) Inhibition of topoisomerase activity by compound 3k in respect to control C2 and positive control etoposide in MCF-7 cell nuclear lysate. In both the figures (a and b) Lane 1 is only plasmid, Lane 2 is plasmid with untreated nuclear lysate, and Lanes 3–7 are for plasmid and nuclear lysate treated with various concentrations of compound 3c/3k (10 nM-75 nM). Lane 8 was treated with (a) C1 and (b) C2, respectively. Lane 9 is lysate treated with etoposide 45 (μM) for both the figures (a and b).
Determination Cell Cycle Profile in Compound 3c and 3k Treated MCF-7 Cells
To investigate the regulation of the cell cycle profile, FACS analysis was carried out in MCF-7 cells after treatment with compounds 3c and 3k. The cells were treated with increasing concentrations of compounds 3c and 3k for 48 h. FACS analysis was performed after staining with PI, and the DNA content of each phase of the cell cycle was measured. It was observed that both the compounds 3c and 3k arrest the cell at G2/M phase. In the case of compound 3c, significant G2/M arrest was observed (80.1%) at IC50 concentration (50 nM) with noticeable apoptosis at higher concentration, i.e., 20.2% apoptosis in 100 nM treatment. Similarly, in the case of compound 3k the G2/M population was found to be 33.2% for 70 nM, and in 100 nM, significant apoptosis (19.9%) occurred. So, this result suggests that the selective compounds arrest the cell at G2/M phase. So, to validate this we compared these compounds with a known compound that arrests the cell at G2/M phase, i.e., combretastatin A-4 (CA-4) (Figure S7). Noticeable G2/M arrests with apoptosis (G0) were observed in CA-4 treated MCF-7 cell lysate. No such profile was obtained for control compounds (C1 and C2), indicating that compounds 3c and 3k arrest the cell at G2/M phase at IC50 concentration with apoptosis at higher concentration, which is in accordance with the expression profile of CDK1 and pCDK1.
Compounds 3c and 3k Inhibit Tubulin Polymerization and Disrupt Microtubule Dynamics
To study possible antitubulin effect of investigated compounds, an in vitro tubulin assembly assay was carried out using breast cancer cells (MCF 7). The compounds 3c and 3k were found to possess good activity in tubulin assembly inhibition (Figure S8a and Table S3). The tubulin assembly inhibition was found to be optimum with IC50 (concentration required to inhibit 50% of tubulin assembly) values of 54 and 60 nM for 3c and 3k, respectively, with comparison to CA-4, which showed IC50 at 2.5 μM. To further validate this result we performed an immunostaining of α tubulin. The left panel represents the PI staining of nucleus. The middle panel represents α tubulin protein assembly in MCF-7 cells conjugated with FITC, and the third panel is the merge image of PI and FITC conjugated α tubulin. The result indicated that the compound 3c induced significant distortion in the tubulin assembly along with the formation of abnormal mitotic spindle in the treated MCF-7 lysate (Figure S8).
Conclusions
A novel series of 2-arylideneimidazo[1,2-a]pyridinones as potential hTopo IIα-targeting anticancer agents was designed by scaffold-hopping of bioactive natural product aurones and consideration of structural feature of hTopo IIα-targeting anticancer N-fused imidazoles previously discovered by our group. Polyphosphoric acid was used as multifunctional activator for a cascade reaction of 2-aminopyridine with 2, 3-epoxyesters to synthesize the designed compounds. Most of the compounds exhibited Topo II-inhibiting activity more than etoposide. The compounds showed properties of hTopo IIα-poison and DNA-binding, while not interfering with hTopo I. In HEK-293T and MCF-7 cells, the compounds exhibited anticancer activity in nanomolar range and dose-dependent cytotoxicities and decrease in colony formation of cancer cells with efficiency extremely higher than etoposide. These compounds were found to be very less toxic to normal cells. The cytotoxic activities occurred due to the apoptotic effects of the compounds. These compounds inhibited the invasiveness of cancer cells. The effect of compounds on inhibition of tubulin assembly, CDK1 and pCDK1, were also found to be significant.
Interestingly, compared to parent aurones, their scaffold-hopped analogues, 2-arylideneimidazo[1,2-a]pyridine-3-ones, were found to be multifold higher potent (IC50: micromolar vs nanomolar) antiproliferative agents against various cancer cells and hTopoIIα-inhibitors in in vitro as well as ex vivo assays. This suggests successful implication of scaffold-hopping strategy on natural product aurones for the discovery of more potent analogues which are outside the patent issues.
Acknowledgments
We gratefully acknowledge financial support from DBT, DST, and CSIR, Government of India, New Delhi, for this investigation. G.P. and S.M.A. are thankful to NIPER for their fellowships. A.N. is thankful to ICMR for providing a fellowship.
Glossary
ABBREVIATIONS
- hTopoIIα
human topoisomerase IIα
- kDNA
kinetoplast DNA
- Nck
nicked
- Rel
relaxed
- Lin
linear
- SC
supercoiled
- TAE
tris-acetate-EDTA
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00242.
Experimental procedure for synthesis and biology, characterization data for new compounds (PDF)
Author Contributions
∥ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Butler M. S. Natural products to drugs: natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural product scaffolds as leads to drugs. Future Med. Chem. 2009, 1, 1415–1427. 10.4155/fmc.09.113. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Morris-Natschke S. L.; Lee K. H. Strategies for the optimization of natural leads to anticancer drugs or drug candidates. Med. Res. Rev. 2016, 36, 32–91. 10.1002/med.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg G. M.; Grothaus P. G.; Newman D. J. Impact of Natural Products on DevelopingNew Anti-Cancer Agents. Chem. Rev. 2009, 109, 3012–3043. 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Schneider P.; Renner S. Scaffold-Hopping: How Far Can You Jump?. QSAR Comb. Sci. 2006, 25, 1162–1171. 10.1002/qsar.200610091. [DOI] [Google Scholar]
- Southall N. T.; Ajay Patent Space Visualization Using Chemical Replacements. J. Med. Chem. 2006, 49, 2103–2109. 10.1021/jm051201m. [DOI] [PubMed] [Google Scholar]
- Sun H.; Tawa G.; Wallqvist A. Classification of Scaffold-Hopping Approaches. Drug Discovery Today 2012, 17, 310–324. 10.1016/j.drudis.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu C.; Ge H.; Song M.; Chen J. H.; Zhou H.; Qi Q.; Wang F.; Ma X.; Yang X.; Zhang G.; Ding Y.; Zhou D.; Peng P.; Shih C. K.; Xu J.; Wu F. Discovery of Imigliptin, a Novel Selective DPP-4 Inhibitor for the Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2014, 5, 921–926. 10.1021/ml5001905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadarshani G.; Amrutkar S. M.; Nayak A.; Banerjee U. C.; Kundu C. N.; Guchhait S. K. Scaffold-hopping of bioactive flavonoids: Discovery of aryl-pyridopyrimidinones as potent anticancer agents that inhibit catalytic role of topoisomerase IIα. Eur. J. Med. Chem. 2016, 122, 43–54. 10.1016/j.ejmech.2016.06.024. [DOI] [PubMed] [Google Scholar]
- Kawamura S.; Unno Y.; Hirokawa T.; Asai A.; Arisawa M.; Shuto S. Rational Hopping of a Peptidic Scaffold into Non-Peptidic Scaffolds: Structurally Novel Potent Proteasome Inhibitors Derived from a Natural Product, Belactosin A. Chem. Commun. 2014, 50, 2445–2447. 10.1039/c3cc48818g. [DOI] [PubMed] [Google Scholar]
- Baviskar A. T.; Madaan C.; Preet R.; Mohapatra P.; Jain V.; Agarwal A.; Guchhait S. K.; Kundu C. N.; Banerjee U. C.; Bharatam P. V. N-Fused Imidazoles As Novel Anticancer Agents That Inhibit Catalytic Activity of Topoisomerase IIα and Induce Apoptosis in G1/S Phase. J. Med. Chem. 2011, 54, 5013–5030. 10.1021/jm200235u. [DOI] [PubMed] [Google Scholar]
- Kashyap M.; Kandekar S.; Baviskar A. T.; Das D.; Preet R.; Mohapatra P.; Satapathy S. R.; Siddharth S.; Guchhait S. K.; Kundu C. N.; Banerjee U. C. Indenoindolone Derivatives as Topoisomerase II-Inhibiting Anticancer Agents. Bioorg. Med. Chem. Lett. 2013, 23, 934–938. 10.1016/j.bmcl.2012.12.063. [DOI] [PubMed] [Google Scholar]
- Baviskar A. T.; Amrutkar S. M.; Trivedi N.; Chaudhary V.; Nayak A.; Guchhait S. K.; Banerjee U. C.; Bharatam P. V.; Kundu C. N. Switch in Site of Inhibition: A Strategy for Structure-Based Discovery of Human Topoisomerase IIα Catalytic Inhibitors. ACS Med. Chem. Lett. 2015, 6, 481–485. 10.1021/acsmedchemlett.5b00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guchhait S. K.; Priyadarshani G. Pd-Catalyzed Ag(I)-Promoted C3-Arylation of Pyrido[1,2-a]pyrimidin-4-Ones with Bromo/Iodo-Arenes. J. Org. Chem. 2015, 80, 8482–8488. 10.1021/acs.joc.5b01573. [DOI] [PubMed] [Google Scholar]
- Guchhait S. K.; Priyadarshani G. Synthesis of 2-Arylpyridopyrimidinones, 6-Aryluracils, and Tri- and Tetrasubstituted Conjugated Alkenes via Pd-Catalyzed Enolic C–O Bond Activation–Arylation. J. Org. Chem. 2015, 80, 6342–6349. 10.1021/acs.joc.5b00771. [DOI] [PubMed] [Google Scholar]
- Haudecoeur R.; Boumendjel A. Recent Advances in the Medicinal Chemistry of Aurones. Curr. Med. Chem. 2012, 19, 2861–2875. 10.2174/092986712800672085. [DOI] [PubMed] [Google Scholar]
- Schoepfer J.; Fretz H.; Chaudhuri B.; Muller L.; Seeber E.; Meijer L.; Lozach O.; Vangrevelinghe E.; Furet P. Structure-Based Design and Synthesis of 2-Benzylidene-benzofuran-3-ones As Flavopiridol Mimics. J. Med. Chem. 2002, 45, 1741–1747. 10.1021/jm0108348. [DOI] [PubMed] [Google Scholar]
- Gao Z. G.; Kim S. K.; Biadatti T.; Chen W.; Lee K.; Barak D.; Kim S. G.; Johnson C. R.; Jacobson K. A. Structural determinants of A3 adenosine receptor activation: nucleoside ligands at the agonist/antagonist boundary. J. Med. Chem. 2002, 45, 4471–4484. 10.1021/jm020211+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballinari D.; Bonomini L.; Ermoli A.; Gude M.; Menichincheri M. J.; Vanotti E.. Aurones as telomerase inhibitors. PCT/EP2002/004191, 2002.
- Kao Y. H.; Hsieh H. P.; Chitlimalla S. K.; Pan W. Y.; Kuo C. C.; Tsai Y. C.; Lin W. H.; Chuang S. E.; Chang J. Y. A novel peroxisome proliferator-activated receptor α/γ agonist, BPR1H0101, inhibits topoisomerase II catalytic activity in human cancer cells. Anti-Cancer Drugs 2008, 19, 151–158. 10.1097/CAD.0b013e3282f28fe. [DOI] [PubMed] [Google Scholar]
- Pinar A.; Yurdakul P.; Ylildiz I.; Temiz-Arpaci O.; Acan N. L.; Aki-Sener E.; Yalcin I. Some fused heterocyclic compounds as eukaryotic topoisomerase II inhibitors. Biochem. Biophys. Res. Commun. 2004, 317, 670–674. 10.1016/j.bbrc.2004.03.093. [DOI] [PubMed] [Google Scholar]
- Tekiner-Gulbas B.; Temiz-Arpaci O.; Yildiz I.; Aki-Sener E.; Yalcin I. 3D-QSAR study on heterocyclic topoisomerase II inhibitorsusingCoMSIA. SAR QSAR Environ. Res. 2006, 17, 121–132. 10.1080/10659360600636105. [DOI] [PubMed] [Google Scholar]
- Guchhait S. K.; Priyadarshani G.; Hura N. α,β-Epoxy Esters in Multiple C–O/C–N Bond-Breaking/Formation with 2-Aminopyridines; Synthesis of Biologically Relevant (Z)-2-Methyleneimidazo[1,2-a]pyridin-3-ones. Synlett 2014, 25, 1692–1696. 10.1055/s-0033-1339105. [DOI] [Google Scholar]
- La Motta C.; Sartini S.; Mugnaini L.; Simorini F.; Taliani S.; Salerno S.; Marini A. M.; Da Settimo F.; Lavecchia A.; Novellino E.; Cantore M. Pyrido [1, 2-a]pyrimidin-4-one derivatives as a novel class of selective aldose reductase inhibitors exhibiting antioxidant activity. J. Med. Chem. 2007, 50, 4917–4927. 10.1021/jm070398a. [DOI] [PubMed] [Google Scholar]
- Sudhakar H.; Mulakayala N. Facile Synthesis of Aurones using Amberlyst-15 as a Reusable Catalyst and their Biological Evaluation. Indian J. Adv. Chem. Sci. 2016, 4, 160–167. [Google Scholar]
- Chaudhary V.; Das S.; Nayak A.; Guchhait S. K.; Kundu C. N. Scaffold-Hopping and Hybridization Based Design and Building Block Strategic Synthesis of Pyridine-Annulated Purines: Discovery of Novel Apoptotic Anticancer Agents. RSC Adv. 2015, 5, 26051–26060. 10.1039/C5RA00052A. [DOI] [Google Scholar]
- Nayak A.; Satapathy S. R.; Das D.; Siddharth S.; Tripathi N.; Bharatam P. V.; Kundu C. N. Nanoquinacrine induced apoptosis in cervical cancer stem cells through the inhibition of hedgehog-GLI1 cascade: Role of GLI-1. Sci. Rep. 2016, 6, 20600. 10.1038/srep20600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousseau Y.; Mollard S.; Qiu H.; Richard L.; Cazal R.; Nizou A.; Vedrenne N.; Remi S.; Baaj Y.; Fourcade L.; Funalot B.; Sturtz F. G. In Vitro 3D Angiogenesis Assay in Egg White Matrix: Comparison to Matrigel, Compatibility to Various Species, and Suitability for Drug Testing. Lab. Invest. 2014, 94, 340–349. 10.1038/labinvest.2013.150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.