

Abstract

Homeobox transcription factor A9 (HoxA9) is overexpressed in 70% of patients diagnosed with acute myeloid leukemia (AML), whereas only a small subset of AML patients respond to current differentiation therapies. A cell line overexpressing HoxA9 was derived from the bone marrow of a lysozyme-GFP mouse. In this fashion, GFP served as an endogenous reporter of differentiation, permitting a high-throughput phenotypic screen against the MLPCN library. Two chemical scaffolds were optimized for activity yielding compound ML390, and genetic resistance and sequencing efforts identified dihydroorotate dehydrogenase (DHODH) as the target enzyme. The DHODH inhibitor brequinar works against these leukemic cells as well. The X-ray crystal structure of ML390 bound to DHODH elucidates ML390s binding interactions.
Keywords: AML, DHODH, HoxA9, ML390
Acute myeloid leukemia (AML) is a disease with a poor survival rate, and there has been no change to the standard of care therapy over the last 40 years.1
The concept of differentiation therapy, in contrast to cytotoxic chemotherapy, aims to restore the normal process of hematopoietic maturation from self-renewing progenitor to terminally differentiated effector cells. Differentiation therapy, e.g., in the form of all-trans retinoic acid, has revolutionized the treatment of patients with acute promyelocytic leukemia (APL), a small (10%) subset of AML.2 We therefore hypothesized that small molecule differentiation therapy to more broadly treat the other 90% of patients with AML would be of great utility.3
Homeobox (Hox) transcription factors are involved in hematopoietic differentiation.4 As cells mature normally, one sees the coordinated and stepwise activation and inactivation of various homeobox genes. In the process of normal myeloid development (i.e., the production of neutrophils and macrophages), the homeobox transcription factor HoxA9 must be turned “OFF” to permit differentiation. Therefore, it is of particular interest that HoxA9 is maintained “ON” in 70% of patients with AML.5 Other studies support the notion that dysregulated HoxA9 expression has particularly deleterious leukemic effects.6−8
With the goal of developing differentiation therapy for AML, we engineered a high-throughput phenotypic screen to identify small molecules that are capable of inducing differentiation in HoxA9-overexpressing progenitors.9 Bone marrow cells from lysozyme-GFP knock-in mice were transduced with a retroviral construct so that they overexpressed an estrogen receptor-HoxA9 fusion protein (ER-HoxA9). The ER-HoxA9 fusion protein results in a protein that is active (HoxA9 “ON”) in the presence of beta-estradiol and inactive (HoxA9 “OFF”) in the absence of beta-estradiol. Thus, active HoxA9 kept the cells frozen in an immature progenitor state, while inactivation of HoxA9 allowed for the normal and synchronous differentiation to mature neutrophils. Important for our screening purposes was that these cells are GFP (green fluorescent protein) negative at baseline but brightly GFP-positive upon maturation. Thus, in essence, our screen asked the question of “What small molecules are capable of triggering green fluorescence (as a surrogate for differentiation) in this model system of AML differentiation arrest?”
We recently published our results showing that inhibitors of dihydroorotate dehydrogenase (DHODH) induced differentiation in our model system as well as multiple other models of AML in vitro and in vivo.10 Importantly, DHODH inhibitors were effective at prolonging survival in animal models of leukemia. Here we present our work in which we optimized the cellular activity of screening hits. This process of chemical optimization and cocrystal structure with our lead compound helped to confirm that DHODH was the active target whose inhibition resulted in the differentiation of acute myeloid leukemia cells.
The lysozyme-GFP-ER-HoxA9 cells were screened against the NIH Molecular Libraries Probe Production Centers Network (MLPCN) library of approximately 330,000 compounds. As no inhibitors of HoxA9 exist, the estrogen receptor antagonist fulvestrant was used as a small molecule positive control. The hits were then tested for toxicity, ER antagonism, and autofluorescence. After multiple rounds of confirmatory testing, only 12 hits showed activity in dose-dependent testing. Two compounds (1 and 2, Figure 1) were chosen for further study based upon their activity in both murine and human cell line models of AML. Furthermore, neither 1 nor the chloro analogue of 2 were considered to be promiscuous binders based upon PubChem data available at the time.11,12
Figure 1.

Chemical structures of HTS hits 1 and 2.
Compound 1, a 2-anilino nicotinic acid, and analogues 1A–1G (Table 1) were synthesized by Ullman coupling, including compound 1F, a known inhibitor of aldo-keto-reductase 1C (AKR1C).13 As there are numerous NSAIDs having the 2-anilino nicotinic acid moiety, NSAIDs niflumic acid, flunixin, and clonixin were tested.14
Table 1. Activity of Compound 1 Analogues.

Calculated from combined data of three experiments; value ± std. error.
Geometric mean ± s.e.m. of at least three experiments.
Due to poorly shaped dose–response curves, unable to calculate std. error for this value.
Compound 2 is no longer commercially available, so the enantiomers of chloro analogue 3 were synthesized separately (Scheme 1), and cellular activity was detected in only the (R)-isomer. Maintaining the N-4-chlorobenzoyl-β-alanine, the (R)-amino tetraline group was replaced with an (R)-indane (4), (R)-phenethyl (5), and achiral cyclohexylamine (6). These compounds were inactive in cells, hence a series of compounds keeping the (R)-amino tetralin was synthesized, varying the substituents on the other phenyl ring, and the linker length between the two amides (Table 2).
Scheme 1. Synthesis of Compound 3 Analogues.

Reagents and conditions: (a) EDAC, Et3N/CH2Cl2; (b) TFA/CH2CH2; (c) ArCOCl, Et3N/CH2Cl2.
Table 2. Activity of Compound 3 Analogues.
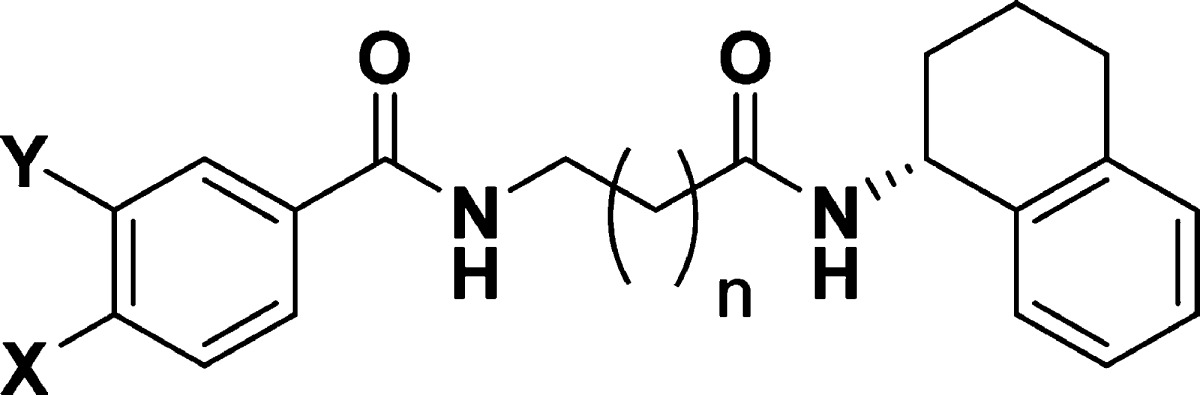
| compd | X | Y | n | ER-HOX-GFP (EC50 μM)c | U937 (EC50 μM)c | THP-1 (EC50 μM)c | DHODH (IC50, μM)d |
|---|---|---|---|---|---|---|---|
| (R)-3 | Cl | H | 1 | 4.5 ± 0.6 | 5.6 ± 0.9 | 3.2 ± 0.5 | 1.3 ± 0.2 |
| 3A | H | H | 1 | NAa | NA | NA | 12.4 ± 0.4 |
| 3B | Me | H | 1 | 8.1b | 4.2b | 2.7b | 1.9 ± 0.2 |
| 3C | OMe | H | 1 | 5.0b | 12b | 5.1b | 3.6 ± 0.2 |
| 3D | CN | H | 1 | NA | NA | NA | 8.9 ± 0.7 |
| 3E | F | H | 1 | NA | NA | NA | 9.5 ± 0.8 |
| 3F | CF3 | H | 1 | 3.6 ± 0.4 | 2.7 ± 0.9 | 2.5 ± 1.1 | 0.91 ± 0.2 |
| 3G | OCF3 | H | 1 | 1.8 ± 0.6 | 8.8 ± 0.8 | 6.5 ± 0.9 | 0.56 ± 0.1 |
| 3H | H | Cl | 1 | 7.9 ± 0.4 | 8.8 ± 0.9 | 6.5 ± 0.4 | 7.4 ± 0.4 |
| 3I | Cl | Cl | 1 | 3.2 ± 0.6 | 2.4 ± 1.0 | 2.5 ± 0.7 | 1.5 ± 0.1 |
| 3J | –OCF2O– | 1 | 4.7 ± 0.2 | 4.4 ± 0.9 | 4.3 ± 0.9 | 2.7 ± 0.5 | |
| 3K | Cl | H | 0 | NA | NA | NA | >20 |
| 3L | Cl | Cl | 0 | NA | NA | NA | >20 |
| 3M | CF3 | H | 0 | NA | NA | NA | >20 |
| 3N | OCF3 | H | 0 | NA | NA | NA | >20 |
| 3O | Cl | H | 2 | NA | NA | NA | >20 |
| 3P | Cl | Cl | 2 | NA | NA | NA | >20 |
| 3Q | CF3 | H | 2 | NA | NA | NA | >20 |
| 3R | OCF3 | H | 2 | NA | NA | NA | >20 |
| Brequinar | 0.51 ± 0.13 | 0.044 ± 0.07 | 0.094 ± 0.08 | 0.004 ± 0.001 | |||
NA = no activity detected at 10 μM.
Curve height minimal.
Calculated from combined data of three experiments; value ± std. error.
Geometric mean ± s.e.m. of at least three experiments.
The cellular activity of the compounds was tested in flow cytometry-based assays of differentiation. The lysozyme-GFP-ER-HoxA9 murine cell line (ER-HOX-GFP) and the THP1 and U937 human AML cell lines were cultured with compounds in a 4-day differentiation assay. At the end of the culture, the cells were stained with antibodies targeting cell-surface differentiation markers (e.g., CD11b/MAC1) and the upregulation of these cell-surface markers (as well as GFP in the murine cells) was measured by flow cytometry.10
In the compound 1 series, the original HTS hit compound 1 was the most active (Table 1). Replacing the chlorine atom with hydrogen (1A) or fluorine (1B) diminished activity, while replacement with a trifluoromethoxy group (1C) retained activity. Analogues where the pyridine nitrogen was replaced with a carbon (1D) or the anilino nitrogen of 1 was replaced with an oxygen (1E) displayed less activity. The AKR1C inhibitor (1F) and all the NSAIDs tested were inactive. The ER-HOX-GFP cell line was always the most sensitive of the three cell lines tested whenever activity was observed.
For the compound 3 series, removing the chlorine atom from (R)-3 (3A) diminished activity. Replacement of chlorine by electron-donating methyl (3B) or methoxy (3C) groups gave compounds with measurable but misleading EC50 values: while the EC50s obtained differ little from those of (R)-3, the maximal increase in GFP fluorescence (ER-HOX) or number of differentiated cells (U937 and THP-1) was marginal.15 Replacement of chlorine by cyano (3D) or fluoro (3E) groups led to inactive compounds, whereas replacement by trifluoromethyl (3F) or trifluoromethoxy (3G) groups improved activity relative to chlorine. Moving the chlorine atom from the para to the meta position (3H) gave a decrease in activity while the 3,4-dichloro analogue (3I) improved activity relative to (R)-3 in all three cell lines. (The purchased ortho-chloro analogue was inactive.) The difluorodioxolane analogue (3J) was active as well. Increasing or decreasing the chain length between the two amides by one methylene unit with the more active substituents led to a complete loss of activity in all cases tested (3K–3R). With the compound 3 series, none of the three cell lines could be considered more or less sensitive than the others.
The most potent compound against our engineered ER-HOX-GFP cell line was compound 3G. Compound 3G has moderate solubility (2.5 μM in phosphate buffered saline (PBS), pH 7.4) and high plasma protein binding (99% murine and human) and is stable in human plasma for 5 h and PBS for 24 h. Compound 3G was designated ML390, as part of the NIH Molecular Libraries Program, as a small-molecule probe active in overcoming differentiation arrest in a model of AML.9
At the time of its probe designation, the mechanism of action of ML390/(R)-3 and compound 1 was unknown. Kinase panels of 1, (R)-3, and (S)-3 yielded no potential kinase targets.16 As previously described,10 four resistant cell lines, both human and murine, were generated for (R)-3 and 1. Cell lines were cross-resistant to the two compounds, suggesting a similar mode of action (or similar mechanism of resistance) for these two chemically distinct scaffolds. RNA sequencing showed that only eight genes were highly (>4-fold) amplified in the resistant cell lines and that only one of these genes expressed a known enzyme, dihydroorotate dehydrogenase (DHODH). DHODH catalyzes the oxidation of dihydroorotate to orotate in de novo pyrimidine synthesis, e.g., uridine 5′-monophosphate.17
DHODH inhibitors have been previously evaluated in a number of therapeutic areas. Brequinar sodium was studied as an anticancer agent in patients with solid tumor malignancies and also studied as an immunosuppressive agent in preclinical models.17 Despite preclinical testing in a murine model of leukemia,18 brequinar was not tested in clinical trials of patients with leukemia. Leflunomide (Arava) is FDA-approved for the treatment of rheumatoid arthritis,19 and its active metabolite teriflunomide (Aubagio) is FDA-approved for the treatment of multiple sclerosis.20
Using a human DHODH enzyme inhibition assay, we confirmed that our active compounds inhibited the recombinant protein in vitro (Tables 1 and 2, last columns). Gratifyingly, both cellularly active (R)-3 and 1 showed activity against DHODH, while the cellularly inactive (S)-3 did not. In each series, the most potent DHODH inhibitor had the greatest effect in cells. The addition of uridine to the cell culture media abrogated the differentiation effects of 1, (R)-3, and ML390, providing further evidence that these compounds’ effects are due to their inhibition of DHODH-catalyzed pyrimidine synthesis.10 Interestingly, 2-anilino nicotinic acids are described in the patent literature as DHODH inhibitors,21 and compounds similar to ML390 with acylated (R)-ring-fused cyclohexylamines (GSK983) inhibit viral DHODH.22 ML390 and (R)-3 do not inhibit DHODH in the P. falciparum parasite, the causative agent of malaria.23
In order to elucidate detailed structural interaction of ML390 with human DHODH, the recombinant protein was crystallized both with and without ML390. Complete data sets were collected and processed to 1.70 and 1.66 Å resolution for DHODH and DHODH-ML390, respectively (Supporting Information Table S1). The atomic coordinates of DHODH and DHODH-ML390 have been deposited in the Protein Data Bank under the accession codes 5K9D and 5K9C, respectively.
The DHODH monomer folds in a well-conserved α/β barrel architecture composed by the N-terminal (Met30-Leu68) and C-terminal (Met78-Arg396) domains, connected by an extended loop as previously described.24 The N-terminal domain is composed of two α helices (α1 and α2), which are part of the putative ubiquinone binding-site, a cavity that gives access to the flavin mononucleotide (FMN) cofactor. The α1 helix composes the entrance of this cavity and we observed a poorly defined electron-density map suggesting high flexibility and a plastic behavior that can regulate the tunnel opening in order to accommodate different sizes of ligands. The canonical α/β barrel contains the reduced FMN and an orotate molecule in the dihydroorotate-binding site. The DHODH-ML390 complex comprises residues 35 to 218 and 226 to 396 and eight solvent sites.
The (R)-amino tetralin group of ML390 is located within the innermost part of the cavity and makes the closest contact, 2.3 Å, with isoalloxazine hydrogen H7M2 from FMN (Figure 2). The two amide groups make hydrogen bond interactions with Arg136 and Thr360 residues. The atomic interactions are ML390(O18)-Arg136(NE), ML390(O18)-Arg136(NH2), and ML390(N19)-Thr(OG1) for the amide of the (R)-amino tetralin group, and ML390(O13)-Arg136(NH2) and ML390(N14)-Thr(O) for the amide from 4-(CF3O)benzamide group (Figure 2). Additionally, the molecule has nonbonded contacts with Met43, Leu46, Gln47, Leu50, Pro52, Ala55, His56, Leu58, Ala59, Phe62, Phe98, Val134, Arg136, Tyr356, Thr360, and Phe361, with the (R)-amino tetralin group stacked with the imidazole ring of His56. Overall, the crystal structure is similar to several other reported structures of DHODH bound to small-molecule inhibitors, the interaction with Arg136 being the most important interaction.24,25
Figure 2.
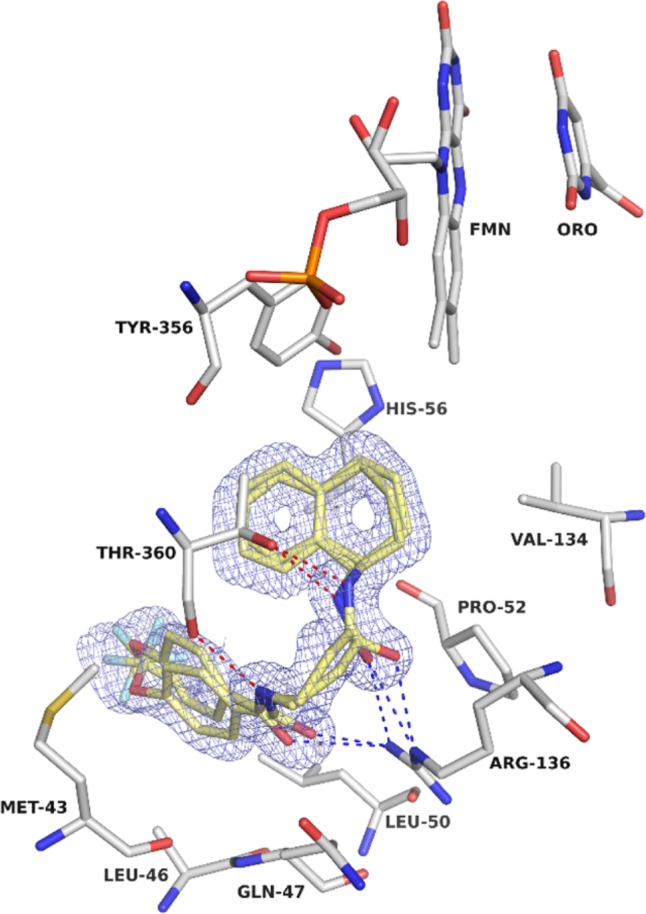
Crystal structure of DHODH-ML390. 2mFo-DFc electron density for ML390 (carbons in yellow) contoured at 1.0 σ. Dashed in blue are the hydrogen bonds formed between ML390 and Arg136; the red dashes represent the hydrogen bonds between ML390 and Thr360.
The X-ray structure indicates that binding to the enzyme may be increased by modifying ML390 with a ring in its central portion to lock the molecule into its binding conformation with the amide substituents cis about the ring. Synthesis of such compounds was unsuccessful, and furthermore, solutions of ML390 did show some decomposition over time. With DHODH established as the target of ML390 and the availability of more potent and well-studied DHODH inhibitors, further optimization of the ML390 scaffold was not pursued. Instead, our attention turned to brequinar, the gold standard of DHODH inhibitors (lit. DHODH IC50 10 nM26), which binds in a similar fashion to DHODH as does ML390.27 Brequinar was very active in our cellular and DHODH assays (Table 2, last row) and was chosen for in vivo experiments where it induced myeloid differentiation and prolonged life in several animal models of AML.10,28
In conclusion, high-throughput phenotypic screening for compounds that cause differentiation in a model of HoxA9-expressing AML led to the identification of two lead scaffolds. Modification of one scaffold did not improve activity, while uncovering the stereospecific activity and chemically optimizing the other series led to a probe that greatly facilitated target identification.
These experiments demonstrate the utility of phenotypic screening and medicinal chemistry in the identification of unexpected pathways (DHODH and pyrimidine synthesis) that may lead to the development of differentiation therapy for patients with acute myeloid leukemia.
Acknowledgments
The authors would like to thank Katrina Maxcy and Alexa Carver at the Massachusetts General Hospital for their technical assistance. The flow cytometry experiments were done with the support of the HSCI-CRM Flow Cytometry Facility. The small-molecule screen was supported by NIH 1R03DA032471-01 and the NIH Molecular Library Program (1 U54 HG005032-1 awarded to S.L.S.). Financial support was provided by Bayer. D.B.S. was supported by grants from the American Society of Hematology, Alex’s Lemonade Stand Foundation, the Leukemia and Lymphoma Society, Harvard Catalyst, and The American Cancer Society Institutional Research Grant. D.T.S. was supported by the Harvard Stem Cell Institute as well as the Amelia Peabody Charitable Fund and the Gerald and Darlene Jordan Chair of Medicine at Harvard University. Structural studies were supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), grant number 2012/25075-0 (to M.C.N.).
Glossary
ABBREVIATIONS
- AML
acute myeloid leukemia
- DHODH
dihydroorotate dehydrogenase
- Hox
homeobox transcription factor
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00316.
Experimental procedures and characterization of all compounds, and crystallographic data including superposition of ML390 and brequinar bound to DHODH (PDF)
Author Contributions
◆ These authors contributed equally to this work. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Yates J. W.; Wallace H. J.; Ellison R. R.; Holland J. F. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother. Rep. 1973, 57, 485–488. [PubMed] [Google Scholar]
- Fenaux P.; Le Deley M. C.; Castaigne S.; Archimbaud E.; Chomienne C.; Link H.; Guerci A.; Duarte M.; Daniel M. T.; Bowen D.; Huebner G.; Bauters F.; Fegueux N.; Fey M.; Sanz M.; Lowenberg B.; Maloisel F.; Auzanneau G.; Sadoun A.; Gardin C.; Bastion Y.; Ganser A.; Jacky E.; Dombret H.; Chastang C.; Degos L. Effect of all transretinoic acid in newly diagnosed acute promyelocytic leukemia. Results of a multicenter randomized trial. European APL 91 Group. Blood 1993, 82, 3241–9. [PubMed] [Google Scholar]
- Petrie K.; Zelent A.; Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr. Opin. Hematol. 2009, 16, 84–91. 10.1097/MOH.0b013e3283257aee. [DOI] [PubMed] [Google Scholar]
- Argiropoulos B.; Humphries R. K. Hox genes in hematopoiesis and leukemogenesis. Oncogene 2007, 26, 6766–76. 10.1038/sj.onc.1210760. [DOI] [PubMed] [Google Scholar]
- Golub T. R.; Slonim D. K.; Tamayo P.; Huard C.; Gaasenbeek M.; Mesirov J. P.; Coller H.; Loh M. L.; Downing J. R.; Caligiuri M. A.; Bloomfield C. D.; Lander E. S. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Lawrence H. J.; Sauvageau G.; Humphries R. K.; Largman C. The role of HOX homeobox genes in normal and leukemic hematopoiesis. Stem Cells 1996, 14, 281–91. 10.1002/stem.140281. [DOI] [PubMed] [Google Scholar]
- Borrow J.; Shearman A. M.; Stanton V. P. Jr.; Becher R.; Collins T.; Williams A. J.; Dube I.; Katz F.; Kwong Y. L.; Morris C.; Ohyashiki K.; Toyama K.; Rowley J.; Housman D. E. The t(7;11)(p15;p15) translocation in acute myeloid leukaemia fuses the genes for nucleoporin NUP98 and class I homeoprotein HOXA9. Nat. Genet. 1996, 12, 159–67. 10.1038/ng0296-159. [DOI] [PubMed] [Google Scholar]
- Drabkin H. A.; Parsy C.; Ferguson K.; Guilhot F.; Lacotte L.; Roy L.; Zeng C.; Baron A.; Hunger S. P.; Varella-Garcia M.; Gemmill R.; Brizard F.; Brizard A.; Roche J. Quantitative HOX expression in chromosomally defined subsets of acute myelogenous leukemia. Leukemia 2002, 16, 186–95. 10.1038/sj.leu.2402354. [DOI] [PubMed] [Google Scholar]
- Sykes D. B.; Haynes M. K.; Waller A.; Garcia M.; Ursu O.; Gouveia K. E.; Sklar L.; Lewis T. A.; Dandapani S.; Munoz B.; Scadden D. T.; Palmer M.; Schreiber S. L.. Discovering Small Molecules that Overcome Differentiation Arrest in Acute Myeloid Leukemia, Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US), 2010. [PubMed] [Google Scholar]
- Sykes D. B.; Kfoury Y. S.; Mercier F. E.; Wawer M. J.; Law J. M.; Jain E.; Haynes M. K.; Lewis T. A.; Schajnovitz A.; Lee D.; Meyer H.; Pierce K. A.; Tolliday N. J.; Waller A.; Ferrara S.; Stoeckigt D.; Maxcy K. L.; Cobert J. M.; Bachand J.; Szekely B. A.; Mukherjee S.; Sklar L. A.; Kotz J. D.; Clish C. B.; Sadrayev R. I.; Clemons P. A.; Janzer A.; Schreiber S. L.; Scadden D. T. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell 2016, 167, 171–186. 10.1016/j.cell.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compound 1 was considered active in 23 of 577 assays, compound 3 (racemic) was considered active in 10 of 341 assays.
- Compounds 1 and the chloro analogue of 2 are designated as C03 and C07, respectively, in ref (10).
- Bauman D. R.; Rudnick S. I.; Szewczuk L. M.; Jin Y.; Gopishetty S.; Penning T. M. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol. Pharm. 2005, 67, 60–68. 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- NSAIDs clonixin, meclofenamic acid, mefenamic acid, tofenamic acid, diclofenac, and celecoxib were inactive.
- see Figure S3B in ref (10) for a graphic example.
- Millipore Kinase Profiler.
- Munier-Lehmann H.; Vidalain P.-O.; Tangy F.; Janin Y. L. On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem. 2013, 56, 3148–3167. 10.1021/jm301848w. [DOI] [PubMed] [Google Scholar]
- Lyons S. D.; Christopherson I. Effects of brequinar and ciprofloxacin on de novo nucleotide biosynthesis in mouse L1210 leukemia. Biochem. Int. 1990, 22, 939–949. [PubMed] [Google Scholar]
- Breedveld F. C.; Dayer J. M. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2000, 59, 841–849. 10.1136/ard.59.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor P.; Wolinsky J. S.; Confavreux C.; Comi G.; Kappos L.; Olsson T. P.; Benzerdjeb H.; Truffinet P.; Wang L.; Miller A.; Freedman M. S. for the TEMSO trial group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N. Engl. J. Med. 2011, 365, 1293–1303. 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- Castro Palomino Laria J. C.; Erra Sola M.; Lozoya Toribio M. E.; Navarro Romero E.. Preparation of aminonicotinic acid derivatives and analogues as DHODH inhibitors. WO 2008077639, 2008.
- Deans R. M.; Morgens D. W.; Okesli A.; Pillay S.; Horlbeck M. A.; Kampmann M.; Gilbert L. A.; Li A.; Mateo R.; Smith M.; Glenn J. S.; Carette J. E.; Khosla C.; Bassik M. C. Parallel shRNA and CRISPER-Cas9 screens enable antiviral drug target identification. Nat. Chem. Biol. 2016, 12, 361–366. 10.1038/nchembio.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedingfiled P. T. P.; Cowen D.; Acklam P.; Cunningham F.; Parsons M. R.; McConkey G. A.; Fishwick G. W. G.; Johnson A. P. Factors influencing the specificity of inhibitor binding to the human and malaria parasite dihydroorotate dehydrogenases. J. Med. Chem. 2012, 55, 5841–5850. 10.1021/jm300157n. [DOI] [PubMed] [Google Scholar]
- Liu S.; Neidhardt E. A.; Grossman T. H.; Ocain T.; Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure 2000, 8, 25–33. 10.1016/S0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- Baumgartner R.; Walloschek M.; Kralik M.; Gotschlich A.; Tasler S.; Mies J.; Leban J. Dual binding mode of a novel series of DHODH inhibitors. J. Med. Chem. 2006, 49, 1239–1247. 10.1021/jm0506975. [DOI] [PubMed] [Google Scholar]
- Knecht W.; Henseling J.; Loffler M. Kinetics of inhibition of human and rat dihydroorotate dehygrogenase by atovaquone, lawsone derivatives, brequinar sodium and polyporic acid. Chem.-Biol. Interact. 2000, 124, 61–76. 10.1016/S0009-2797(99)00144-1. [DOI] [PubMed] [Google Scholar]
- See SI Figure 4 for crystal comparisons of ML390 and brequinar bound to DHODH.
- Less potent but well-studied DHODH inhibitors teriflunomide and leflunomide displayed little activity in our cellular assays.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.