

Abstract
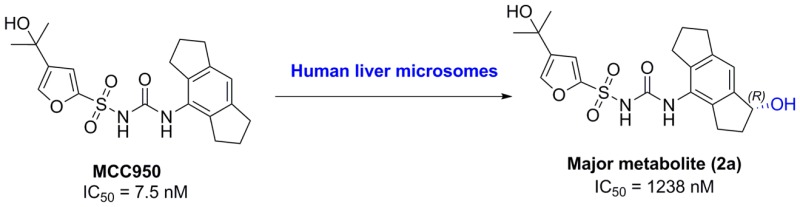
MCC950 is an orally bioavailable small molecule inhibitor of the NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome that exhibits remarkable activity in multiple models of inflammatory disease. Incubation of MCC950 with human liver microsomes, and subsequent analysis by HPLC–MS/MS, revealed a major metabolite, where hydroxylation of MCC950 had occurred on the 1,2,3,5,6,7-hexahydro-s-indacene moiety. Three possible regioisomers were synthesized, and coelution using HPLC–MS/MS confirmed the structure of the metabolite. Further synthesis of individual enantiomers and coelution studies using a chiral column in HPLC–MS/MS showed the metabolite was R-(+)- N-((1-hydroxy-1,2,3,5,6,7-hexahydro-s-indacen-4-yl)carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide (2a). Incubation of MCC950 with a panel of cytochrome P450 enzymes showed P450s 2A6, 2C9, 2C18, 2C19, 2J2, and 3A4 catalyze the formation of the major metabolite 2a, with a lower level of activity shown by P450s 1A2 and 2B6. All of the synthesized compounds were tested for inhibition of NLRP3-induced production of the pro-inflammatory cytokine IL-1β from human monocyte derived macrophages. The identified metabolite 2a was 170-fold less potent than MCC950, while one regioisomer had nanomolar inhibitory activity. These findings also give first insight into the SAR of the hexahydroindacene moiety.
Keywords: NLRP3, inflammasome, MCC950, microsome, metabolite, cytochrome P450
The NLRP3 inflammasome acts as a key mediator of inflammatory responses through the activation of caspase-1 leading to processing and release of the pro-inflammatory cytokines interleukin-1β (IL-1β) and interleukin-18 (IL-18).1,2 NLRP3 is associated with several diseases such as cryopyrin-associated periodic syndrome (CAPS), type 2 diabetes, atherosclerosis, asthma, gouty arthritis, and inflammatory central nervous system (CNS) diseases.3−5 Recently, a potent and selective small molecule inhibitor of the NLRP3 inflammasome, MCC950 (Figure 1), was reported (IC50 7.5 nM) with early promise for treatment of inflammatory diseases.6
Figure 1.

(A) (+)-QTOF-ESI-MS TIC. (B) XIC chromatogram of major human liver microsomal metabolite reaction showing the structure of MCC950 and the major metabolite.
The detection and identification of in vivo metabolites is an important part of the drug discovery and development process.7 Metabolically labile sites can be modified to improve the lead compounds ADME properties. The majority of drugs are eliminated from the body by metabolism as a detoxification process. However, metabolites may have pharmacological activity that influence the efficacy of the therapeutic or can be responsible for potential toxicity and drug–drug interaction issues. Elucidating the metabolic fate of drug candidates is a key requirement of regulatory bodies.8−10 Sometimes these metabolites guide the development of new and improved drugs.11 In this Letter, we report the identification, synthesis, and NLRP3 inhibitory activity of the major human microsomal metabolite of the NLRP3 inflammasome inhibitor MCC950 and identify the P450 enzymes, which catalyze its formation.
For the metabolite identification, MCC950 was incubated at 37 °C with human liver microsomes and the cofactor NADPH for 120 min. The reaction was quenched with ice cold CH3CN, vortexed, and centrifuged. The supernatant was analyzed using (+)- and (−)-QTOF-ESI-MS/MS, and one major metabolite was identified (Figure 1). In the (−)-ESI-MS the major metabolite showed a strong [M–H]− mass ion peak at m/z 419.1, while the (+)-ESI-MS showed a reduced intensity for the [M + H]+ mass ion peak and two facile losses of water (SI). The (−)-ESI-MS/MS spectra of MCC950 and its metabolite showed common fragments for the furan sulfonamide (m/z 204) (SI), which indicated that the oxygen was in the 1,2,3,5,6,7-hexahydro-s-indacene unit. The facile double dehydration of the metabolite ruled out phenolic oxidation, and three possible metabolite structures 1–3 were proposed and synthesized (Figure 2).
Figure 2.

Proposed structures of the major human microsomal metabolite of MCC950.
The synthesis of compounds 1–3 (Figure 2) required key intermediates 8 and 9 (Scheme 1); these intermediates were synthesized by procedures reported by Urban et al.12 Initially, 3-chloropropionyl chloride was prepared from 3-chloropropionic acid using oxalyl chloride with a catalytic amount of DMF in anhydrous DCM at room temperature.13 The resulting 3-chloropropionyl chloride was subsequently reacted with indane in the presence of aluminum trichloride in anhydrous DCM at room temperature to give chloroketone 5 in 85% yield. Chloroketone 5 was cyclized by heating in the presence of concentrated sulfuric acid at 55–60 °C for 48 h and was found to be a mixture of ketone 6 with small amounts of regioisomer 7, as confirmed by 1H NMR spectroscopy. These regioisomers were difficult to separate at this stage, and therefore, the mixture was used for the next step. Nitration of the regioisomeric ketones was achieved under standard conditions by reacting with concentrated H2SO4/HNO3 (1:1) at 0 °C. Three nitrated products were separated by column chromatography and identified as major isomer 8, and minor isomers 9 and 10. During the analysis of compounds 8–10, surprisingly we noticed some discrepancies between our 1H NMR data and that previously reported by Urban et al.12 We confirmed the structures of the three isomers 8–10 by 2D NMR analysis and single crystal X-ray crystallography for each compound (SI) and showed that 9 was incorrectly assigned by Urban et al.12
Scheme 1. Synthesis of Key Intermediates 8 and 9.

Reagents and conditions: (a) 3-chloropropionyl chloride, AlCl3, DCM, 2 h, rt, 85%; (b) H2SO4, 48 h, 55–60 °C; (c) H2SO4 and HNO3 (1:1), 1 h, 0–5 °C, 42%, 11%, 5% of 8, 9, and 10, respectively.
For the synthesis of compound 1 (Scheme 2), 8-nitro-3,5,6,7-tetrahydro-s-indacen-1(2H)-one (8) was reduced to the corresponding amine 11 by hydrogenation using 10% Pd/C as catalyst under hydrogen atmosphere (1 atm) at room temperature.14 Amine 11 was converted to the isocyanate intermediate 12 using Boc2O and DMAP in CH3CN at room temperature, then reacted in situ with 4-(2-hydroxypropan-2-yl)furan-2-sulfonamide 13 using NaH as base in anhydrous THF at room temperature to give sulfonylurea intermediate 14.15 Reduction of the ketone intermediate using NaBH4 in MeOH at room temperature for 3 h gave desired product 1 as a racemic mixture.
Scheme 2. Synthesis of Compound 1.

Reagents and conditions: (a) 10% Pd/C, H2 (1 atm), MeOH, rt, 2 h, 93%; (b) Boc2O, DMAP, CH3CN, rt 30 min; (c) NaH, THF, rt, 16 h; 67% (2-steps); (d) NaBH4, MeOH, rt, 3 h, 86%.
Compound 2 racemate (Figure 2) was synthesized via the same procedures used for compound 1 (Scheme 2) but using 4-nitro-3,5,6,7-tetrahydro-s-indacen-1(2H)-one (9) as the starting material.
Our initial route to synthesis of compound 3 is shown in Scheme 3. The ketone group of 8-nitro-3,5,6,7-tetrahydro-s-indacen-1(2H)-one (8) was reduced and dehydrated as before to give the corresponding olefin 16.16 However, conversion to alcohol intermediate 18 directly from olefin 16 was challenging. Several methods were investigated, without success, including TiCl4/NaBH4 and hydroboration using BH3·THF followed by oxidation with H2O2/NaOH.17,18 An alternative method proceeded via the epoxide 17, which was successfully produced by oxidation of olefin 16 using m-CPBA in DCM at room temperature.19 To open the epoxide ring regioselectively, toward desired alcohol 18, we followed a procedure reported by Finkielsztein et al. using ZnI2/NaCNBH4 in DCE under reflux conditions for 4 h.20 Intermediate 18 was then reduced to the aniline 19 and converted to the isocyanate 20 as before. Subsequent reaction with sulfonamide intermediate 13 gave the desired product 3.
Scheme 3. Synthesis of Compound 3.

Reagents and conditions: (a) NaBH4, MeOH, rt, 2 h, 97%; (b) p-TSA, toluene, reflux, 2 h, 80%; (c) m-CPBA, DCM, rt, 16 h, 82%; (d) ZnI2/NaCNBH4, DCE, reflux, 4 h, 86%; (e) 10% Pd/C, H2 (1 atm), MeOH, 16 h, 69%; (f) Boc2O, DMAP, CH3CN, rt, 30 min; (g) NaH, THF, rt, 16 h, 39% (2-steps).
To confirm the structure of the major metabolite of MCC950, initially we coeluted the major metabolite with each of the synthesized compounds 1–3 (Figure 2) using HPLC-(−)-ESI-SRM-MS/MS. MCC950 was incubated with human liver microsomes and NADPH, then sampled at t = 0; three further samples were taken at t = 120 min, and each were spiked with one of the synthesized compounds. Only compound 2 coeluted with the major metabolite of MCC950, thereby confirming the structure of the major metabolite formed in human liver microsomes.
To confirm the stereochemistry of the metabolite, we synthesized individual enantiomers (2a and 2b, Scheme 4) to use in coelution studies. Enantioselective reduction of intermediate 9 (Scheme 4) with (−) or (+) CBS catalyst and BH3·Me2S reducing agent gave the corresponding alcohols 21a and 21b.21,22 The (S)-(−)-CBS reagent produced (R)-alcohol (21a, yield 84%) in enantiomeric ratio 96:4 and (R)-(+)-CBS reagent produced (S)-alcohol (21b) in enantiomeric ratio 80:20 as determined by chiral HPLC. The (S)-alcohol (21b, yield 30%) was further purified by chiral semipreparative HPLC to 99:1 enantiomeric ratio. Compounds 21a and 21b were reduced to their corresponding amines by hydrogenation using 10% Pd/C catalyst under hydrogen atmosphere (1 atm) at room temperature. Subsequent conversion to the isocyanate and in situ reaction with 4-(2-hydroxypropan-2-yl)furan-2-sulfonamide 13, as before, give desired products 2a and 2b in 22% and 7% yield, respectively (see SI).
Scheme 4. Synthesis of Compound 2a and 2b.

Reagents and conditions: (a) (S)-(−)-CBS, BH3·Me2S, THF, 0 °C, 30 min, 84%; (b) (R)-(+)-CBS, BH3·Me2S, THF, 0 °C, 30 min, 30%. Further details in SI.
To confirm the structure of the major metabolite of MCC950, we coeluted with each of the synthesized compounds 2a and 2b (Scheme 4) using a chiral column in HPLC-(−)-ESI-SRM-MS/MS. Only compound 2a (R-isomer) coeluted thereby confirming stereochemistry of the major metabolite.
The major P450 isoforms responsible for the metabolism of MCC950 were then investigated. Metabolism of MCC950 by recombinant P450 enzymes was analyzed by incubation at 37 °C using enzymes coexpressed with NADPH-cytochrome P450 reductase (hCPR) in Escherichia coli membrane fractions. The following forms were assessed: P450 1A2, 2A6, 2A13, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 2S1, 2U1, 2W1, 3A4, 3A5, and 4A11. Membranes containing hCPR but no P450 were used as a negative control. The results detailed in Table 1 show eight of the 17 tested isoforms can form the metabolite, particularly CYP2A6 and 2C9.
Table 1. P450 Isoforms That Catalyze Formation of Major Metabolite.
| P450 isoform | major metabolite 2a (%), t = 2 h |
|---|---|
| 1A2 | 0.6 |
| 2A6 | 2.5 |
| 2B6 | 0.1 |
| 2C9 | 4.9 |
| 2C18 | 1.3 |
| 2C19 | 1.1 |
| 2J2 | 1.1 |
| 3A4 | 1.3 |
The ability of the synthesized compounds to inhibit NLRP3 inflammasome activity was investigated (Table 2). NLRP3-induced production of the pro-inflammatory cytokine IL-1β from LPS primed human monocyte derived macrophages, stimulated with ATP, was tested in the presence and absence of increasing concentrations of compounds 1–3, 2a, and 2b. The determined IC50 values were then compared to that for MCC950 used in this assay as positive control. The major metabolite 2a (IC50 1238 nM) was 170-fold less active than MCC950 but more active than the S-isomer 2b (IC50 6352 nM). Comparing the regioisomers, compound 3 (IC50 1828 nM) had almost similar activity to compound 2 (IC50 1971 nM), while interestingly compound 1 had IC50 in the nanomolar range (232 nM). The cytotoxicity of MCC950 and the hydroxylated isomers 1–3 was tested using human kidney and liver cell lines HEK293 or HepG2, and no evidence of toxicity was found. These results indicate, in the hexahydroindacene moiety, hydroxylation is generally detrimental to NLRP3 inhibitory activity but is better tolerated in compound 1 and could be further explored in future studies.
Table 2. NLRP3 Inflammasome Inhibition Activity (IL-1β IC50) and Cytotoxicity of MCC950 and Compounds 1–3.
| cytotoxicity |
|||
|---|---|---|---|
| compd | IL-1β IC50 (nM) | CC50 Hek293 (μM) | CC50 HepG2 (μM) |
| MCC950 | 7.5 | >62.5 | >62.5 |
| 1 | 232 | >62.5 | >62.5 |
| 2 | 1971 | >62.5 | >62.5 |
| 2a | 1238 | ||
| 2b | 6352 | ||
| 3 | 1828 | >62.5 | >62.5 |
This work gives the first indication as to the likely metabolic fate of MCC950 while simultaneously revealing SAR of the hexahydroindacene moiety. Future work will focus on characterizing MCC950 metabolism in vivo and examining the effects of MCC950 and the metabolite 2a on CYP inhibition and CYP induction processes.
In conclusion, the metabolism of MCC950, a potent and specific inhibitor of NLRP3 inflammasome, was investigated in human liver microsomes. MCC950 formed a major metabolite, the structure of which was proposed based on the analysis of (+)- and (−)-UPLC-QTOF-ESI-MS. Three possible regioisomers were synthesized, and coelution with the microsome product confirmed the metabolite structure as N-((1-hydroxy-1,2,3,5,6,7-hexahydro-s-indacen-4-yl) carbamoyl)-4-(2-hydroxypropan-2-yl)furan-2-sulfonamide (2). Synthesis of individual enantiomers 2a and 2b followed by coelution with microsomal metabolite using a chiral column in HPLC-(−)-ESI-SRM-MS/MS confirmed the stereochemistry of the metabolite as R enantiomer 2a. The R enantiomer 2a was 170-fold less potent as an NLRP3 inhibitor than MCC950 in human cell-based assay. MCC950 incubated with a panel of P450 enzymes showed isoforms 1A2, 2A6, 2B6, 2C9, 2C18, 2C19, 2J2, and 3A4 are likely to be responsible for formation of the major metabolite in human tissues, with the relative importance of individual forms likely to depend on their expression level in different tissues and subjects.
Acknowledgments
We would like to thank David Edwards (IMB, UQ) for advice and support in analytics and purification. We would like to acknowledge The Australian Red Cross for their supply of buffy coat tested blood for biological assays. We thank the IMB Mass Spectroscopy facility manager Alun Jones for his support.
Glossary
ABBREVIATIONS
- ADME
absorption, distribution, metabolism, and excretion
- NADPH
nicotinamide adenine dinucleotide phosphate
- HepG2
human liver hepatocellular carcinoma G2 cells
- HEK293
human embryonic kidney 293 cells
- IL-1β
interleukin-1β
- XIC
extracted-ion chromatogram
- SI
- Pd/C
palladium on carbon
- MeOH
methanol
- m-CPBA
meta-chloroperoxybenzoic acid
- TiCl4
titanium tetrachloride
- NaH
sodium hydride
- NaBH4
sodium borohydride
- NaCNBH4
sodium cyanoborohydride
- ZnI2
zinc iodide
- BH3·THF
borane tetrahydrofuran complex
- CBS
Corey–Bakshi–Shibata
- BH3·Me2S
borane dimethyl sulfide complex
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00198.
Experimental procedures, characterization data for all intermediates and final compounds, descriptions of biological assays, X-ray crystal structures of intermediates 8, 9 and 10, and LC–MS and LC–MS/MS spectra of MCC950 and its major metabolite 2a (PDF)
The authors are thankful to National Health and Medical Research Council (NHMRC) for the financial support in terms of funding (NHRMRC grant APP1086786). M.S. is supported by an UQ International Scholarship (UQI) Ph.D. scholarship by The University of Queensland. M.A.C. is an NHMRC Principal Research Fellow (APP1059354).
The authors declare no competing financial interest.
Supplementary Material
References
- Schroder K.; Tschopp J. The inflammasomes. Cell 2010, 140, 821–832. 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Gross O.; Thomas C. J.; Guarda G.; Tschopp J. The inflammasome: an integrated view. Immunol. Rev. 2011, 243, 136–151. 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- Strowig T.; Henao-Mejia J.; Elinav E.; Flavell R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- Walsh J. G.; Muruve D. A.; Power C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. 10.1038/nrn3638. [DOI] [PubMed] [Google Scholar]
- Guo H.; Callaway J. B.; Ting J. P. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll R. C.; Robertson A. A.; Chae J. J.; Higgins S. C.; Munoz-Planillo R.; Inserra M. C.; Vetter I.; Dungan L. S.; Monks B. G.; Stutz A.; Croker D. E.; Butler M. S.; Haneklaus M.; Sutton C. E.; Nunez G.; Latz E.; Kastner D. L.; Mills K. H.; Masters S. L.; Schroder K.; Cooper M. A.; O’Neill L. A. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. 10.1038/nm.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J. H.; Lu A. Y. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol. Rev. 1997, 49, 403–449. [PubMed] [Google Scholar]
- Kumar G. N.; Surapaneni S. Role of drug metabolism in drug discovery and development. Med. Res. Rev. 2001, 21, 397–411. 10.1002/med.1016. [DOI] [PubMed] [Google Scholar]
- Pellegatti M. Preclinical in vivo ADME studies in drug development: a critical review. Expert Opin. Drug Metab. Toxicol. 2012, 8, 161–172. 10.1517/17425255.2012.652084. [DOI] [PubMed] [Google Scholar]
- Hop C. E. Role of ADME studies in selecting drug candidates: Dependence of ADME parameters on physicochemical properties. Encyclopedia of Drug Metabolism and Interactions 2012, 1–43. 10.1002/9780470921920.edm049. [DOI] [Google Scholar]
- Kang M. J.; Song W. H.; Shim B. H.; Oh S. Y.; Lee H. Y.; Chung E. Y.; Sohn Y.; Lee J. Pharmacologically active metabolites of currently marketed drugs: potential resources for new drug discovery and development. Yakugaku Zasshi 2010, 130, 1325–1337. 10.1248/yakushi.130.1325. [DOI] [PubMed] [Google Scholar]
- Urban F. J.; John Jasys V.; Raggon J. W.; Buzon R. A.; Hill P. D.; Eggler J. F.; Weaver J. D. Novel synthesis of 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl]urea, an anti-inflammatory agent. Synth. Commun. 2003, 33, 2029–2043. 10.1081/SCC-120021029. [DOI] [Google Scholar]
- Sariola E.; Kotiaho A.; Tkachenko N. V.; Lemmetyinen H.; Efimov A. Mono-, bis- and tetrahydroxy phthalocyanines as building blocks for monomolecular layer assemblies. J. Porphyrins Phthalocyanines 2010, 14, 397–411. 10.1142/S1088424610002185. [DOI] [Google Scholar]
- Koike H.; Oda K.; Nishino Y.; Kakinuma M.. Process for producing triterpene derivative. US Patent 7,223,882, 2007.
- Knölker H. J.; Braxmeier T.; Schlechtingen G. A novel method for the synthesis of isocyanates under mild conditions. Angew. Chem., Int. Ed. Engl. 1995, 34, 2497–2500. 10.1002/anie.199524971. [DOI] [Google Scholar]
- Xu H.; Wolf C. Asymmetric synthesis of chiral 1, 3-diaminopropanols: Bisoxazolidine-catalyzed C–C bond formation with α-keto amides. Angew. Chem., Int. Ed. 2011, 50, 12249–12252. 10.1002/anie.201105778. [DOI] [PubMed] [Google Scholar]
- Kano S.; Tanaka Y.; Hibino S. Formation of alcohols from alkenes with TiCl4-NaBH4. J. Chem. Soc., Chem. Commun. 1980, 9, 414–415. 10.1039/C39800000414. [DOI] [Google Scholar]
- Zaveri N.; Chao W.-R.; Bensari A.. Analogues of green tea polyphenols as chemotherapeutic and chemopreventive agents. US Patent 7,122,573, 2006.
- Gross M. F.; Beaudoin S.; McNaughton-Smith G.; Amato G. S.; Castle N. A.; Huang C.; Zou A.; Yu W. Aryl sulfonamido Indane inhibitors of the Kv1.5 ion channel. Bioorg. Med. Chem. Lett. 2007, 17, 2849–2853. 10.1016/j.bmcl.2007.02.052. [DOI] [PubMed] [Google Scholar]
- Finkielsztein L.; Aguirre J.; Lantano B.; Alesso E.; Iglesias G. M. ZnI2/NaCNBH3 as an efficient reagent for regioselective ring opening of the benzylic epoxide moiety. Synth. Commun. 2004, 34, 895–901. 10.1081/SCC-120028362. [DOI] [Google Scholar]
- Corey E. J.; Helal C. J. Reduction of carbonyl compounds with chiral oxazaborolidine catalysts: a new paradigm for enantioselective catalysis and a powerful new synthetic method. Angew. Chem., Int. Ed. 1998, 37, 1986–2012. . [DOI] [PubMed] [Google Scholar]
- Teitelbaum A. M.; Meissner A.; Harding R. A.; Wong C. A.; Aldrich C. C.; Remmel R. P. Synthesis, pH-dependent, and plasma stability of Meropenem prodrugs for potential use against drug-resistant tuberculosis. Bioorg. Med. Chem. 2013, 21, 5605–5617. 10.1016/j.bmc.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.