

Abstract

GPR142 has been identified as a potential glucose-stimulated insulin secretion (GSIS) target for the treatment of type 2 diabetes mellitus (T2DM). A class of triazole GPR142 agonists was discovered through a high throughput screen. The lead compound 4 suffered from poor metabolic stability and poor solubility. Lead optimization strategies to improve potency, efficacy, metabolic stability, and solubility are described. This optimization led to compound 20e, which showed significant reduction of glucose excursion in wild-type but not in GPR142 deficient mice in an oral glucose tolerance test (oGTT) study. These studies provide strong evidence that reduction of glucose excursion through treatment with 20e is GPR142-mediated, and GPR142 agonists could be used as a potential treatment for type 2 diabetes.
Keywords: GPR142 agonists, type 2 diabetes mellitus (T2DM), triazole, glucose-stimulated insulin secretion (GSIS), solubility
Type 2 diabetes mellitus (T2DM) is a disease derived from multiple causative factors and characterized by elevated levels of plasma glucose (hyperglycemia) in the fasting state or after administration of glucose during an oral glucose tolerance test (oGTT).1 Type 2 diabetic patients have increased risk of coronary heart disease, peripheral vascular disease, hypertension, nephropathy, and retinopathy.2 Therefore, safe and effective treatments of T2DM are highly desirable. Recently there has been a focus on the development of novel agents that promote glucose-stimulated insulin secretion (GSIS) from pancreatic β-cells, such as DPP-4 inhibitors,3 which could minimize the potential risk of hypoglycemia and other adverse effects associated with many current therapies.4
GPR 142 is a Gq-coupled, family A seven transmembrane G-protein-coupled receptor (GPCR) that is highly expressed in pancreatic β-cells and has been identified as a potential GSIS target for the treatment of T2DM.5,6 Considerable interest is thus centered on the generation of potent and selective small molecule ligands as possible therapeutic agents for T2DM patients.7−11 For example, Yu et al. have reported the discovery of potent aminopyrazole–phenylalanine based GPR142 agonist 1 (Figure 1), which showed GPR142 dependent stimulation of insulin secretion in isolated mouse islets.9 Toda et al. reported GPR142 agonist 2, which showed potent glucose lowering effects during an oral glucose tolerance test in mice and monkeys.10 More recently, Wilson et al. reported GPR142 agonist 3 (h-GPR142, FLIPR, EC50 = 63 nM), which was active in oGTT study in wide type mice, but not in GPR142 deficient mice.11
Figure 1.

Examples of GPR142 agonists.
Herein, we report our hit-to-lead efforts in discovering novel, potent and orally bioavailable triazole GPR142 agonists and their pharmacological effects in mice. Triazole 4 was identified as a GPR142 agonist hit from a high throughput screen (HTS) (Figure 2).12 It exhibited modest human GPR142 agonist activity in a FLIPR assay (h-GPR142, FLIPR, EC50 = 115 nM) and an inositol phosphate (IP) accumulation assay (h-GPR142, IP, EC50 = 252 nM).13 Additionally, compound 4 had potent mouse GPR142 activity (m-GPR142, FLIPR, EC50 = 37 nM; m-GPR142, IP, EC50 = 23 nM). Unfortunately, 4 only showed 16% reduction of glucose excursion at a 10 mg/kg oral dose in a mouse oral glucose tolerance test (oGTT).14 Given that 4 has relatively low mouse microsomal stability (32% remaining after incubation for 30 min in mouse microsomes), and low solubility in phosphate buffer solution (PBS) at pH = 2 and 7, a better tool compound for in vivo proof-of-concept studies was sought.
Figure 2.

Structure and profile of HTS hit 4.
We first investigated whether the amide could be replaced by an amine, a reverse amide, or a urea linker (analogues 10a–10c, Table 1). The synthesis of 10a–10c started with alcohol 5 (Scheme 1). Mesylation of 5 followed by azide displacement gave azide 6. A copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction of azide 6 with known alkyne 7(12) furnished triazole 8 in good yield. Deprotection of the Boc group of 8 afforded amine 9, which was subsequently converted to 10a–10c.
Table 1. SAR on the Amide and Naphthyl Replacementsa.

nd = not determined; assay protocols: see ref (15).
Scheme 1. Synthesis of Triazole Analogues 10a–10c.

Reagents and conditions: (a) MsCl, Et3N, DCM; (b) NaN3, 15-crown-5, DMF, 90 °C; (c) CuSO4, sodium ascorbate, t-BuOH/H2O; (d) HCl, DCM/MeOH; (e) 1-bromonaphthalene, NaOtBu, BrettPhos Pd G1, dioxane, 60 °C; (f) 1-isocyanatonaphthalene, Et3N, DCE; (g) 1-naphthoyl chloride, TEA, DCE.
FLIPR assays were used to first assess GPR142 potency given their higher throughput. Potent compounds of interest from the FLIPR assay were then tested in GPR142 Inositol phosphate (IP) assays as secondary assays.13 Amine 10a, reverse amide 10b, and urea 10c lost GPR142 potency in the FLIPR assay, suggesting that a rigid amide moiety was required for GPR142 potency.
We then set out to replace the flat naphthyl group (Table 1, 10d–10g) to improve the physicochemical properties of the current lead. The synthesis of 10d–10g was initiated with ethyl 2-bromoacetate 11 (Scheme 2). Azide displacement of the bromide followed by the CuAAC reaction afforded triazole 13. Ester hydrolysis of 13 gave acid 14. Amide coupling of amines with 14 gave the final analogues 10d–10g. A very tight structure–activity relationship (SAR) was observed. The aliphatic tetrahydronaphthalene analogue 10d lost GPR142 activity. While 2,3-difluorophenyl analogue 10e was considerably less potent than 4, the more lipophilic analogues, 2,3-dimethylphenyl 10f and 2,3-dichlorophenyl 10g, maintained GPR142 activity in both FLIPR and IP assays.
Scheme 2. Synthesis of Triazole Amide Analogues 10d–10g.

Reagents and conditions: (a) NaN3, 15-crown-5, DMF, 90 °C; (B) CuSO4, sodium ascorbate, t-BuOH/H2O; (c) LiOH, THF/MeOH/H2O; (d) amine, HATU, DIEA, DMF.
After we discovered that 2,3-dichlorophenyl could serve as a replacement for the naphthyl group, we explored the SAR on the middle phenyl ring, and the left-hand side imidazole ring (Table 2). The general synthesis of 20a–20f is shown in Scheme 3. Displacement of the 4-fluoro substituent of aryl aldehyde 16 with imidazole 15 afforded aldehyde 17. Alkynylation of aldehyde 17 with the Bestmann–Ohira reagent afforded alkyne 18, which was reacted with azide 19 to give triazole products 20a–20f. A similar route was used to prepare analogue 20g.
Table 2. SAR on the Left Side Imidazole Phenyl Moietiesa.
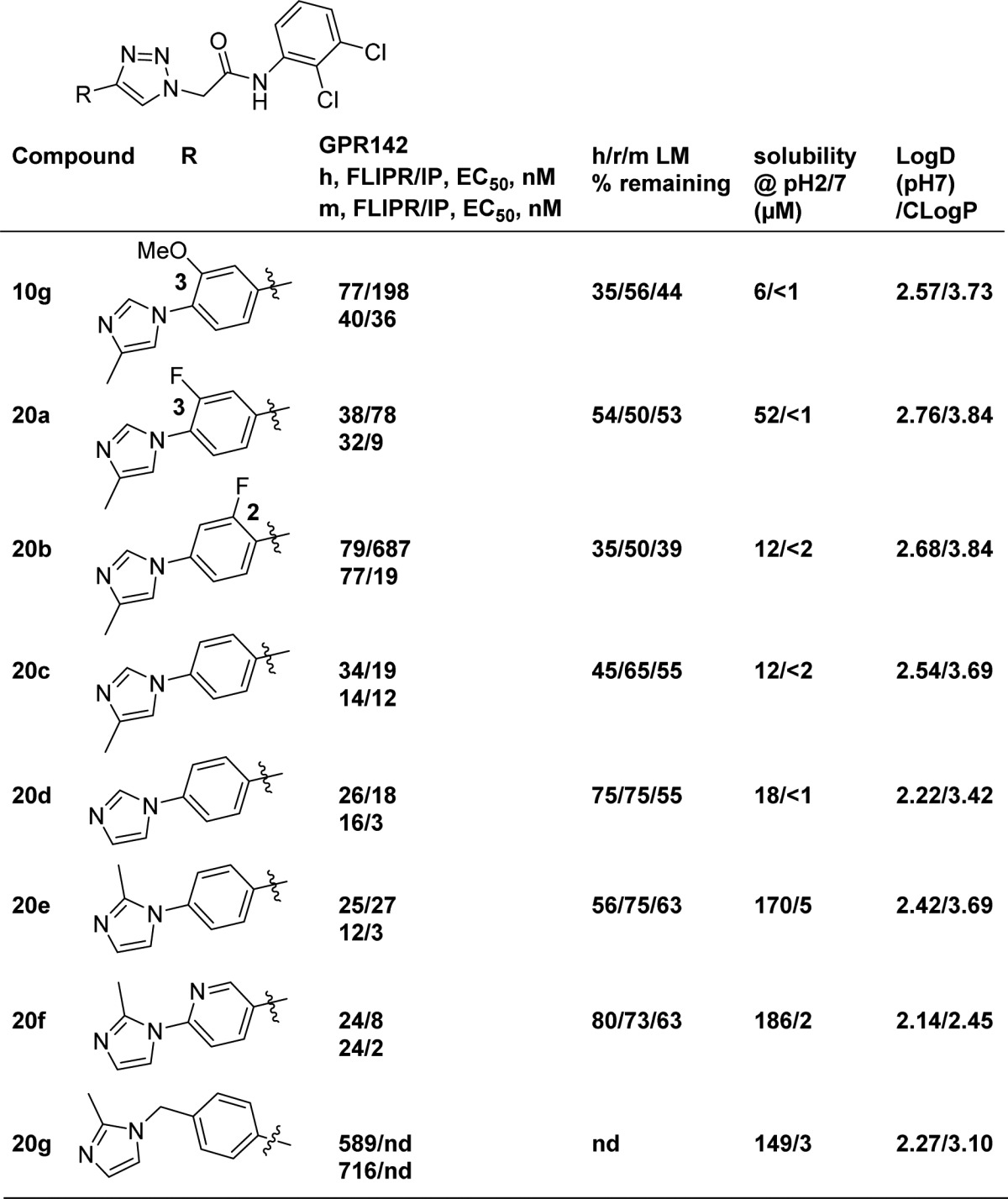
nd = not determined; h/r/m LM % remaining = percentage of compound remaining intact after incubation in human/rat/mouse liver microsomes at 1 μM for 30 min at 37 °C.
Scheme 3. General Synthesis of Triazole Analogues 20a–20f.

Reagents and conditions: (a) K2CO3, DMF, heat; (b) K2CO3, MeOH; (c) CuSO4, sodium ascorbate, t-BuOH/H2O.
As shown in Table 2, 10g demonstrated moderately low microsome stabilities across various species and poor solubility. Replacing OMe with F resulted in modestly improved potency and liver microsome stability (20a). However, 2-fluoro substitution 20b on the middle phenyl ring reduced potency. It is noteworthy that the unsubstituted analogues 20c and 20d maintained good GPR142 potency and showed balanced human and mouse GPR142 potency in both FLIPR and IP assays.
Although the potency and metabolic stability were improved, 20c and 20d still suffered from poor solubility in phosphate buffered saline (PBS). It is well-known that poor solubility of drug candidates presents challenges during drug development.15 Improving aqueous solubility was therefore viewed as essential to identify a promising tool compound or potential drug from this series.
The Yalkowsky solubility equation (Log[solubility (M)] = 0.5 – (Log P) – 0.01{[melting point (°C)] – 25}) demonstrates that LogP and melting point are two key parameters that determine solubility.16 In addition, as summarized by Ishikawa, one effective strategy to reduce melting point is disrupting molecular planarity to decrease crystal packing.17 We hypothesized that the degree of planarity of the linear triaryl would influence crystal packing. Conformational analysis using density functional theory of compound 20d indicated that the dihedral angle between the phenyl and terminal imidazole was 38°, while the preferred angle was increased to 47° with 2-methyl imidazole analogue 20e.18 To our satisfaction, 20e improved solubility in PBS dramatically at pH = 2 and modestly at pH = 7, while maintaining good GPR142 potency. It is worth noting that these dihedral angles are rather similar and that the influence on solubility may be small. The substitutions on the molecules modulate the physicochemical properties and influence the solubility and metabolic stability, which may explain the poor solubility of 10g, even with a dihedral angle of 44°.18 Since the pH = 7 solubility of 20e was still low, the aza analogue 20f was designed and synthesized in hope of improving solubility by reducing cLogP. Although the improvement in solubility was not significant with this modification, the GPR142 activity was well tolerated, and human liver microsome stability was further improved. To further disrupt the planarity, analogue 20g with an additional methylene linker was prepared. However, this modification resulted in significant loss of GPR142 activity.
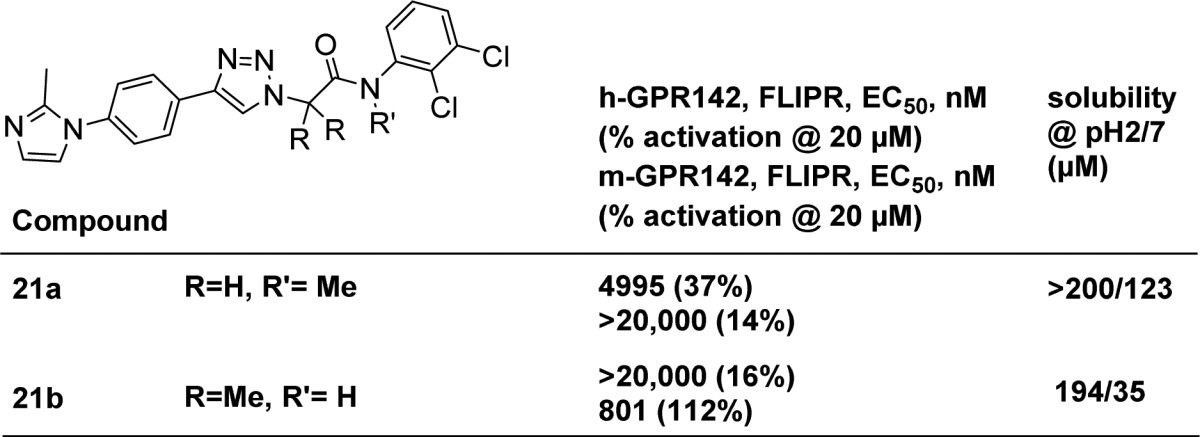
Inspired by the methyl effect in 20e, we were also interested in whether a methyl group could be incorporated into the right-hand side of the molecule (Table 3). Indeed the solubilities of methylated analogues 21a and 21b were significantly improved, potentially by disrupting the crystal packing of the right-hand of the molecules.18 Unfortunately, those modifications were not tolerated for GRP142 activity.
Table 3. Methylated Analogues of 20e.
Given their improved GPR142 potency and metabolic stability, compounds 20a, 20e, and 20f were further profiled (Table 4). These compounds exhibited acceptable selectivity profiles in CYPs 3A4 and 2D6 and in cardiac ion channels, and good permeability (Papp) in LLC-PK1 cells. These compounds were also evaluated in rat pharmacokinetic studies (rat PK). All three analogues showed low to modest plasma clearance (Clp) and good oral bioavailabilities (F%). When corrected for plasma protein binding, however, compounds 20e and 20f exhibited lower unbound clearance (Clu = Clp/fu; fu = unbound or free fraction of compound in plasma) in rats compared to 20a.
Table 4. Profile of 20a, 20e, and 20f.
Compound 20e was selected for evaluation of its ability to potentiate glucose-stimulated insulin secretion (GSIS) ex vivo in freshly isolated mouse islets (Figure 3).20 While treatment at low glucose concentration (2 mM) did not show any effect on insulin secretion, at high glucose concentration (16 mM), treatment with 20e showed a modest effect on increasing insulin secretion. In the same experiment, GLP-1 showed a significant effect on enhancing insulin secretion in a glucose-dependent manner as anticipated.
Figure 3.
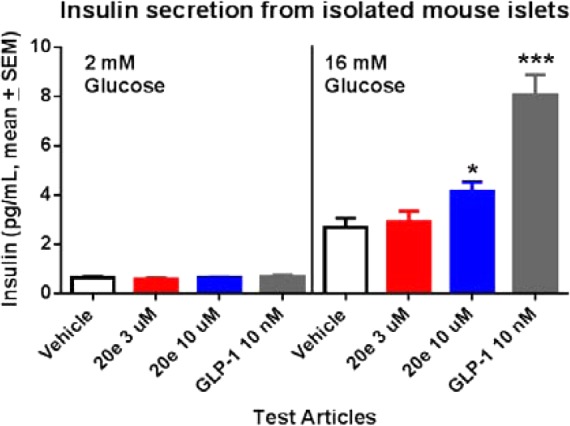
Compound 20e and GLP-1 showed glucose-dependent insulin secretion in isolated mouse islets. *p < 0.05; *** p < 0.0001 versus vehicle (1-way ANOVA test).
Compound 20e was further evaluated in vivo in lean mice in an oral glucose tolerance test (oGTT).20 In Figure 4, the black bar represents the glucose excursion after the mice were orally challenged with 5 g/kg dextrose. Compound 20e showed a dose-dependent reduction of glucose excursion at oral doses from 1 to 30 mpk. At 30 mpk, 20e achieved 89% glucose AUC0–120 min reduction (11.5 μM of total 20e and 149 nM of free 20e @ 2.5 h postdose). To demonstrate that the observed glucose lowing effect by 20e was GPR142 mediated, 20e was tested in GPR142 KO mice in the same oGTT paradigm, and the compound did not reduce glucose excursion in these KO mice (Figure 5). This suggests that the glucose lowering effect of 20e is mediated through its GPR142 agonist activity.
Figure 4.
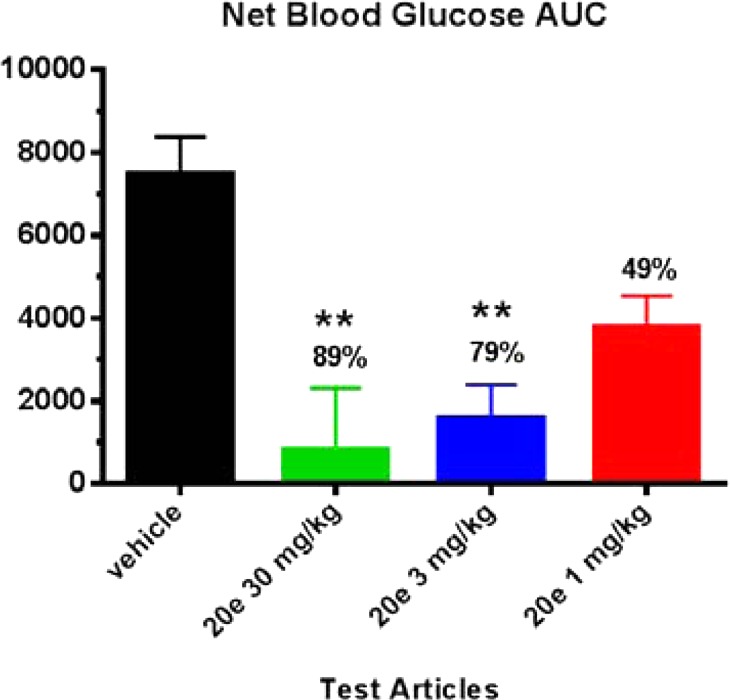
Compound 20e showed a dose-dependent reduction of glucose excursion in mouse oGTT; N = 7. **p < 0.001 versus vehicle group via ANOVA and Dunnett’s post-test; total plasma drug levels @ 2.5 h postdosing, 30 mg/kg (11.5 μM), 3 mg/kg (0.274 μM), and 1 mg/kg (0.044 μM).
Figure 5.
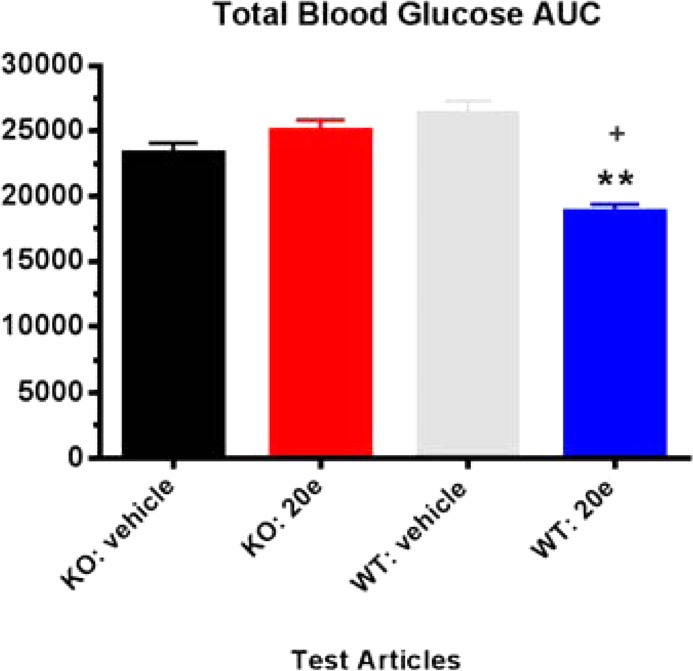
Glucose AUC0–120 min following glucose challenge post 20e treatment in both wild type and GPR142 KO mice; N = 7. +p < 0.01 vs KO treatments; **p < 0.001 vs WT veh + dex.
In summary, a novel class of triazole GPR142 agonists was discovered from HTS. Optimization of an initial HTS hit focused on improving not only GPR142 potency, but also metabolic stability and solubility. Compound 20e demonstrated robust reduction of glucose excursion in wild-type but not in GPR142 KO mice. This mechanism-based glucose reduction provided strong evidence that GPR142 agonists could be potentially used in type 2 diabetes treatment. Further development of this class of compounds will be reported in due course.
Acknowledgments
The authors gratefully acknowledge Dr. Eric Meade, Dr. Andrew Stamford, and Dr. Jeffrey Hale for reviewing and providing comments on this manuscript; and Jason L. Hoar for liver microsome stability studies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00314.
Syntheses and characterization data for compound 20e, assay protocols, and computational modeling (PDF)
Author Contributions
⊥ Project leads.
The authors declare no competing financial interest.
Supplementary Material
References
- Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2009, 32, S62–S67. 10.2337/dc09-S062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skyler J. S. Diabetes Mellitus: Pathogenesis and TreatmentStrategies. J. Med. Chem. 2004, 47, 4113–4117. 10.1021/jm0306273. [DOI] [PubMed] [Google Scholar]
- Weber A. E. Dipeptidyl Peptidase IV Inhibitors for the Treatment of Diabetes. J. Med. Chem. 2004, 47, 4135–4141. 10.1021/jm030628v. [DOI] [PubMed] [Google Scholar]
- Bodmer M.; Meier C.; Krähenbühl S.; Jick S. S.; Meier C. R. Metformin, Sulfonylureas, or Other Antidiabetes Drugs and the Risk of Lactic Acidosis or Hypoglycemia, A nested case-control analysis. Diabetes Care 2008, 31 (11), 2086–2091. 10.2337/dc08-1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suesens U.; Hermans-Borgmeyer I.; Urny J.; Schaller H. C. Characterisation and differential expression of two very closely related G-protein-coupled receptors, GPR139 and GPR142, in mouse tissue and during mouse development. Neuropharmacology 2006, 50, 512–520. 10.1016/j.neuropharm.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Wang J.; Carrillo J. J.; Lin H. V. GPR142 Agonists Stimulate Glucose-Dependent Insulin Secretion via Gq-Dependent Signaling. PLoS One 2016, 11 (4), e0154452. 10.1371/journal.pone.0154452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizarzaburu M.; Turcotte S.; Du X.; Duqutte J.; Fu A.; Houze J.; Li L.; Liu J.; Oda K.; Okuyama R.; Reagan J. D.; Yu M.; Medina J. C. Discovery and optimization of a novel series of GPR142 agonists for the treatment of type 2 diabetes mellitus. Bioorg. Med. Chem. Lett. 2012, 22, 5942–5947. 10.1016/j.bmcl.2012.07.063. [DOI] [PubMed] [Google Scholar]
- Du X.; Kim Y.-J.; Lai S.; Chen X.; Lizarzaburu M.; Turcotte S.; Oda K.; Okuyama R.; Nara F.; Murakoshi M.; Fu A.; Reagan J.; Liu Q.; Zhang Y.; Motani A.; Fan P.; Xiong Y.; Shen W.; Li L.; Houze J.; Medina J. C. Phenylalanine derivatives as GPR142 agonists for the treatment of Type II diabetes. Bioorg. Med. Chem. Lett. 2012, 22, 6218–6223. 10.1016/j.bmcl.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Yu M.; Lizarzaburu M.; Motani A.; Fu Z.; Du X.; Liu J.; Jiao X.; Lai S.; Fan P.; Fu A.; Liu Q.; Murakoshi M.; Nara F.; Oda K.; Okuyama R.; Reagan J. D.; Watanabe N.; Yamzaki M.; Xiong Y.; Zhang Y.; Zhuang R.; Lin D. C.; Houze J. B.; Medina J. C.; Li L. Aminopyrazole-Phenylalanine Based GPR142 Agonists: Discovery of Tool Compound and in Vivo Efficacy Studies. ACS Med. Chem. Lett. 2013, 4, 829–834. 10.1021/ml4000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda N.; Hao X.; Ogawa Y.; Oda K.; Yu M.; Fu Z.; Chen Y.; Kim Y.; Lizarzaburu M.; Lively S.; Lawlis S.; Murakoshi M.; Nara F.; Watanabe N.; Reagan J. D.; Tian H.; Fu A.; Motani A.; Liu Q.; Lin Y.-J.; Zhuang R.; Xiong Y.; Fan P.; Medina J.; Li L.; Izumi M.; Okuyama R.; Shibuya S. Potent and Orally Bioavailable GPR142 Agonists as Novel Insulin Secretagogues for the Treatment of Type 2 Diabetes. ACS Med. Chem. Lett. 2013, 4, 790–794. 10.1021/ml400186z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. E.; Kurukulasuriya R.; Sinz C.; Lombardo M.; Bender K.; Parker D.; Sherer E. C.; Costa M.; Dingley K.; Li X.; Mitelman S.; Tong S.; Bugianesi R.; Ehrhardt A.; Priest B.; Ratliff K.; Ujjainwalla F.; Nargund R.; Hagmann W. K.; Edmondson S. Discovery and development of benzo-[1,2,4]-triazolo-[1,4]-oxazepine GPR142 agonists for the treatment of diabetes. Bioorg. Med. Chem. Lett. 2016, 26, 2947–2951. 10.1016/j.bmcl.2016.04.018. [DOI] [PubMed] [Google Scholar]
- Compound 4 originated from a class of γ-secretase modulators:Fischer C.; Zultanski S. L.; Zhou H.; Methot J. L.; Shah S.; Nuthall H.; Hughes B. L.; Smotrov N.; Hill A.; Szewczak A. A.; Moxham C. M.; Bays N.; Middleton R. E.; Munoz B.; Shearman M. S. Triazoloamides as potent γ-secretase modulators with reduced hERG liability. Bioorg. Med. Chem. Lett. 2012, 22, 3140–3146. 10.1016/j.bmcl.2012.03.054. [DOI] [PubMed] [Google Scholar]
- Protocols for GPR142 FLIPR assay and GPR142 IP inositol phosphate (IP) accumulation assay can be found in the Supporting Information.
- For details on the glucose tolerance test models in mice, please see:Tan C. P.; Feng Y.; Zhou Y.-P.; Eiermann G. J.; Petrov A.; Zhou C.; Lin S.; Salituro G.; Meinke P.; Mosley R.; Akiyama T. E.; Einstein M.; Kumar S.; Berger J. P.; Mills S. G.; Thornberry N. A.; Yang L.; Howard A. D. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes 2008, 57 (8), 2211–2219. 10.2337/db08-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegemann S.; Leveiller F.; Franchi D.; de Jong H.; Lindén H. When Poor Solubility Becomes an Issue: From Early Stage to Proof of Concept. Eur. J. Pharm. Sci. 2007, 31, 249–261. 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- Jain N.; Yalkowsky S. H. Estimation of the Aqueous SolubilityI: Application to Organic Nonelectrolytes. J. Pharm. Sci. 2001, 90, 234–252. . [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Hashimoto Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 2011, 54, 1539–1554. 10.1021/jm101356p. [DOI] [PubMed] [Google Scholar]
- Details of the synthesis of 20e and computational modeling can be found in the Supporting Information.
- He H.; Lyons K.; Shen X.; Yao Z.; Bleasby K.; Chan G.; Hafey M.; Li X.; Xu S.; Salituro G. M.; Cohen L. H.; Tang W. Utility of unbound plasma drug levels and p-glycoprotein transport data in prediction of central nervous system exposure. Xenobiotica 2009, 39, 687–693. 10.1080/00498250903015402. [DOI] [PubMed] [Google Scholar]
- For details on oGTT and GSIS study models in mice, please see the Supporting Information, ref (14), and the following references:; a Shah S.; He S.; Guo L.; Truong Q.; Qi H.; Du W.; Lai Z.; Liu J.; Jian T.; Hong Q.; Dobbelaar P.; Ye Z.; Sherer E.; Feng Z.; Yu Y.; Wong F.; Samuel K.; Madiera M.; Karanam B. V.; Reddy V. B.; Mitelman S.; Tong S. X.; Chicchi G. G.; Tsao K.-L.; Trusca D.; Feng Y.; Wu M.; Shao Q.; Trujillo M. E.; Eiermann G. J.; Li C.; Pachanski M.; Fernandez G.; Nelson D.; Bunting P.; Morissette P.; Morissette P.; Volksdorf S.; Kerr J.; Zhang B. B.; Howard A. D.; Zhou Y.-P.; Pasternak A.; Nargund R. P.; Hagmann W. K. Discovery of MK-1421, a potent, selective sstr3 antagonist, as a development candidate for type 2 diabetes. ACS Med. Chem. Lett. 2015, 6, 513–517. 10.1021/ml500514w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lacy P. E.; Kostianovsky M. Method for the isolation of intact islet of Langerhans from the rat pancreas. Diabetes 1967, 16, 35–39. 10.2337/diab.16.1.35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.