

Abstract

Current treatment of toxoplasmosis targets the parasite’s folate metabolism through inhibition of dihydrofolate reductase (DHFR). The most widely used DHFR antagonist, pyrimethamine, was introduced over 60 years ago and is associated with toxicity that can be largely attributed to a similar affinity for parasite and human DHFR. Computational analysis of biochemical differences between Toxoplasma gondii and human DHFR enabled the design of inhibitors with both improved potency and selectivity. The approach described herein yielded TRC-19, a promising lead with an IC50 of 9 nM and 89-fold selectivity in favor of Toxoplasma gondii DHFR, as well as crystallographic data to substantiate in silico methodology. Overall, 50% of synthesized in silico designs met hit threshold criteria of IC50 < 10 μM and >2-fold selectivity favoring Toxoplasma gondii, further demonstrating the efficiency of our structure-based drug design approach.
Keywords: Toxoplasmosis, DHFR inhibitor, in silico screening, molecular modeling
Parasites of the phylum apicomplexa are associated with pathogeneses that constitute some of the most widespread threats to human health.1−3 These diseases include malaria, cryptosporidiosis, and toxoplasmosis. Apicomplexan parasites uniquely utilize a bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) fusion enzyme for folate metabolism and pyrimidine biosynthesis, which are essential processes for cellular growth and proliferation. Dual functionality accelerates metabolic processing via substrate channeling enabled by intradomain cross-talk and allosteric modulation.4 In contrast, the majority of eukaryotic organisms enlist two distinct enzymes to carry out an identical set of reactions, further exemplifying the parasite’s evolutionary advantage toward rapid proliferation. These characteristics make protozoan TS-DHFR an attractive target for pharmacologic intervention. Thus, competitive inhibition of the DHFR domain has been used extensively in the clinic for the treatment of parasitic infections, including toxoplasmosis.2,5,6
Toxoplasmosis results from infection by the Toxoplasma gondii parasite and is the second leading cause of death from a foodborne illness in the U.S., where an estimated 60 million individuals are carriers of T. gondii, most of whom are asymptomatic. Exposure typically occurs through ingestion of undercooked meat from infected livestock, or consumption of food or water contaminated with the excrement of infected felines. The parasite proliferates intracellularly until suppression is mediated by host immune response.7−9 In an effort to evade immune defense mechanisms, T. gondii can undergo a physiological transformation into a latent, encysted form, effectively shielding itself from immunological detection.1 Cysts have the propensity to populate within longer lived tissue types, particularly those of the brain, eye, and skeletal muscle.10,11 Rupture of tissue cysts releases the encapsulated parasite, leading to re-emergence of acute infection. Acute toxoplasmosis can cause severe illness, particularly in immunocompromised patients for whom the disease can be life threatening.12−14 Three predominant manifestations of toxoplasmosis include toxoplasmic encephalitis, ocular, and congenital toxoplasmosis. Despite the relatively high prevalence of T. gondii infection in the U.S. and abroad, the CDC lists toxoplasmosis as a neglected parasitic disease, which is in large part the result of a paucity in industry-sponsored research programs for new treatments.
The current standard of care for acute toxoplasmosis is a combination therapy consisting of DHFR and dihydropteroate synthase (DHPS) inhibitors that synergistically target de novo folate biosynthesis.2 The most widely utilized DHFR inhibitor, the 2,4-diaminopyrimidine derivative pyrimethamine (PYR), was approved by the FDA for the treatment of toxoplasmosis in 1958. Originally heralded for its ability to combat malaria, PYR’s efficacy against toxoplasmosis was first demonstrated in a mouse model of the disease in 1952, while the concept of coadministering a DHPS inhibitor was introduced the following year.15,16 Treatment with PYR has been associated with a number of dose-limiting toxicities, including myelosuppression, that minimize value as a monotherapy.12 Such issues are exacerbated by additional toxicological consequences associated with the majority of DHPS inhibitors, consisting almost exclusively of sulfa drugs that tend to cause allergic reactions.17 These issues collectively threaten compliance and complicate treatment options for a significant proportion of toxoplasmosis patients.
Given that PYR displays modest selectivity for T. gondii DHFR (TgDHFR) over human DHFR (hDHFR), it comes as little surprise that mechanism-based toxicity is observed with escalating doses.18 More selective target engagement is a logical approach to maximizing the efficacy and tolerability of a folate-based toxoplasmosis treatment. While various research groups have attempted to improve the pharmacological profile of DHFR inhibitors for T. gondii and other pathogens,19−24 a candidate with ideal physiochemical properties, in addition to optimized potency and selectivity for TgDHFR, has yet to be realized.
The past success of virtual screens using DHFR crystal structures from different microbial species support the notion that these molecular models are physiologically relevant and that structure-based drug design could be a viable means to ameliorate the clinical utility of a TgDHFR inhibitor.25 This lent credence to the idea that if sequence and structural discrepancies exist between TgDHFR and hDHFR active sites, appropriate rational design could facilitate the discovery of more selective inhibitors for improved toxoplasmosis treatment in patients by mitigating on-target toxicity.
We began by examining the chemical structures of DHFR inhibitors with well-documented activity and analyzing conserved binding features using cocrystal structures. The vast majority of successful inhibitors contain either 2,4-diamino-1,3,5-triazine or 2,4-diaminopyrimidine rings. These moieties share some common features with the pterin heterocycle of the natural DHFR substrate, dihydrofolic acid (DHF, Figure 1a), accounting for their ability to act as competitive inhibitors of the enzyme. DHFR species selectivity can be fine-tuned by appending additional molecular segments to these common core functionalities.
Figure 1.

Similar residue environment in the binding pocket of TgDHFR and hDHFR. (a) Structures of PYR and DHF, with common features highlighted in red. (b) Structural alignment of the DHFR domain of TgTS-DHFR (gray; PDB: 4KYA) and hDHFR (blue; PDB: 4M6K). The substrate DHF is shown in yellow, with its aligned binding sites magnified. (c) Aligned TgDHFR and hDHFR displaying the common hydrogen bonding contacts (green) made in the PYR binding site (obtained by superposition with PDB: 4KM0). (d) Aligned TgDHFR and hDHFR displaying the conserved hydrophobic interactions made by PYR. Alignments performed using Molecular Operating Environment (MOE) software from CCG, Inc.
Homology between TgDHFR and hDHFR active sites was assessed via sequence alignment and subsequent structural superposition of folate binding pocket residues (Figure 1b). Analysis revealed a high degree of sequence and structural conservation among residues comprising the folate binding site. Similarity was particularly striking in the portion of the pocket occupied by PYR (Figures 1c,d), which is consistent with modest selectivity. Further examination of sequence homology in regions directly adjacent to the PYR binding site revealed two potentially exploitable differences between the human and T. gondii DHFR sequences. First, a glycine residue (G22) in TgDHFR replaces an aspartic acid residue (D21) in hDHFR, creating a cavity that could be occupied by a small molecule to increase TgDHFR selectivity (Figure 2a). Furthermore, this solvent-exposed cavity contains a hydrophobic leucine residue (L23) adjacent to G22 (Figure 2b). Inhibitor occupancy within this region might permit entropically favorable displacement of water molecules lining the active-site pocket, while simultaneously acquiring advantageous hydrophobic, van der Waals interactions. Our hypothesis proved consistent with computational analysis of solvent occupancy using a three-dimensional reference interaction site model (3D-RISM). In contrast, the polar aspartic acid side chain (D21) of hDHFR might encounter unfavorable electrostatic and steric interactions with hydrophobic moieties within this site. Second, TgDHFR contains two phenylalanine side chains (F32 and F91) flanking one end of the substrate cavity, creating an opportunity for an inhibitor to participate in favorable π–π stacking (Figure 2c). Such an interaction is unlikely with hDHFR, where an asparagine residue (N64) is present in place of F91 (Figure 2d).
Figure 2.

Structural differences between TgDHFR and hDHFR targeted for rational inhibitor design. (a) Structural comparison of TgDHFR (gray; PDB: 4KYA) and hDHFR (blue; PDB: 4M6K), indicating the presence of a glycine residue (G22) in TgDHFR relative to an aspartic acid (D21) in hDHFR. (b) Presence of a leucine residue (L23) adjacent to G22 creates a solvent-exposed, hydrophobic cavity. Water molecules are shown occupying the cavity as predicted by 3D-RISM (MOE, CCG, Inc.). (c) F32 and F91 in TgDHFR are appropriately positioned for participation in π–π interactions with small molecule ligands. (d) Presence of an asparagine (N64) in hDHFR (blue) at the same position as F91 in TgDHFR (gray) indicates that achieving π–π stacking could modulate species specificity.
We then used computational methods (see Supporting Information for details) to design ligands possessing structural features conserved across known DHFR inhibitors as well as any additional modifications capable of exploiting the aforementioned species differences. We iteratively constructed small-molecule libraries in silico based on accessible synthetic disconnection points incorporating common DHFR pharmacophores (see Supporting Information, Figure S1). Following combinatorial enumeration, libraries were filtered using selection criteria for optimal physiochemical properties. Filtered libraries were then docked into TgDHFR, and the output poses generated for the top scoring members were examined manually to assess agreement with the aforementioned selectivity criteria. A subset of the top scoring ligands was subsequently docked into hDHFR in an in silico counter-screen. Screening libraries totaled >1.5 million unique, small molecules.
Selection of an initial chemical probe set was determined on the basis of TgDHFR docking score, selectivity over hDHFR, enrichment of scaffold class, a (more stringent) threshold of physiochemical properties, structural diversity, and shape and pharmacophoric diversity. It was anticipated that screening results from this small subset of compounds would provide sufficient preliminary SAR to facilitate optimization by combining structure and ligand-based computation with medicinal chemistry. Databases consisting of hundreds of close analogues were compiled throughout the course of this process, further enabling rapid optimization via selection from clustered subsets.
The synthesized probe set (see Supporting Information for synthetic chemistry and Figure S2 for compound structures) was evaluated in an enzymatic assay for both TgDHFR and hDHFR inhibition26 (see Supporting Information for assay details), with PYR and methotrexate serving as controls. Results from these functional assays (Table 1) achieved a 50% hit rate based on predetermined criteria (TgDHFR IC50 < 10 μM, >2-fold selectivity).
Table 1. Screening Results from Probe Set.
| compda | TgDHFR IC50 (μM) | hDHFR IC50 (μM) | Tg fold selectivity |
|---|---|---|---|
| methotrexateb | 0.071 ± 0.004 | 0.0046 ± 0.0002 | 0.066 ± 0.003 |
| PYRb | 0.23 ± 0.01 | 4.3 ± 0.2 | 20 ± 1 |
| 1 | >10 | >10 | ND |
| 2 | >10 | >10 | ND |
| 3 | 0.98 ± 0.04 | >10 | >10 |
| 4 | 2.7 ± 0.03 | >10 | >3.5 |
| 5 | 80 ± 5 | 9.2 ± 0.4 | 0.12 ± 0.01 |
| 6 | 2.4 ± 0.1 | >10 | >4 |
| 7 | 1.1 ± 0.01 | 1.1 ± 0.004 | 1.0 ± 0.08 |
| 8 | >10 | >10 | ND |
| 9 | 0.37 ± 0.02 | 2.1 ± 0.2 | 6.5 ± 1 |
| 10 | 3.0 ± 0.03 | >10 | >3.3 |
| 11 | 0.20 ± 0.004 | 0.92 ± 0.01 | 4.7 ± 0.05 |
| 12b | 0.95 ± 0.1 | >75 | >80 |
| 13 | 0.91 ± 0.06 | 2.5 ± 0.03 | 2.9 ± 0.2 |
| 14 | 0.46 ± 0.001 | 2.8 ± 0.2 | 6.1 ± 0.4 |
| 15b | 0.017 ± 0.003 | 0.092 ± 0.007 | 10 ± 2 |
| 16b | 0.0087 ± 0.004 | 0.76 ± 0.03 | 89 ± 4 |
| 17 | >10 | >10 | ND |
| 18 | >10 | >10 | ND |
| 19 | >10 | >10 | ND |
| 20 | >10 | 4.2 ± 0.07 | <1 |
| 21 | >10 | >10 | ND |
| 22 | >10 | >10 | ND |
IC50 values reported as average of n = 2 experiments ± SEM, unless noted otherwise.
IC50 values reported as average of n ≥ 3 experiments ± SEM. ND = not determined.
The most informative screening results indicated that substitution of the 2,4-diaminopyrimidine at the 5′-positon with a piperazine linker facilitates extension toward the G22 pocket. 4-Phenyl substitution on the piperazine ring enhances potency by favorably occupying the available cavity, as evidenced by the low nanomolar IC50 of 15. Introduction of a second aryl ring to give the meta-biphenyl analogue 16 (TRC-19) further extends the side-chain toward F91, affording still greater selectivity for TgDHFR. Notably, molecular modeling indicated that the chlorine atoms in 15 fail to meaningfully extend into the region near F91, while the distal phenyl ring of 16 appears to position itself in a π–π stacking orientation as displayed in Figure 3d. Interestingly, placing a methylene spacer between the pyrimidine and piperazine moieties tends to abolish both potency and selectivity, as 7 has an IC50 close to 1 μM with little preference for either species. This is likely attributable to rotational freedom of the sp3 carbon linker minimizing entropic gains stemming from a more rigid scaffold. While we attempted to constrain the conformational flexibility by introducing a carbonyl group into the spacer, this proved to be even less acceptable, likely due to proximity near several hydrophobic side chains. The benzyloxy derivative 12 appears to be an exception within this series, suggesting a unique binding pose when additional conformational flexibility is conferred upon the portions of the ligand extending toward F91.
Figure 3.
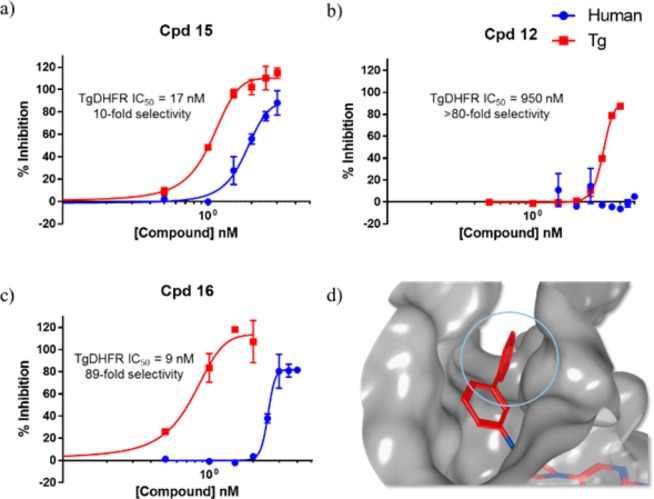
Initial leads selected for further optimization. (a–c) Representative inhibition curves for compounds 12, 15, and 16. (d) Docking pose of 16 (red) into TgDHFR (gray; PDB: 4KYA).
To facilitate lead optimization, we sought to obtain crystallographic confirmation of inhibitor binding modes. This yielded a cocrystal structure of 15 complexed with TgDHFR at 3.12 Å resolution (see Supporting Information for details). Figure 4 depicts the predicted docking pose of 15 (PDB: 4KYA) relative to the experimentally determined cocrystal structure of 15 with TgDHFR (PDB: 5T0L). As evidenced by structural alignment, there are no significant induced-fit effects observed between 5T0L and 4KYA, which is consistent with previously reported observations of inhibitors bound to DHFR of other species.25 As hypothesized during the initial design process, the dichlorophenyl ring extends into a hydrophobic pocket adjacent to G22, with ligand binding closely mirroring the predicted docking pose.
Figure 4.
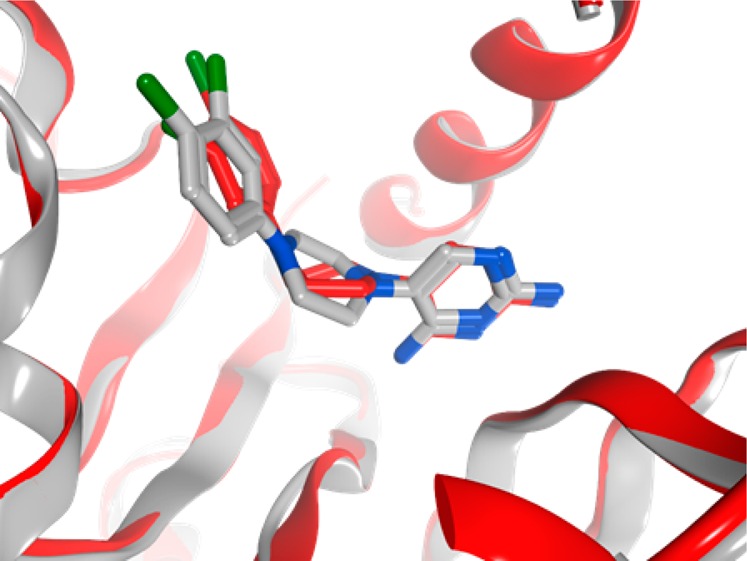
Co-crystal structure of 15 bound to TgDHFR. Experimentally determined complex of 15 (red; PDB: 5T0L) aligned with predicted docking pose (gray; PDB: 4KYA).
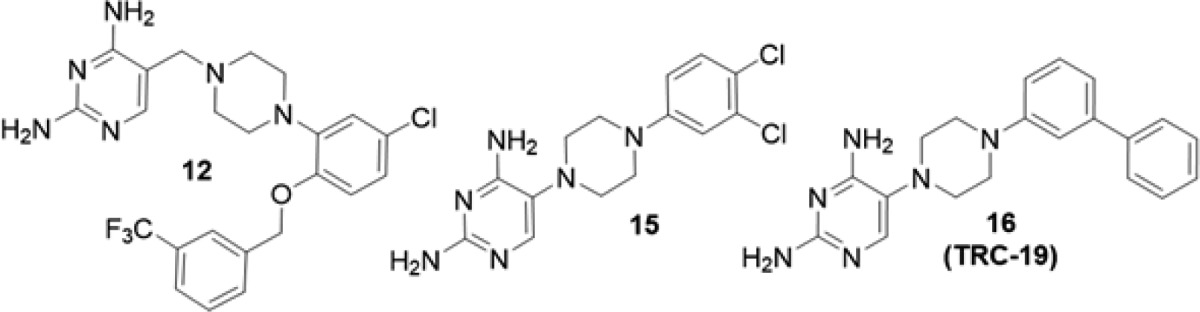
In summary, although PYR has served as the backbone of toxoplasmosis treatment for over 60 years, therapeutic improvements are long overdue. Modern drug discovery methodologies have enabled the identification of compounds with enhanced potency and selectivity for TgDHFR relative to PYR. While previous efforts have exploited unique topological features of TgDHFR in the rational design of more selective trimethoprim analogues,20,21 none have taken advantage of the more recently published crystal structures by targeting the specific three-dimensional differences between TgDHFR and hDHFR discussed above. Herein, we report the discovery of novel lead chemical matter by testing only 22 ligands designed in silico. The high success rate achieved by screening a relatively small subset of compounds can be attributed to a robust computational design strategy, leveraging de novo combinatorial library construction and iterative docking with multicomponent in silico analyses. The lead series reported is further differentiated from prior searches for pathogen-selective DHFR inhibitors by displaying excellent predicted physiochemical properties, including favorable size, solubility, and permeability (Table 2). Ongoing work is focused on the optimization of these lead series and advancement toward an improved treatment for toxoplasmosis.
Table 2. Predicted Properties of Selected Compounds.
| physiochemical propertya | 12 | 15 | 16 |
|---|---|---|---|
| molecular weight | 492.9 | 339.2 | 346.4 |
| LogP | 2.98 | 1.31 | 1.70 |
| H-bond acceptors | 7 | 6 | 6 |
| H-bond donors | 4 | 4 | 4 |
| LogS | –5.50 | –3.58 | –4.13 |
| LogD | 2.48 | 1.17 | 1.56 |
| TPSA | 93.5 | 84.3 | 84.3 |
| molar refractivity | 12.5 | 8.81 | 10.4 |
| rotational bond count | 8 | 4 | 5 |
| number of non-H atoms | 34 | 22 | 26 |
| # N atoms + # O atoms | 7 | 6 | 6 |
Physiochemical properties were calculated using chemical descriptors of molecules in their neutral state (MOE, CCG, Inc.).
Glossary
ABBREVIATIONS
- DHFR
dihydrofolate reductase
- TS
thymidylate synthase
- PYR
pyrimethamine
- DHPS
dihydropteroate synthase
- DHF
dihydrofolate
- MOE
molecular operating environment
- 3D-RISM
three-dimensional reference interaction site model
- SAR
structure–activity relationship
- ND
not determined
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00328.
Experimental procedures for molecular modeling, crystallography, biological assays, and chemical synthesis of all compounds (PDF)
Accession Codes
Coordinate and structure factors have been deposited in the Protein Data Bank with accession code 5T0L.
Author Present Address
€ Atoms2Applications, Jersey City, NJ 07306.
Author Contributions
M.E.W. designed and performed the computational experiments. M.E.W., S.B.T., and G.A. designed the in vitro pharmacology experiments. G.P. performed and analyzed the in vitro pharmacology experiments. S.B.T., M.E.W., Z.J., G.Y., and Y.Y. oversaw and designed the synthetic chemistry. Z.J., G.Y., and Y.Y. performed the synthetic chemistry. Y.L., H.L., and Z.C. performed all crystallographic experiments. S.B.T. and M.E.W. wrote the manuscript with input from all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Sacks D.; Sher A. Evasion of innate immunity by parasitic protozoa. Nat. Immunol. 2002, 3 (11), 1041–7. 10.1038/ni1102-1041. [DOI] [PubMed] [Google Scholar]
- Boyer K.; McLeod R. Toxoplasmosis. Principles and Practice of Pediatric Infectious Diseases 2007, 3, 1303–1322. [Google Scholar]
- Bargieri D.; Lagal V.; Andenmatten N.; Tardieux I.; Meissner M.; Menard R. Host cell invasion by apicomplexan parasites: the junction conundrum. PLoS Pathog. 2014, 10 (9), e1004273. 10.1371/journal.ppat.1004273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma H.; Landau M. J.; Vargo M. A.; Spasov K. A.; Anderson K. S. First three-dimensional structure of Toxoplasma gondii thymidylate synthase-dihydrofolate reductase: insights for catalysis, interdomain interactions, and substrate channeling. Biochemistry 2013, 52 (41), 7305–17. 10.1021/bi400576t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A. C. Targeting DHFR in parasitic protozoa. Drug Discovery Today 2005, 10 (2), 121–8. 10.1016/S1359-6446(04)03308-2. [DOI] [PubMed] [Google Scholar]
- Rosowsky A.; Forsch R. A.; Sibley C. H.; Inderlied C. B.; Queener S. F. New 2,4-diamino-5-(2′,5′-substituted benzyl)pyrimidines as potential drugs against opportunistic infections of AIDS and other immune disorders. Synthesis and species-dependent antifolate activity. J. Med. Chem. 2004, 47 (6), 1475–86. 10.1021/jm030438k. [DOI] [PubMed] [Google Scholar]
- Barragan A.; Sibley L. D. Migration of Toxoplasma gondii across biological barriers. Trends Microbiol. 2003, 11 (9), 426–30. 10.1016/S0966-842X(03)00205-1. [DOI] [PubMed] [Google Scholar]
- Dubey J. P.; Lindsay D. S.; Speer C. A. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin Microbiol Rev. 1998, 11 (2), 267–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenter A. M.; Heckeroth A. R.; Weiss L. M. Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 2000, 30 (12–13), 1217–58. 10.1016/S0020-7519(00)00124-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. A Bradyzoite is a Bradyzoite is a Bradyzoite?. Trends Parasitol. 2015, 31 (12), 610–2. 10.1016/j.pt.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan W. J. Jr.; Jeffers V. Mechanisms of Toxoplasma gondii persistence and latency. FEMS Microbiol Rev. 2012, 36 (3), 717–33. 10.1111/j.1574-6976.2011.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsheikha H. M. Congenital toxoplasmosis: priorities for further health promotion action. Public Health 2008, 122 (4), 335–53. 10.1016/j.puhe.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Remington J. S. Immunopathogenesis of toxoplasmic encephalitis. J. Infect. Dis. 1994, 170 (5), 1057–67. 10.1093/infdis/170.5.1057. [DOI] [PubMed] [Google Scholar]
- Luft B. J.; Remington J. S. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 1992, 15 (2), 211–22. 10.1093/clinids/15.2.211. [DOI] [PubMed] [Google Scholar]
- Eyles D. E.; Coleman N. Tests of 2,4-diaminopyrimidines on toxoplasmosis. Public Health Rep 1952, 67 (3), 249–52. 10.2307/4588050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyles D. E.; Coleman N. Synergistic effect of sulfadiazine and daraprim against experimental toxoplasmosis in the mouse. Antibiot Chemother (Northfield) 1953, 3 (5), 483–90. [PubMed] [Google Scholar]
- McLeod R.; Khan A. R.; Noble G. A.; Latkany P.; Jalbrzikowski J.; Boyer K. Toxoplasmosis Study, G., Severe sulfadiazine hypersensitivity in a child with reactivated congenital toxoplasmic chorioretinitis. Pediatr Infect Dis J. 2006, 25 (3), 270–2. 10.1097/01.inf.0000202070.59190.9a. [DOI] [PubMed] [Google Scholar]
- Allegra C. J.; Kovacs J. A.; Drake J. C.; Swan J. C.; Chabner B. A.; Masur H. Potent in vitro and in vivo antitoxoplasma activity of the lipid-soluble antifolate trimetrexate. J. Clin. Invest. 1987, 79 (2), 478–82. 10.1172/JCI112837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb K. M.; N G. D.; Wright D. L.; Anderson A. C. Elucidating features that drive the design of selective antifolates using crystal structures of human dihydrofolate reductase. Biochemistry 2013, 52 (41), 7318–26. 10.1021/bi400852h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelphrey P. M.; Popov V. M.; Joska T. M.; Beierlein J. M.; Bolstad E. S.; Fillingham Y. A.; Wright D. L.; Anderson A. C. Highly efficient ligands for dihydrofolate reductase from Cryptosporidium hominis and Toxoplasma gondii inspired by structural analysis. J. Med. Chem. 2007, 50 (5), 940–50. 10.1021/jm061027h. [DOI] [PubMed] [Google Scholar]
- Chio L. C.; Queener S. F. Identification of highly potent and selective inhibitors of Toxoplasma gondii dihydrofolate reductase. Antimicrob. Agents Chemother. 1993, 37 (9), 1914–23. 10.1128/AAC.37.9.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brophy V. H.; Vasquez J.; Nelson R. G.; Forney J. R.; Rosowsky A.; Sibley C. H. Identification of Cryptosporidium parvum dihydrofolate reductase inhibitors by complementation in Saccharomyces cerevisiae. Antimicrob. Agents Chemother. 2000, 44 (4), 1019–28. 10.1128/AAC.44.4.1019-1028.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mui E. J.; Schiehser G. A.; Milhous W. K.; Hsu H.; Roberts C. W.; Kirisits M.; Muench S.; Rice D.; Dubey J. P.; Fowble J. W.; Rathod P. K.; Queener S. F.; Liu S. R.; Jacobus D. P.; McLeod R. Novel triazine JPC-2067-B inhibits Toxoplasma gondii in vitro and in vivo. PLoS Neglected Trop. Dis. 2008, 2 (3), e190. 10.1371/journal.pntd.0000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirawaraporn W.; Sertsrivanich R.; Booth R. G.; Hansch C.; Neal R. A.; Santi D. V. Selective inhibition of Leishmania dihydrofolate reductase and Leishmania growth by 5-benzyl-2,4-diaminopyrimidines. Mol. Biochem. Parasitol. 1988, 31 (1), 79–85. 10.1016/0166-6851(88)90147-8. [DOI] [PubMed] [Google Scholar]
- Wyss P. C.; Gerber P.; Hartman P. G.; Hubschwerlen C.; Locher H.; Marty H. P.; Stahl M. Novel dihydrofolate reductase inhibitors. Structure-based versus diversity-based library design and high-throughput synthesis and screening. J. Med. Chem. 2003, 46 (12), 2304–12. 10.1021/jm020495y. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Zhang M.; Zhu L.; Liao R. P.; Mutai C.; Hafsat S.; Sherman D. R.; Wang M. W. High-throughput screening and sensitized bacteria identify an M. tuberculosis dihydrofolate reductase inhibitor with whole cell activity. PLoS One 2012, 7 (6), e39961. 10.1371/journal.pone.0039961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.