

Abstract
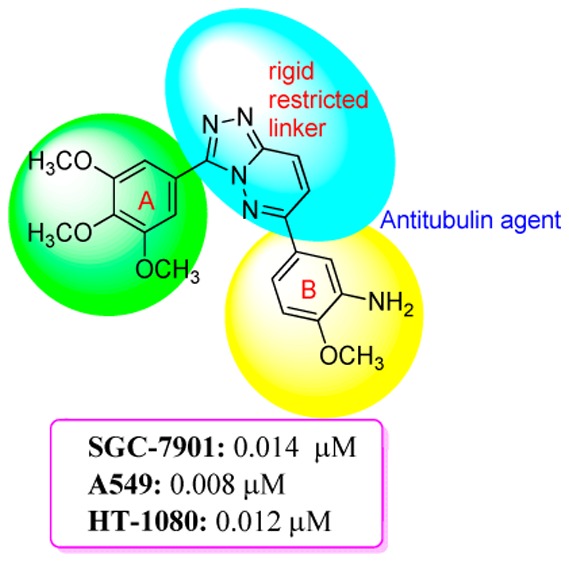
A series of 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines were designed as a class of vinylogous CA-4 analogues. The easily isomerized (Z,E)-butadiene linker of vinylogous CA-4 was replaced by a rigid [1,2,4]triazolo[4,3-b]pyridazine scaffold. Twenty-one target compounds were synthesized and exhibited moderate to potent antiproliferative activity. The compound 4q with a 3-amino-4-methoxyphenyl moiety as the B-ring, comparable to CA-4 (IC50 = 0.009–0.012 μM), displayed the highly active antiproliferative activity against SGC-7901, A549, and HT-1080 cell lines with IC50 values of 0.014, 0.008, and 0.012 μM, respectively. Tubulin polymerization experiments indicated that 4q effectively inhibited tubulin polymerization, and immunostaining assay revealed that 4q significantly disrupted tubulin microtubule dynamics. Moreover, cell cycle studies revealed that compound 4q dramatically arrested cell cycle progression at G2/M phase in A549 cells. Molecular modeling studies showed that 4q could bind to the colchicine binding site on microtubules.
Keywords: [1,2,4]Triazolo[4,3-b]pyridazine; combretastatin A-4; tubulin; colchicine binding site; molecular modeling
Tubulin, which plays a key role in cell mitosis, is one of the most effective molecular targets for anticancer drugs discovery.1,2 Microtubule targeting drugs disrupt tubulin/microtubule dynamics by binding to distinct sites such as taxol, vinca alkaloid, and colchicine binding sites and arrests cells during mitosis, leading to cell death.3,4 Combretastatin A-4 (CA-4, 1, Figure 1) is a well-known antitubulin agent that binds to the colchicine site.5 In recent years, a wide variety of CA-4 analogues have been developed for the structure–activity relationships study.6,7 In brief, the existence of the 3,4,5-trimethoxy group on the A-ring and the Z-restricted configuration have been reported as preconditions for effective antiproliferative activity.8 However, the cis-olefin bond of CA-4 easily isomerizes to its inactive transform (E geometry) under light, heat, and protic media resulting in a sharp reduction in both antiproliferative and antitubulin activities.9 Therefore, to restrict the compounds in the desired Z-restricted configuration, a wide variety of strategies have been designed, particularly by rigidifying the olefin bond with heterocyclic moieties (five-membered and six-membered rings, Figure 1).10−13 Interestingly, when the cis-olefin bridge of CA-4 was replaced by a four carbons (Z,E)-butadiene linker, the resulting vinylogous CA-4 (2, Figure 1) actually retained potent bioactivity.14 Furthermore, vinylogous CA-4 analogue 3 whose B-ring was a phenyl group was discovered to be more active than CA-4 in antitubulin activity.14 These dienic analogues of CA-4 also tend to isomerization to the more stable but inactive (E,E)-isomeric derivatives.14,15
Figure 1.

Design strategy and structures for the target compounds.
Over the years, researchers have been highly interested in compounds containing the [1,2,4]triazolo[4,3-b]pyridazine scaffold, on account of their characteristic chemical structure and various biological properties including anticancer activity.16−19 As part of our search for new antitubulin agents,20−24 we hypothesized that the replacement of the easily isomerized (Z,E)-butadiene linker of vinylogous CA-4 analogues with a rigid [1,2,4]triazolo[4,3-b]pyridazine scaffold could be a successful strategy to unravel a class of antitubulin agents (Figure 1). Thus, a series of 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines were synthesized and distinguished into two types. Type I target compounds (4a–q) include a 3,4,5-trimethoxyphenyl unit as the A-ring, which is an essential component to induce cytotoxicity in the compounds based on CA-4 pharmacophores, while type II target compounds (5a–d) include a 2,3,4-trimethoxy substitution on the A-ring for comparison (Figure 1).25 To explore the SAR of target compounds, various substitutions with electron-withdrawing (F, Cl, Br, CF3, CN, and NO2) and electron-donating (CH3, OCH3, SCH3, and NH2) groups were introduced at different positions of the B-ring. In addition, the most potent compound 4q was selected to investigate its mechanism of activity and the possible binding mode of 4q on tubulin.
Target compounds 4a–q and 5a–d were prepared as outlined in Scheme 1. In brief, substituted acetophenones 6 were reacted with glyoxylic acid in acetic acid and then treated with hydrazine to afford the desired pyridazinones 7 in 80–95% yield. Subsequently, pyridazinones 7 were treated with phosphorus oxychloride to afford the key intermediates 8. However, the substituted benzoic acids 9 were reacted with excess methanol by using concentrated sulfuric acid as catalyst to give the corresponding esters 10, which were further reacted with 80% hydrazine monohydrate in methanol to get hydrazides 11 in 60–90% yield. Finally, intermediates 8 were reacted with hydrazides 11 to afford the desired compounds in n-butyl alcohol under microwave irradiation (150 W, 120 °C) in the absence of catalysts.17 The nitro-compounds 4g, 4m, 4p, and 5c were reduced by hydrazine hydrate to obtain corresponding amino-compounds 4h, 4n, 4q, and 5d.20
Scheme 1.

Reagents and conditions: (a) (1) glyoxylic acid, AcOH, reflux, (2) NH4OH, (3) N2H4·H2O, reflux; (b) POCl3, 100 °C, 2 h; (c) MeOH, H2SO4, reflux; (d) N2H4·H2O, MeOH, reflux; (e) n-butyl alcohol, MW, 120 °C.
To explore the ability of various 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines to inhibit cancer cells, the target compounds and reference compounds CA-4 (1) were screened for antiproliferative activity against gastric adenocarcinoma SGC-7901 cells, lung adenocarcinoma A549 cells, and fibrosarcoma HT-1080 cells. As illustrated in Table 1, the majority of these target compounds exhibited moderate to potent antiproliferative activity. When ring-A was a 3,4,5-trimethoxyphenyl, a comparison of compounds 4a–j revealed an electronic properties substitution effect at para-substitution of ring-B. The compounds with electron-donating groups such as CH3 (4a), OCH3 (4c), NH2 (4h), and SCH3 (4i) on para-substitution of ring-B gave higher activities than those with electron-withdrawing groups such as CF3 (4b), NO2 (4g), and CN (4j) with the exception of compounds 4d–f with para-halogen substituted electron-withdrawing groups (e.g., F, Cl, Br). For example, compound 4e with a chlorine substituent on para-substitution of ring-B displayed potent antiproliferative activity which may be due to chlorine being a relative small and weak electron-withdrawing group in contrast with other electron-withdrawing groups. However, for the electronic effects to meta-substitution of ring-B, only compound 4n with an electron-donating group NH2 exhibited moderate antiproliferative activity (4k–m vs 4n). Furthermore, to explore the effect of disubstitution on the B-ring, methoxyl, fluorine, nitro, and amino groups were introduced in the meta- and para-positions (4o–q). Interestingly, the compound 4q with a 3-amino-4-methoxyphenyl moiety as the B-ring, comparable to CA-4 (IC50 = 0.009–0.012 μM), displayed the highly active antiproliferative activity against SGC-7901, A549, and HT-1080 cell lines with IC50 values of 0.014, 0.008, and 0.012 μM, respectively. Moreover, replacement of the 3,4,5-trimethoxy-substituted A-ring with 2,3,4-trimethoxy-substituted A-ring (5a–d) induced a reduction in antiproliferative activity of resulting compound (4a vs 5a, 4c vs 5b, 4p vs 5c, 4q vs 5d). Compound 5b showed moderate antiproliferative activity, and this result was consistent with the data in the reported literature.19
Table 1. Antiproliferative Activity of Target Compounds 4a–q, 5a–d, and CA-4.
| IC50 (μM)a |
|||
|---|---|---|---|
| compd | SGC-7901 | A549 | HT-1080 |
| 4a | 0.016 ± 0.009 | 0.023 ± 0.012 | 0.14 ± 0.07 |
| 4b | 2.21 ± 0.11 | 10.4 ± 1.3 | 1.86 ± 0.22 |
| 4c | 0.26 ± 0.21 | 0.051 ± 0.009 | 0.017 ± 0.011 |
| 4d | 2.02 ± 0.07 | 2.18 ± 0.06 | 0.021 ± 0.010 |
| 4e | 0.025 ± 0.006 | 0.051 ± 0.009 | 0.45 ± 0.03 |
| 4f | 1.28 ± 0.03 | 0.78 ± 0.05 | 0.15 ± 0.07 |
| 4g | 2.38 ± 0.05 | 3.85 ± 0.07 | 4.94 ± 0.09 |
| 4h | 1.23 ± 0.09 | 2.35 ± 0.08 | 1.62 ± 0.06 |
| 4i | 0.098 ± 0.012 | 0.28 ± 0.03 | 0.073 ± 0.013 |
| 4j | 30.9 ± 0.5 | 51.6 ± 0.9 | 33.5 ± 2.1 |
| 4k | 63.1 ± 0.7 | 47.9 ± 1.3 | 60.5 ± 1.4 |
| 4l | 59.0 ± 0.9 | 52.2 ± 1.7 | 54.5 ± 1.6 |
| 4m | 86.4 ± 2.3 | 90.5 ± 3.4 | 50.0 ± 1.5 |
| 4n | 0.98 ± 0.06 | 2.98 ± 0.05 | 0.39 ± 0.07 |
| 4o | 4.17 ± 0.07 | 4.23 ± 0.08 | 1.69 ± 0.03 |
| 4p | 2.28 ± 0.01 | 4.57 ± 0.03 | 6.85 ± 0.05 |
| 4q | 0.014 ± 0.002 | 0.008 ± 0.006 | 0.012 ± 0.004 |
| 5a | 4.53 ± 0.06 | 10.4 ± 0.07 | 0.17 ± 0.09 |
| 5b | 0.42 ± 0.06 | 6.98 ± 0.06 | 5.57 ± 0.09 |
| 5c | 9.14 ± 0.07 | 11.4 ± 1.1 | 18.2 ± 0.7 |
| 5d | 0.91 ± 0.09 | 2.62 ± 0.05 | 0.92 ± 0.04 |
| CA-4b | 0.012 ± 0.003 | 0.009 ± 0.002 | 0.009 ± 0.006 |
IC50: 50% inhibitory concentration (determined by standard MTT assay). Each experiment was carried out in triplicate.
Used as a positive control.
To investigate whether the antiproliferative activity of these new compounds was due to interaction with tubulin, the highly active compound 4q was evaluated for its inhibition of tubulin polymerization. CA-4 (1) and paclitaxel were employed as positive and negative controls, respectively. As shown in Figure 2, 4q displayed antitubulin activity with an IC50 value of 1.80 μM, which is less active than that of CA-4 (IC50 = 0.64 μM). Moreover, 4q and CA-4 inhibited tubulin polymerization in a concentration-dependent manner (Figure 2). The tubulin polymerization experiment strongly implicated a direct interaction of 4q with tubulin.
Figure 2.
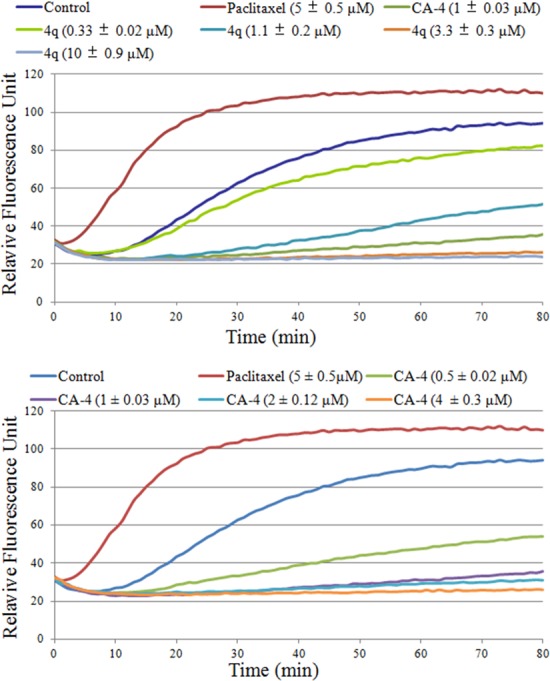
Effects of 4q and CA-4 on tubulin polymerization. Tubulin had been incubated with 4q (0.33, 1.1, 3.3, and 10 μM), paclitaxel (5.0 μM), CA-4 (0.5, 1.0, 2.0, and 4.0 μM), or DMSO (vehicle control) at room temperature. Values are the mean ± SD of three different experiments performed in triplicates.
To further confirm the influence of inhibition of tubulin polymerization in cells, we explored microtubule structure and distribution in cultured A549 cells by using the indirect immunofluorescence assay. A549 cells treated with DMSO showed a normal arrangement and organization throughout the cells (Figure 3). Instead, A549 cells treated with compound 4q and CA-4 (at their respective 2-fold IC50 concentrations) demonstrated a destruction of the tubulin network (Figure 3). In short, the results further confirmed that tubulin was the molecular target for compound 4q.
Figure 3.
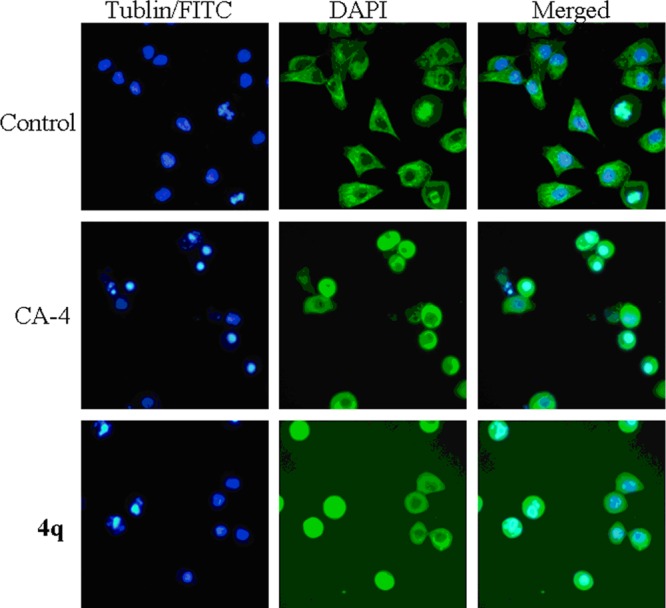
Effects of 4q (0.016 μM) and CA-4 (0.018 μM) on inhibition of tubulin polymerization in A549 cells by immunofluorescence. The left and middle panels represent the tubulin assembly stained with FITC and DAPI, and the right panel represents a merge of the corresponding left and middle panels.
To investigate whether the potent compound 4q could arrest cell cycle distribution, the effect of compound 4q on the cell cycle was analyzed by flow cytometry. First, A549 cells were treated with 4q of different concentrations (0.5-, 1-, and 2-fold IC50) for 12 h (Figure 4A). However, A549 cells were treated with 4q or CA-4 (at their 2-fold IC50 concentrations, respectively) for 0, 12, 24, and 48 h, respectively (Figure 4B). Cell cycle analysis revealed that 4q caused a significant cell cycle arrest at the G2/M phase in both concentration- and time-dependent manners.
Figure 4.
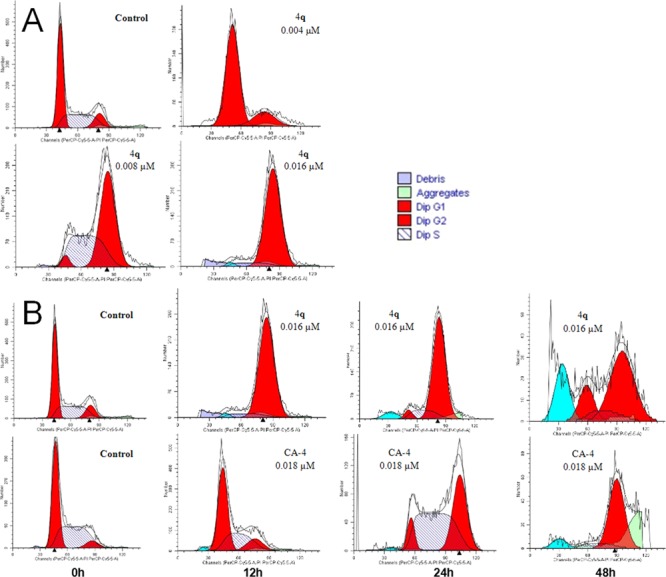
(A) Compound 4q caused G2/M phase arrest in a concentration-dependent manner. A549 cells were treated with different concentrations (0.004 to 0.016 μM) of 4q for 12 h, then stained with PI and subjected to flow cytometric analysis. (B) Compound 4q and CA-4 induced G2/M phase arrest in a time-dependent manner. A549 cells were treated with 4q or CA-4 for time 12, 24, and 48 h, then stained with PI and subjected to flow cytometric analysis.
To further understand the possible binding mode of these new compounds with the colchicine binding site of tubulin, molecular modeling study of the most potent compound 4q, CA-4, and vinylogous CA-4 analogue 3 were performed by using CDOCKER protocol in Discovery Studio 3.0 software package (PDB: 1SA0). The docking study (Figure 5A) showed that compound 4q superimposed well with CA-4 and 3 in colchicine binding site, 4q displays a distorted conformation similar to CA-4 and 3, which may be responsible for its bioactivity. For compound 4q, the oxygen atom of the para-methoxy group on A-ring formed a hydrogen bond with the thiol group of Cys β241, and the amino nitrogen atom on B-ring formed another hydrogen bond with the sulfur atom of Met β259 (Figure 5B). Additionally, the Ala β250 residue formed a direct hydrogen bond with the [1,2,4]triazolo[4,3-b]pyridazine linker. The docking study and tubulin polymerization assay suggested that 4q could bind at the colchicine binding site of tubulin.
Figure 5.
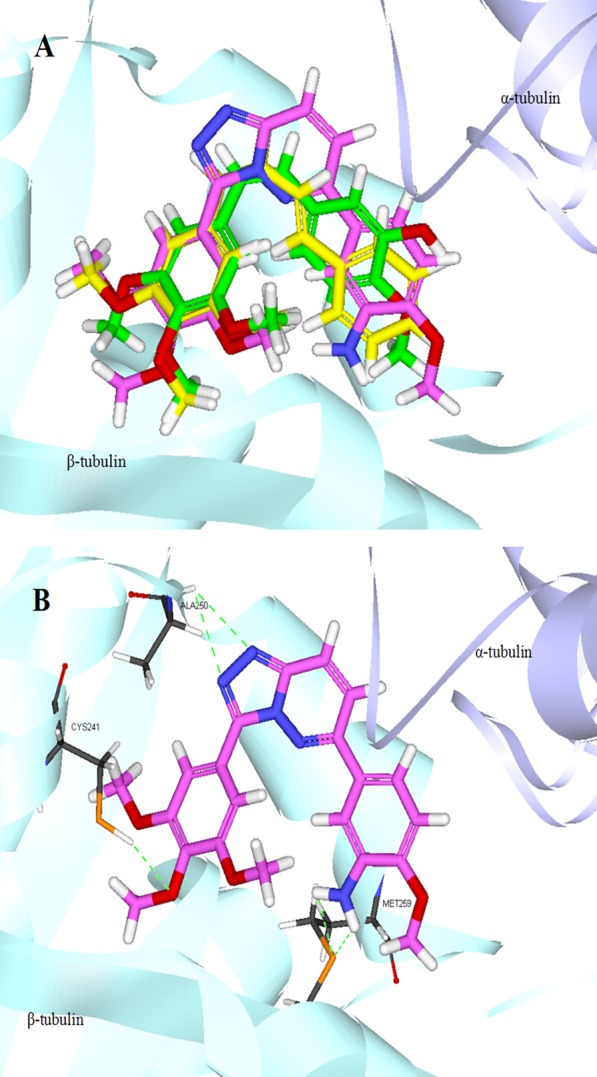
(A) Possible binding mode of compound 4q (violet), CA-4 (green), and vinylogous CA-4 analogue 3 (yellow) in the colchicine binding site. (B) Overlay of 4q in the binding site. Hydrogen bonds are displayed by green dashed lines (hydrogen bond distance <3 Å).
In conclusion, we designed and synthesized a set of 3,6-diaryl-[1,2,4]triazolo[4,3-b]pyridazines as a new class of vinylogous CA-4 analogues. The structure of these compounds are unique as they involved a rigid [1,2,4]triazolo[4,3-b]pyridazine scaffold as the linker to fix the Z,E-diene configuration of A-ring and B-ring. These target compounds exhibited moderate to potent antiproliferative activity with IC50 values from 0.008 to 90.5 μM. Interestingly, the compound 4q with a 3-amino-4-methoxyphenyl moiety as the B-ring, comparable to CA-4 (IC50 = 0.009–0.012 μM), displayed the highly active antiproliferative activity against SGC-7901, A549, and HT-1080 cell lines with IC50 values of 0.014, 0.008, and 0.012 μM, respectively. The tubulin polymerization assay suggested that 4q effectively inhibited tubulin polymerization, and immunofluorescence staining experiment revealed that 4q significantly disrupted tubulin microtubule dynamics. Furthermore, cell cycle analysis studies revealed that compound 4q significantly arrested cell cycle progression at G2/M phase in A549 cells. Additionally, the results of docking study together with the other two in vitro tubulin experiments showed that 4q may bind to colchicine binding site of tubulin. Our work not only expands the exploration of the linker modification of tubulin inhibitor CA-4 but also provides a set of rigid analogues of vinylogous CA-4 with potent antiproliferative activity.
Acknowledgments
We gratefully acknowledge Program for Innovative Research Team of the Ministry of Education and Program for Liaoning Innovative Research Team in University.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00252.
Experimental procedures and data for compounds (PDF)
Author Contributions
Q.X., Y.W., J.X., M.S., H.T., and D.Z. performed the experiments. Q.X., Q.G., K.B., Y.W., and W.Z. analyzed and interpreted the data. Q.X., K.B., and W.Z. wrote the paper.
This work was funded by the National Natural Science Foundation of China (81502932) and State Key Laboratory of Natural Medicines and Active Substance (No. GTZK201603).
The authors declare no competing financial interest.
Supplementary Material
References
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- Ferrara R.; Pilotto S.; Peretti U.; Caccese M.; Kinspergher S.; Carbognin L.; Karachaliou N.; Rosell R.; Tortora G.; Bria E. Tubulin inhibitors in non-small cell lung cancer: looking back and forward. Expert Opin. Pharmacother. 2016, 17, 1113. 10.1517/14656566.2016.1157581. [DOI] [PubMed] [Google Scholar]
- Rohena C. C.; Mooberry S. L. Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. 10.1039/c3np70092e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur R.; Kaur G.; Gill R. K.; Soni R.; Bariwal J. Recent developments in tubulin polymerization inhibitors: An overview. Eur. J. Med. Chem. 2014, 87, 89–124. 10.1016/j.ejmech.2014.09.051. [DOI] [PubMed] [Google Scholar]
- Pettit G. R.; Singh S. B.; Hamel E.; Lin C. M.; Alberts D. S.; Garcia-Kendall D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209–211. 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- Patil P. O.; Patil A. G.; Rane R. A.; Patil P. C.; Deshmukh P. K.; Bari S. B.; Patil D. A.; Naphade S. S. Recent advancement in discovery and development of natural product combretastatin-inspired anticancer agents. Anti-Cancer Agents Med. Chem. 2015, 15, 955–969. 10.2174/1871520615666150526141259. [DOI] [PubMed] [Google Scholar]
- Mikstacka R.; Stefański T.; Różański J. Tubulin-interactive stilbene derivatives as anticancer agents. Cell Mol. Biol. Lett. 2013, 18, 368–397. 10.2478/s11658-013-0094-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tron G. C.; Pirali T.; Sorba G.; Pagliai F.; Busacca S.; Genazzani A. A. Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 2006, 49, 3033–3044. 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- Nam N. H. Combretastatin A-4 analogues as antimitotic antitumor agents. Curr. Med. Chem. 2003, 10, 1697–1722. 10.2174/0929867033457151. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Chen J.; Xiao M.; Li W.; Miller D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. 10.1007/s11095-012-0828-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Woods K. W.; Li Q.; Barr K. J.; McCroskey R. W.; Hannick S. M.; Gherke L.; Credo R. B.; Hui Y. H.; Marsh K.; Warner R.; Lee J. Y.; Zielinski-Mozng N.; Frost D.; Rosenberg S. H.; Sham H. L. Potent, orally active heterocycle-based combretastatin A-4 analogues: synthesis, structure-activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J. Med. Chem. 2002, 45, 1697–1711. 10.1021/jm010523x. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Zhong Q.; Mottamal M.; Zhang Q.; Zhang C.; Lemelle E.; McFerrin H.; Wang G. Design, synthesis, and biological evaluation of novel pyridine-bridged analogues of combretastatin-A4 as anticancer agents. J. Med. Chem. 2014, 57, 3369–3381. 10.1021/jm500002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaninetti R.; Cortese S. V.; Aprile S.; Massarotti A.; Canonico P. L.; Sorba G.; Grosa G.; Genazzani A. A.; Pirali T. A concise synthesis of pyrazole analogues of combretastatin A1 as potent anti-tubulin agents. ChemMedChem. 2013, 8, 633–643. 10.1002/cmdc.201200561. [DOI] [PubMed] [Google Scholar]
- Kaffy J.; Pontikis R.; Florent J. C.; Monneret C. Synthesis and biological evaluation of vinylogous combretastatin A-4 derivatives. Org. Biomol. Chem. 2005, 3, 2657–2660. 10.1039/b505955k. [DOI] [PubMed] [Google Scholar]
- Ty N.; Kaffy J.; Arrault A.; Thoret S.; Pontikis R.; Dubois J.; Morin-Allory L.; Florent J. C. Synthesis and biological evaluation of cis-locked vinylogous combretastatin-A4 analogues: derivatives with a cyclopropyl-vinyl or a cyclopropyl-amide bridge. Bioorg. Med. Chem. Lett. 2009, 19, 1318–1322. 10.1016/j.bmcl.2009.01.062. [DOI] [PubMed] [Google Scholar]
- Guan L. P.; Sui X.; Deng X. Q.; Quan Y. C.; Quan Z. S. Synthesis and anticonvulsant activity of a new 6-alkoxy-[1,2,4]triazolo[4,3-b]pyridazine. Eur. J. Med. Chem. 2010, 45, 1746–1752. 10.1016/j.ejmech.2009.12.077. [DOI] [PubMed] [Google Scholar]
- Albright J. D.; Moran D. B.; Wright W. B. Jr.; Collins J. B.; Beer B.; Lippa A. S.; Greenblatt E. N. Synthesis and anxiolytic activity of 6-(substituted-phenyl)-1,2,4-triazolo[4,3-b]pyridazines. J. Med. Chem. 1981, 24, 592–600. 10.1021/jm00137a020. [DOI] [PubMed] [Google Scholar]
- Kaur R.; Dwivedi A. R.; Kumar B.; Kumar V. Recent Developments on 1,2,4-Triazole Nucleus in Anticancer Compounds: A Review. Anti-Cancer Agents Med. Chem. 2016, 16, 465–489. 10.2174/1871520615666150819121106. [DOI] [PubMed] [Google Scholar]
- Cai S.; Tian E. Y.; Dong H.; Yu Z.; Chen L.; Wu L.; Liu L.; Yin F.. Preparation of 3-aryl-6-aryl-[1,2,4]triazolo[4,3-b]pyridazine derivatives as cell proliferation inhibitors. Int. Appl. WO 2012094966A1 20120719, 2012.
- Wen Z.; Xu J.; Wang Z.; Qi H.; Xu Q.; Bai Z.; Zhang Q.; Bao K.; Wu Y.; Zhang W. 3-(3,4,5-Trimethoxyphenylselenyl)-1H-indoles and their selenoxides as combretastatin A-4 analogues: microwave-assisted synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 27, 184–194. 10.1016/j.ejmech.2014.11.024. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Qi H.; Sun M.; Zuo D.; Jiang X.; Wen Z.; Wang Z.; Wu Y.; Zhang W. Synthesis and Biological Evaluation of 3-Alkyl-1,5-Diaryl-1H-Pyrazoles as Rigid Analogues of Combretastatin A-4 with Potent Antiproliferative Activity. PLoS One 2015, 10, e0128710. 10.1371/journal.pone.0128710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z.; Li X.; Zuo D.; Lang B.; Wu Y.; Jiang M.; Ma H.; Bao K.; Wu Y.; Zhang W. Ultrasound-promoted two-step synthesis of 3-arylselenylindoles and 3-arylthioindoles as novel combretastatin A-4 analogues. Sci. Rep. 2016, 6, 23986. 10.1038/srep23986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Q.; Han C.; Zuo D.; Zhai M.; Li Z.; Zhang Q.; Zhai Y.; Jiang X.; Bao K.; Wu Y.; Zhang W. Synthesis and evaluation of benzimidazole carbamates bearing indole moieties for antiproliferative and antitubulin activities. Eur. J. Med. Chem. 2014, 87, 306–315. 10.1016/j.ejmech.2014.09.071. [DOI] [PubMed] [Google Scholar]
- Guan Q.; Yang F.; Guo D.; Xu J.; Jiang M.; Liu C.; Bao K.; Wu Y.; Zhang W. Synthesis and biological evaluation of novel 3,4-diaryl-1,2,5-selenadiazol analogues of combretastatin A-4. Eur. J. Med. Chem. 2014, 87, 1–9. 10.1016/j.ejmech.2014.09.046. [DOI] [PubMed] [Google Scholar]
- Negi A. S.; Gautam Y.; Alam S.; Chanda D.; Luqman S.; Sarkar J.; Khan F.; Konwar R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. 10.1016/j.bmc.2014.12.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.