

Abstract
Dipeptidyl nitroalkenes are potent reversible inhibitors of cysteine proteases. Inhibitor 11 resulted to be the most potent one with Ki values of 0.49 and 0.44 nM against rhodesain and cruzain, respectively. According to enzymatic dilution and dialysis experiments, as well as computational and NMR studies, dipeptidyl nitroalkenes are tightly binding covalent reversible inhibitors.
Keywords: Rhodesain, cruzain, inhibitors, Chagas’ disease, sleeping sickness
The target proteases of the presented study, rhodesain and cruzain, are parasite proteases, which belong to the papain-family of cysteine proteases.1,2 They are related to human cathepsins.1,2 Rhodesain is expressed by the protozoa Trypanosoma brucei rhodesiense, which causes the African sleeping sickness.3,6,7 Cruzain is expressed by T. cruzi, the parasite causing Chagas’ disease occurring in South and Central America.4,5 Both proteases are essential for the life cycles of the pathogens. Consequently, their inhibition is an important strategy for the treatment of these diseases.8,9
K11777 (Figure 1a), a dipeptidyl vinyl sulfone, irreversibly inactivates cysteine proteases10 by conjugate addition of the thiolate of the cysteine at the active site to the double bond. The resulting carbanion is subsequently protonated driving the process thermodynamically to the more stable enzyme–inhibitor complex (Figure 1b).
Figure 1.
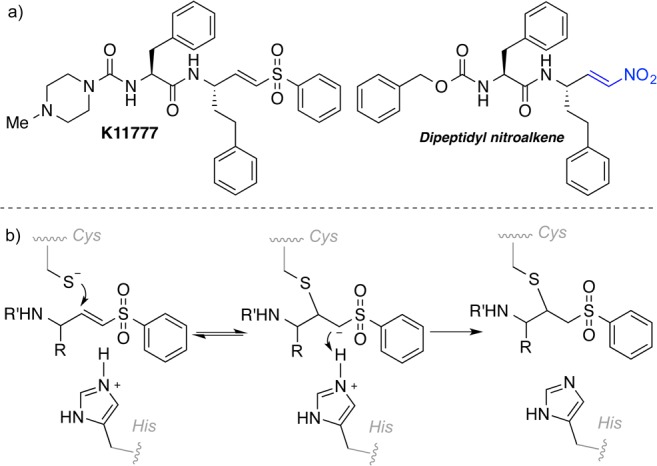
(a) Structures of vinyl sulfone K11777 and a dipeptidyl nitroalkene. (b) Inhibition mechanism by vinyl sulfones.
Irreversible inhibitors can give rise to undesired side reactions. Turning dipeptidyl vinyl sulfones into compounds that reversibly react with thiols by introducing thioethers or halogen atoms into the structure has been reported.11 In case of the halogenated vinylsulfones this leads to covalent reversible inhibition.12
We envisioned dipeptidyl nitroalkenes as inhibitors for cysteine proteases. Based upon the inhibition mechanism of vinyl sulfones, protonation of the carbanion might be less favored in the case of the arising nitroalkane carbanion since the acidity of the corresponding acid, namely, the nitroalkane, is higher, and thus, the basicity of the carbanion is lower (Figure 1b).13
Dipeptidyl nitroalkenes were prepared through a short and efficient synthetic route. The first step of the synthesis was a nitroaldol reaction between the corresponding N-protected amino aldehyde and nitromethane (or nitroethane) which afforded a mixture of nitroaldols 1–5. The next step was the deprotection and coupling with the corresponding N-protected amino acid giving rise to compounds 6–10. Finally dehydration of dipeptidyl nitroaldols through methanesulfonates yielded the desired dipeptidyl nitroalkenes 11–20 (Scheme 1).14
Scheme 1. Synthesis of Dipeptidyl Nitroalkenes.

Reagents and conditions: (a) nitromethane or nitroethane, triethylamine, CH2Cl2, 0 °C to rt, overnight, 85–95%; (b) TFA, CH2Cl2,0 °C, 30 min then DIPEA, HOBT, EDC, CbzPhe or CbzLeu, 69–86%; (c) methanesulfonyl chloride, DIPEA, CH2Cl2, 0 °C to rt, overnight, 64–93%.
The dipeptidyl nitroalkenes 11–20 were screened against rhodesain and cruzain (Table 1), and Ki determinations showed that the compounds are potent reversible inhibitors of these enzymes. Inhibitory activity was dependent on the peptidic framework. Compound 11 with l-alanine at the P1 position and l-phenylalanine at P2 proved to be the most potent inhibitor displaying a Ki in the picomolar range. Compounds 15 and 20, having a methyl group at the α-position of the double bond, were considerably less active. Compounds were also tested against human cysteine proteases cathepsins B and L, also belonging to the papain-family (Table 1). In all cases Ki values were one or two orders of magnitude higher compared with the parasitic enzymes.
Table 1. Inhibition Data for Compounds 11–20 (Ki in nM).
| inhibitor | rhodesain (RD) | cruzain (CRZ) | cathepsin B (CB) | cathepsin L (CL) |
|---|---|---|---|---|
| 11 | 0.49 | 0.44 | 8 | 11 |
| 12 | 6.5 | 16 | 68 | 30 |
| 13 | 29 | 130 | 310 | 110 |
| 14 | 4.2 | 11 | 34 | 19.6 |
| 15 | 50 | NDa | 6300 | 280 |
| 16 | 30 | 130 | 310 | 200 |
| 17 | 18.4 | 25 | 210 | 46 |
| 18 | 18 | 28 | 710 | 49 |
| 19 | 14 | 17 | 72 | 27 |
| 20 | 544 | ND | 3200 | 2020 |
| K11777b | 20 | ND | 390 | 80 |
ND = Not determined.
ksecond values [M–1 s–1] for inhibition by K11777: RD: 6.6 × 105; CL: 9.2 × 105; CB: 2.3 × 104.
To confirm the reversibility of inhibition, dilution assays (see Supporting Information) with inhibitors 11, 12, 18, 17 (CL), 16, and 17 (CB), as well as dialysis assays (Figure 2) with compounds 12, 17, and 19 (RD) were performed. The assays revealed that the enzymes’ activities recovered quite fast in the case of compounds 18 and 19, while slower regeneration of the enzyme activities was found with 11, 12, 16, and 17.
Figure 2.
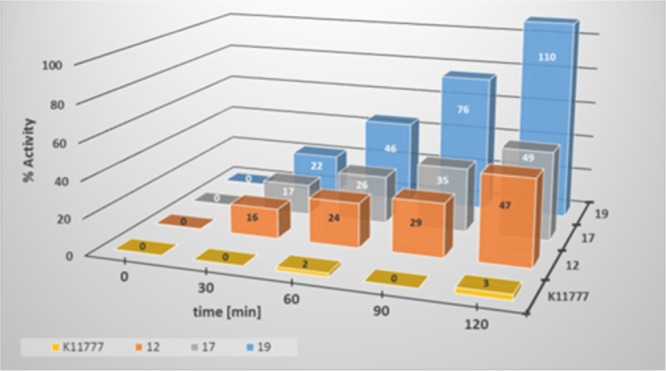
Experimental verification of the reversibility of the inhibition via dialysis assays. Enzyme and inhibitor (final concentration 1 μM) were incubated for 5 min, and the reaction mixture was subjected to dialysis with assay buffer using our published dialysis apparatus.15 The residual enzyme activity was measured after 30, 60, 90, and 120 min by adding the substrate (final concentration 10 μM).
The fact that the dipeptidyl nitroalkenes are reversible inhibitors may have several reasons: first, it could be a consequence of the orientation of the warhead within the active site, which would prevent nucleophilic attack at the double bond. However, this stands in contrast to the high inhibition potencies of the compounds, which can hardly be explained by an only noncovalent binding of the recognition units to the binding pockets. Additionally, the docking experiments indicate that a reaction, i.e., a nucleophilic attack at the double bond, should be possible. The reversibility could also result from a weaker exothermicity of the covalent inhibition step enabling the back reaction. To test this possibility we computed the reaction energy of the addition reaction with the model warheads (CH3)HCβ=Cα(NO2)R. We computed both the reaction for R=H and R=CH3 in order to investigate if the weaker inhibition potencies of the compounds 15 and 20 with R=CH3 are due to differences in the reaction energies of the covalent inhibition step. Even if the docking investigations (see below) indicate that the addition of the thiolate group of Cys25 takes place at Cβ while the proton is transferred to Cα, we computed both possibilities, i.e., attack of the thiolate at Cα and Cβ (see Supporting Information). The reaction energies were obtained from B3LYP calculations16,17 in combination with the cc-pVTZ18 basis set. Possible influences of a polarizable environment were tested by additional COSMO computations employing ε = 78.39.19 The computations were performed with the TURBOMOLE program package.20 The computations for a polar environment predict that the reaction energies for α- and β-attack are only about −12 kcal/mol for R=H. This indeed indicates that the addition reaction is reversible. The corresponding value for K11777 is far below −20 kcal/mol. For R=CH3 the reaction is even less exothermic (−10 and −7 kcal/mol for the sulfur attack at Cβ and Cα, respectively). This small difference compared to R=H indicates that the weaker inhibition potencies of compounds 15 and 20 do not solely stem from the less exothermic reaction energy, but should mainly result from steric effects.
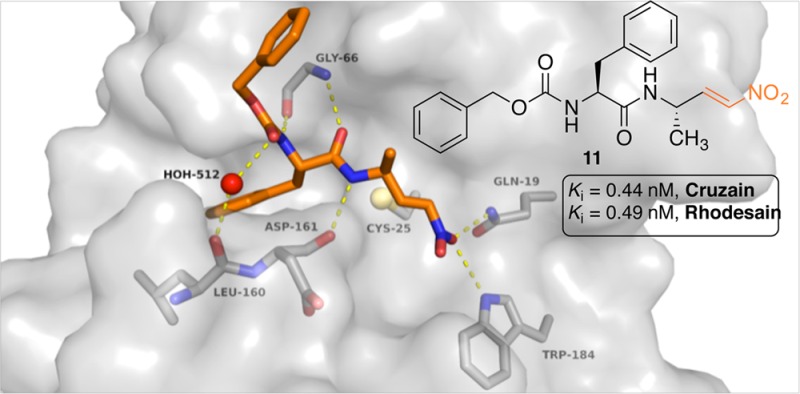
This is underlined by the docking studies that were performed with inhibitors 11, 14, and 15. For the docking studies we used the X-ray structure of rhodesain in complex with K11777 as template (pdb 2P7U) and performed noncovalent (FlexX/LeadIT)21 as well as covalent (DOCKTITE)22 docking. The results show that the dipeptidyl nitroalkenes should bind in a comparable manner as the corresponding vinylsulfones. Similar hydrogen bonds should be formed. Notably the key interactions of the warhead to the enzyme (Gln19, Trp184) are nearly identical (Figure 3). The vinylic β-carbon atom comes in close proximity to the Cys25 sulfur (2.77–2.99 Å). The docking scores (Hyde score, kJ/mol/FlexX score: −22/–24.06 (11), −19/–21.39 (14), −11/–23.34 (15)) reveal that inhibitor 11 should form the most favorable noncovalent interactions with the enzyme, which is in accordance with the experimental results (Figure 3, see also Supporting Information).
Figure 3.
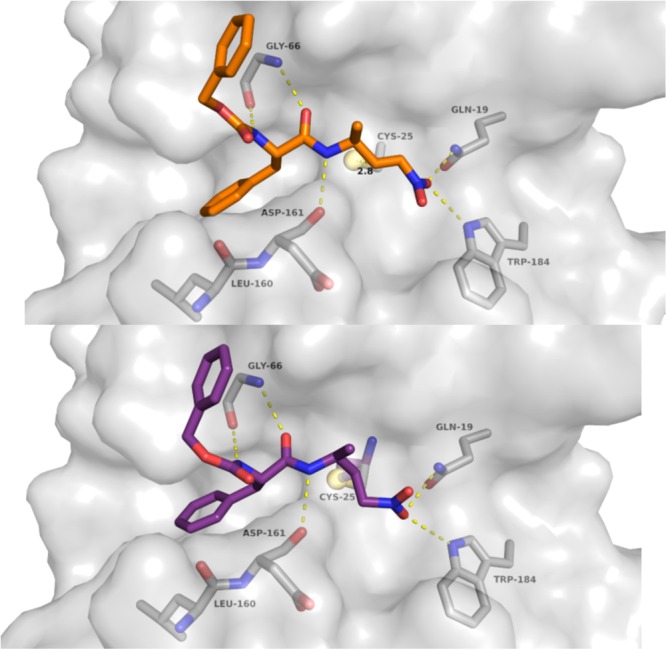
Proposed binding geometry and hydrogen bonding for inhibitor 11: within the noncovalent enzyme–inhibitor complex (above) and within the covalent enzyme–inhibitor complex (below).
15N-Rhodesain was expressed (see Supporting Information) and 1H–15N-TROSY-HSQC spectra were recorded to measure the interaction of rhodesain with inhibitor 12. First a 1:1 mol/mol mixture (protein/inhibitor) was incubated for 15 min and measured. Then the inhibitor 12 concentration was increased to a 1:3 molar ratio (rhodesain/inhibitor) by adding 12 in DMSO-d6 and further incubation for 7 min before measurement. As observed in the spectra (Figure 4), full saturation of 12 binding to rhodesain is already achieved at a 1:1 molar ratio, which denotes inhibitor 12 to bind very tightly to rhodesain (Figure 4).
Figure 4.
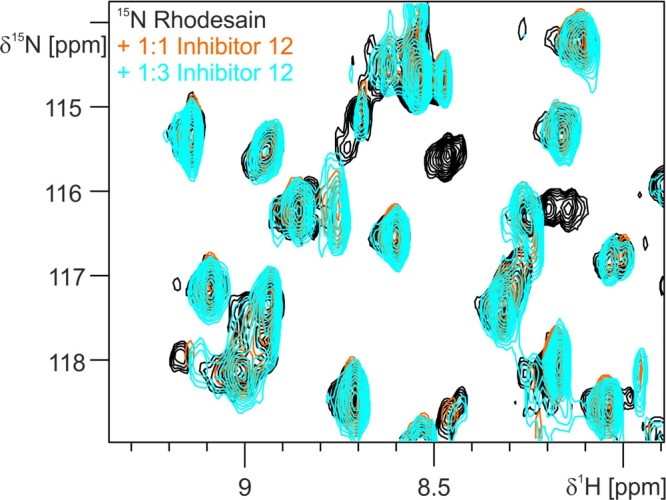
15N NMR spectra of rhodesain in complex with inhibitor (12); rhodesain (black), 1:1 molar ratio (orange), and 1:3 molar ratio (blue).
The activity of the most potent rhodesain inhibitors 11, 12, and 14 against T. brucei brucei was also determined (Table 2).23,24 All three compounds were found to exhibit good antitrypanosomal activity in the low micromolar range. The increasing EC50 values during time may be a result of instability of the compounds in the medium after >24 h.
Table 2. Antitrypanosomal Activity against Trypanosoma brucei brucei (EC50 in μM).
| inhibitor | EC50 (24 h) | EC50 (48 h) | EC50 (72 h) |
|---|---|---|---|
| 11 | 5.64 | 7.17 | 10.82 |
| 12 | 0.99 | 1.31 | 2.06 |
| 14 | 1.62 | 2.10 | 3.45 |
| chlorhexidineb | 0.43 | 0.41 | 0.43 |
Values are mean values from two independent assays performed each in triplicate; the values of all experiments incl. standard deviations are given in the SI.
Used as a control.
In summary, we report dipeptidyl nitroalkenes as a new type of highly potent covalent reversible inhibitors of cysteine proteases. The compounds exhibit certain selectivity for rhodesain and cruzain. According to the computations (Docking, QM), the reversibility of the inhibition, which is a difference to the related vinylsulfones, is a result of a weaker exothermicity and is not due to a different binding mode preventing nucleophilic attack of the active site Cys residue at the activated double bond.
Glossary
ABBREVIATIONS
- RD
rhodesain
- CRZ
cruzain
- CB
cathepsin B
- CL
cathepsin L
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00276.
Experimental procedures for the preparation of inhibitors, spectroscopic data, enzyme assays, antitrypanosomal activity, computational studies and protein NMR (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
We thank Fundación Española para la Ciencia y la Tecnología (Fecyt) and Generalitat Valenciana (AICO/2016/32) for financial support. T S. and B.E. thank the DFG (Deutsche Forschungsgemeinschaft) in the framework of the SFB630 for financial support. We thank Universitat Jaume I for technical suppport and funding. U.A.H. acknowledges support by the Carl-Zeiss foundation as well as the Center of Biomolecular Magnetic Resonance (BMRZ) funded by the state of Hesse.
The authors declare no competing financial interest.
Dedication
We dedicate this work to Prof. Dr. Gerhard Bringmann, Institute of Organic Chemistry, University of Würzburg, Germany, on the occasion of his 65th birthday.
Supplementary Material
References
- McKerrow J. H.; James M. N. G. In Perspectives in Drug Discovery and Design; Anderson P. S., Kenyon G. L., Marshall G. R., Eds.; ESCOM Science Publishers: Leiden, 1996; Vol. 6. [Google Scholar]
- Otto H.-H.; Schirmeister T. Cysteine Proteases and Their Inhibitors. Chem. Rev. 1997, 97, 133–172. 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- Eakin A. E.; McGrath M. E.; McKerrow J. H.; Fletterick R. J.; Craik C. S. Production of crystallizable cruzain, the major cysteine protease from Trypanosoma cruzi. J. Biol. Chem. 1993, 268, 6115–6118. [PubMed] [Google Scholar]
- McKerrow J. H.; Sun E.; Rosenthal P. J.; Buvier J. The proteases and pathogenicity of parasitic protozoa. Annu. Rev. Microbiol. 1993, 47, 821–853. 10.1146/annurev.mi.47.100193.004133. [DOI] [PubMed] [Google Scholar]
- Godal T.; Nagera J. In WHO Division of Control in Tropical Diseases; World Health Organization: Geneva; pp 12–13. [Google Scholar]
- Molyneux D. H. In Trypanosomiasis and Leishmaniasis: Biology and Control; Hide G., Mottram J. C., Coombs G. H., Holmes P. H., Eds.; CAB Int.: Oxford, 1997; pp 39–50. [Google Scholar]
- Control and surveillance of African trypanosomiasis. World Health Org. Technol. Rep. Ser. 1998, 881. [PubMed] [Google Scholar]
- Lecaille F.; Kaleta J.; Brömme D. Human and Parasitic Papain-Like Cysteine Proteases: Their Role in Physiology and Pathology and Recent Developments in Inhibitor Design. Chem. Rev. 2002, 102, 4459–4488. 10.1021/cr0101656. [DOI] [PubMed] [Google Scholar]
- Nicoll-Griffith D. A. Use of cysteine-reactive small molecules in drug discovery for trypanosomal disease. Expert Opin. Drug Discovery 2012, 7, 353–366. 10.1517/17460441.2012.668520. [DOI] [PubMed] [Google Scholar]
- Palmer J. T.; Rasnick D.; Klaus J. L.; Brömme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. 10.1021/jm00017a002. [DOI] [PubMed] [Google Scholar]
- Schneider T. H.; Rieger M.; Ansorg K.; Sobolev A. N.; Schirmeister T.; Engels B.; Grabowsky S. Vinyl sulfone building blocks in covalently reversible reactions with thiols. New J. Chem. 2015, 39, 5841–5853. 10.1039/C5NJ00368G. [DOI] [Google Scholar]
- Schirmeister T.; Kesselring J.; Jung S.; Schneider T.; Weickert A.; Becker J.; Lee W.; Bamberger D.; Wich P.; Distler U.; Tenzer Stefan; Johe Patrick; Hellmich U. A.; Engels B. Quantum chemical-based protocol for the rational design of covalent inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. 10.1021/jacs.6b03052. [DOI] [PubMed] [Google Scholar]
- pKa in DMSO for nitroalkane = 17.2; for sulfone = 29.
- Slight degree of racemization (15/1 to 5/1) during nitroaldol reaction was observed for some cases.
- Ludewig S.; Kossner M.; Schiller M.; Baumann K.; Schirmeister T. Enzyme kinetics and hit validation in fluorimetric protease assays. Curr. Top. Med. Chem. 2010, 10, 368–382. 10.2174/156802610790725498. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5653. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C. T.; Yang W. T.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Weigend F. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schüürmann G. A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- TURBOMOLE, V6.1 2009; University of Karlsruhe and Forschungszentrum Karlsruhe GmbH: Karlsruhe, Germany, 2007. [Google Scholar]
- LeadIT/FlexX, Version 2.1.3; BioSolveIT GmbH: St. Augustin, Germany, 2012. [Google Scholar]
- Scholz C.; Knorr S.; Hamacher K.; Schmidt B. J. Chem. Inf. Model. 2015, 55, 398–406. 10.1021/ci500681r. [DOI] [PubMed] [Google Scholar]
- Crouch S. P. M.; Kozlowski R.; Slater K. J.; Fletcher J. The Use of ATP bioluminescence as a measure of cell proliferation and cytotoxicity. J. Immunol. Methods 1993, 160, 81–88. 10.1016/0022-1759(93)90011-U. [DOI] [PubMed] [Google Scholar]
- Fueller F.; Jehle B.; Putzker K.; Lewis J. D.; Krauth-Siegel R. L. High throughput screening against the peroxidase cascade of African trypanosomes identifies antiparasitic compounds that inactivate tryparedoxin. J. Biol. Chem. 2012, 287, 8792–8802. 10.1074/jbc.M111.338285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.