

Abstract
Ribosome crystal structures have revealed that two small subunit proteins, S9 and S13, have C-terminal tails, which, together with several features of 16S rRNA, contact the anticodon stem-loop of P-site tRNA. To test the functional importance of these protein tails, we created genomic deletions of the C-terminal regions of S9 and S13. All of the tail deletions, including double mutants containing deletions in both S9 and S13, were viable, showing that Escherichia coli cells can synthesize all of their proteins by using ribosomes that contain 30S P sites composed only of RNA. However, these mutants have slower growth rates, indicating that the tails may play a supporting functional role in translation. In vitro analysis shows that 30S subunits purified from the S13 deletion mutants have a generally decreased affinity for tRNA, whereas deletion of the S9 tail selectively affects the binding of tRNAs whose anticodon stem sequences are most divergent from that of initiator tRNA.
Extensive evidence from more than three decades of experiments (1) supports the idea that ribosomal function is based primarily on rRNA (2, 3). This is most clearly seen in ribosome crystal structures, which show that the functional region of the ribosome, at the subunit interface, is composed mostly of rRNA, whereas the ribosomal proteins are found mainly at the periphery of the ribosome (4–7). However, there are a number of instances where ribosomal proteins contact tRNA (6). One of these occurs in the 30S P site, which plays a major role in initiation of protein synthesis, binding the initiator tRNA and positioning the start codon of mRNA. The 30S P site is also responsible for binding and positioning the anticodon stem-loop (ASL) of peptidyl-tRNA during polypeptide elongation and for maintaining the translational reading frame when the A site is vacant. Binding of the ASL depends not only on base-pairing with the P-site codon of mRNA, but also on interactions with the 30S subunit itself. In addition to contacts involving several elements of 16S rRNA, the extended C-terminal tails of two proteins, S9 and S13, penetrate the 30S P site within contact distance of the ASL (5, 6). The universally conserved C-terminal arginine of S9 appears to contact phosphate 35 at the apex of the anticodon loop of the P-site tRNA (Fig. 1). The C-terminal tail of S9 is phylogenetically invariant in length, an observation that is explained by the crystal structure, which shows that the P-site tRNA would physically block extension of the S9 tail. The tail of S13 runs parallel to the anticodon stem, crossing the tRNA backbone between positions 29 and 30 and is thus more tolerant of variations in length (Fig. 1). Although more variable in sequence, the S13 tail always contains several basic side chains, which can make electrostatic interactions with tRNA phosphates. The interactions of the S9 and S13 protein tails with P-site tRNA and their phylogenetic conservation suggest that they are directly involved in the function of the 30S P site.
Fig. 1.

Interaction of the C-terminal tails of S9 and S13 with tRNA (red) in the Thermus thermophilus 30S subunit P site (5, 6). Large (S9Δ26, S13Δ36) and small (S9Δ3, S13Δ5) tail deletions are red and yellow, respectively; a C-terminal extension of S13 present in T. thermophilus but not in E. coli is gray. The rest of the subunit is represented in a semitransparent rendering.
Early in vitro reconstitution studies provided evidence for the participation of S9 and S13 in ribosome function. Omission of S9 from 30S subunits reconstituted in vitro from purified RNA and protein components resulted in slower-sedimenting particles that were defective in functional tests, including P-site binding and factor-dependent initiator tRNA binding (8). However, when 30S subunits were reconstituted from CsCl-derived core particles and split proteins, omission of S9 yielded particles that were fully active in in vitro protein synthesis and binding tRNA to 70S ribosomes, but partially deficient in binding tRNA to isolated 30S subunits (9) and deficient in ribosome-stimulated EF-Tu- and EF-G-catalyzed hydrolysis of GTP (10, 11). By contrast, omission of S13 in reconstitutions carried out with purified components yielded particles whose sedimentation was indistinguishable from WT 30S subunits, but slightly defective in functional tests (8). In early genetic screens, Dabbs and coworkers reported deletions of both S9 (12, 13) and S13 (14). The S9 deletion was obtained in a search for dependence on kasugamycin, an antibiotic that inhibits binding of fMet-tRNAfMet during initiation.
Here, we directly test the functional importance of the tail domains of S9 and S13. We constructed Escherichia coli strains with precise chromosomal deletions in rpsI and rpsM, in which all of the cellular ribosomes contain S9 and/or S13 with truncated tails, eliminating all potential interactions with P-site tRNA, according to the crystal structure. All of the deletion mutants, including a double mutant containing tail deletions in both S9 and S13, are viable, indicating that the tails are not essential, although they show various phenotypes, including slower growth rates and cold sensitivity. In vitro assays indicate that the C termini of S9 and S13 contribute significantly to binding of P tRNA to isolated 30S subunits, although the importance of the tails is different for different tRNA species.
Materials and Methods
Chromosomal Deletions. Strains containing precise deletions within the rpsI and rpsM genes were made by using the vector pKO3 as described (15). First, the L13 and α operons were amplified by PCR from E. coli K12 genomic DNA and cloned into pKO3 to yield pLH2 and pLH6, respectively. Next, deletions of the 3′ ends of the rpsM and rpsI genes were engineered by QuikChange mutagenesis (Stratagene) as described (16). Plasmids pLH22, pLH17, and pLH24, containing the ΔS9, S9Δ26, and S9Δ3 deletions, respectively, were generated from pLH2. Mutation ΔS9 was engineered by using primers lh63 (5′-ATCTAATCGGGATTATAGGCATAATTGGCTTCTGCTCCGGCA) and lh64 (5′-TGCCGGAGCAGAAGCCAATTATGCCTATAATCCCGATTAGAT); S9Δ26 with primers lh15 (5′-CTGCGTAAAGCTGGCTTCGTTTAATTGGCTTCTGCTCCGGCAGAA) and lh16 (5′-TTCTGCCGGAGCAGAAGCCAATTAAACGAAGCCAGCTTTACGCAG); and S9Δ3 with primers lh69 (5′-GCACGTCGTCGTCCGCAGTTCTAATTGGCTTCTGCTCC GGCA) and lh70 (5′-TGCCGGAGCAGAAGCCAATTAGAACTGCGGACGACGACGTGC). Plasmid pLH19 containing S13Δ36 and pLH25 containing S13Δ5 were generated from pLH6. Mutation S13Δ36 was engineered by using primers lh26 (5′-GAGCATCAAGCGCCTGATGGATTAATCGGGGTGATTGAATAATGG) and lh27 (5′-CCATTATTCAATCACCCCGATTAATCCATCAGGCGCT TGATGCTC); and S13Δ5 was engineered by using primers lh65 (5′-CGTACCCGTAAGGGTCCGCGCTAATCGGGGTGATTGAATAATGG) and lh66–2(5′-CCATTATTCAATCACCCCGATTAGCGCGGACCCTTACGGGTACG). Finally, each of these plasmids (pLH22, pLH17, pLH24, pLH19, and pLH25) was used to replace the WT rpsI or rpsM gene of CSH142 (17) with the deletion allele (15). PCR was used to verify each allelic replacement. To construct the S9Δ3/S13Δ5 double mutant, kefB20::Tn10(kan) (E. coli Genetic Stock Center http://cgsc.biology.yale.edu), which is linked to rpsM, was moved by P1 transduction into the rpsMΔ5 strain (encoding S13Δ5). Then, the resulting strain [rpsMΔ5 kefB20::Tn10(kan)] was used as a donor to transduce the rpsIΔ3 strain, and transductants were screened by PCR for the presence of both rpsMΔ5 rpsIΔ3 mutations. Thus, the double rpsMΔ5 rpsIΔ3 mutant additionally contains kefB20::Tn10(kan). Control experiments indicate that kefB20::Tn10(kan) confers no growth defect (data not shown).
Growth Rates. LB medium (50 ml) was equilibrated to 30°C, 37°C, or 42°C and inoculated with 50 μl of saturated overnight culture. One-milliliter aliquots were taken every 30 min and measured for absorbance at 550 nm. Doubling rates for each strain were calculated from at least four growth curves at each temperature.
Toeprinting. mRNAs were made by in vitro transcription with T7 RNA polymerase as described (18). All tRNAs were purchased from Sigma except for overproduced tRNAThr, which was a gift from H. Asahara (University of California, Santa Cruz). 32P-labeled primer kf132 (5′-CTTTATCTTCAGAAGAAAAACC) was annealed to mRNA (0.7 μM final concentration) in 50 mM Tris·HCl (pH 7.5) and 100 mM NH4Cl by heating to 68°C for 5 min and placing on ice. MgCl2 was added to 20 mM, heat-activated 30S subunits (19) were added to 0.7 μM, and tRNA (0.7 μM when present) was added to fill the P site by incubation at 37°C for 20 min. A 2-μl aliquot was removed and added to a 10-μl extension mix containing 10 mM Tris·HCl (pH 7.5), 10 mM MgCl2, 60mMNH4Cl, 375 μM of each dNTP, 6 mM 2-mercaptoethanol, and 2.6 units of avian myeloblastosis virus reverse transcriptase (Seikagaku Kogyo, Tokyo). Reactions were extended for 10 min and stopped with 10 μl of stop buffer to give a final concentration of 47.5% (wt/vol) formamide, 5 mM EDTA, 0.025% (wt/vol) bromophenol blue, and 0.025% xylene cyanol FF. Two microliters was loaded onto a 6% polyacrylamide gel.
Nitrocellulose Filter Binding. tRNAfMet and tRNAVal (Sigma) were aminoacylated and acetylated as described (20). Either 100 nM tRNAfMet or 200 nM tRNAVal and 5 μM sm291 mRNA (5′-UAAAAAGGAAAUAAAAAUGUUUGUAUACAAAUC) were titrated with 0–2.5 μM heat-activated 30S subunits and incubated for 10 min at 37°C in 50 mM K-Hepes (pH 7.5), 20 mM MgCl2, 100 mM NH4Cl, and 1 mM DTT in a total volume of 10 μl for tRNAfMet binding reactions or 20 μl for tRNAVal. Binding reactions were spotted onto 0.45-μm HA nitrocellulose membrane filters (Millipore) and washed three times with 5 ml of cold binding buffer. Filters were submerged in 4 ml of Bio-Safe II counting mixture (Research Products International, Mount Prospect, IL) and counted in a liquid scinitillation counter (Beckman model LS5801).
Titration curves were plotted from data averaged from three experiments and fit to the following equation (21) by using the graphing program kaleidagraph (Abelbeck Software, Reading, PA) to estimate KD: 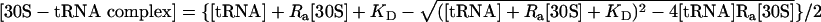 , where Ra = [active 30S]/[total 30S]. The equation-derived value for Ra is in agreement with 30S functional assays (data not shown).
, where Ra = [active 30S]/[total 30S]. The equation-derived value for Ra is in agreement with 30S functional assays (data not shown).
Results
Genomic sequences coding for the tail regions of S9 and S13 were precisely deleted by using the method of Link et al. (15) as described in Materials and Methods. The short deletions, which removed the C-terminal three amino acid residues from S9 (S9Δ3) and five residues from S13 (S13Δ5), were designed as minimal deletions that would eliminate contact with P-site tRNA (Fig. 1). More extensive deletions of 26 residues from S9 (S9Δ26) and 36 residues from S13 (S13Δ36), which completely eliminate the extended tails (Fig. 1), were also constructed. The mutant strains were all viable, but exhibited altered growth phenotypes, which correlated with the size of the deletion (Fig. 2). The large deletions of S9 and S13, S9Δ26 and S13Δ36, confer growth rates about half that of WT at all temperatures tested (Fig. 2B). At 37°C and 42°C, the small deletion alleles of S9 and S13, S9Δ3 and S13Δ5, show only modest growth defects (Fig. 2B and Table 1), whereas the S9Δ3 strain shows a cold-sensitive phenotype, growing substantially slower than WT at 30°C. The double mutant, S9Δ3/S13Δ5, shows an additive growth defect compared to each of the individual deletion mutants at both 37°C and 42°C. Interestingly, however, the cold-sensitive phenotype observed for S9Δ3 is suppressed by the S13Δ5 mutation (Fig. 2B).
Fig. 2.

Growth phenotypes of S9 and S13 deletion mutants. (A) Growth on LB plates at 37°C. (B) Doubling rates in LB liquid culture at 30°C, 37°C, and 42°C, relative to WT. ○, WT; ▪ ,S9Δ3; ▿, S13Δ5; •, S9Δ3/S13Δ5; ▵,S9Δ26; ▴, S13Δ36;□, ΔS9.
Table 1. In vivo and in vitro effects of S9 and S13 deletions: Doubling times (minutes).
| 30°C | 37°C | 42°C | |
|---|---|---|---|
| WT | 38 ± 4 | 24 ± 2 | 22 ± 3 |
| S9Δ26 | 109 ± 12 | 41 ± 3 | 42 ± 7 |
| S13Δ36 | 96 ± 14 | 51 ± 7 | 45 ± 5 |
| S9Δ3 | 83 ± 17 | 29 ± 2 | 26 ± 2 |
| S13Δ5 | 52 ± 4 | 29 ± 1 | 26 ± 1 |
| S9Δ3/S13Δ5 | 60 ± 7 | 39 ± 1 | 38 ± 7 |
S9 has been shown to contribute to proper assembly of the 30S subunit, whereas S13 has no detectable role in subunit assembly (8). A complete chromosomal deletion of S9 (ΔS9) was constructed and found to be viable (Fig. 2). The strong growth phenotype of ΔS9 is consistent with a partial defect in ribosome assembly. Although Dabbs and coworkers (12–14) have previously reported S9 and S13 deletion strains, the method of Link et al. (15) provided a means to create precise chromosomal deletions. We did not attempt to generate an S13 deletion strain.
The in vivo phenotypes of the S9 and S13 mutant alleles suggest that the C-terminal tails of these proteins contribute to ribosome function. To test how each mutation affected the P site, 30S ribosomal subunits were purified from each deletion strain and tested for their ability to bind tRNA by using a toeprinting assay (22). In this assay, 30S–tRNA–mRNA complexes are formed and the position of mRNA in the ribosome is mapped by primer extension of the mRNA from a downstream primer. When 30S subunits from the S13 deletion mutants (S13Δ5 or S13Δ36) were used, decreased toeprint intensities were observed for all of the tRNA species tested, suggesting that truncation of S13 confers a general defect in tRNA binding (Fig. 3).
Fig. 3.

Toeprinting 30S–tRNA complexes on m291 (A and B) and m301 (C–E) mRNAs. 30S subunits from each of the five tail deletion mutants were tested for their ability to stably bind initiator (fM), Cys (C), Val (V), Lys (K), Phe (F), yeast Phe (yF), Tyr (Y), and Thr (T) tRNAs to the 30S P site. Band numbering (+16, +18, etc.) indicates the downstream positions of the reverse transcriptase stops (19). (E) As in D except complexes were formed with 70S ribosomes.
In contrast, when 30S subunits containing truncated S9 were used, the toeprinting results varied according to the species of tRNA. Whereas modest decreases in toeprinting intensities were observed when tRNAfMet, tRNACys, or tRNAPhe were bound to the S9Δ3 or S9Δ26 mutant 30S subunits, almost no P-site toeprints were seen when tRNAVal, tRNALys, 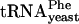 , tRNA-Tyr, or tRNAThr were tested with these subunits (Fig. 3 B and D). Binding of tRNAVal, tRNALys,
, tRNA-Tyr, or tRNAThr were tested with these subunits (Fig. 3 B and D). Binding of tRNAVal, tRNALys, 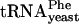 , tRNATyr, or tRNAThr to subunits containing the S9Δ3/S13 Δ5 double deletion is also selectively destabilized relative to that of tRNAfMet, tRNACys, or tRNAPhe (Fig. 3 B and D). To confirm that low toeprint signals are caused by weak tRNA binding, we titrated S9Δ3 30S subunits with increasing amounts of tRNAVal and observed enhanced toeprints as tRNAVal is increased (data not shown).
, tRNATyr, or tRNAThr to subunits containing the S9Δ3/S13 Δ5 double deletion is also selectively destabilized relative to that of tRNAfMet, tRNACys, or tRNAPhe (Fig. 3 B and D). To confirm that low toeprint signals are caused by weak tRNA binding, we titrated S9Δ3 30S subunits with increasing amounts of tRNAVal and observed enhanced toeprints as tRNAVal is increased (data not shown).
The selective loss of binding of this subset of tRNAs to 30S subunits containing S9 deletions is suppressed by the 50S subunit. Association of 30S subunits from the different deletion mutants with 50S subunits to form 70S ribosomes restored the toeprinting intensities (Fig. 3E).
The toeprinting assay reflects the ability of bound tRNA to position mRNA stably in the ribosomal complex, and thus provides an indirect measure of tRNA binding. To obtain a direct, quantitative measure, binding of radioactively labeled tRNA was assayed in a nitrocellulose filter-binding assay. The affinities of formyl-[35S]Met-tRNAfMet and N-acetyl-[14C]Val-tRNAVal for WT, S9Δ3, and S13Δ5 30S subunits were compared. Consistent with the results of the toeprinting assay, S9Δ3 and S13Δ5 are only negligibly different in binding f-[35S]Met-tRNAfMet, but S9Δ3 shows a 350-fold lower affinity for N-acetyl-[14C]Val-tRNAVal, whereas only a 2-fold difference is measured for S13Δ5 (Fig. 4 and Table 2). These data indicate that the C terminus of S9 differentially affects the binding of different tRNAs to the 30S subunit P site.
Fig. 4.

Binding affinities of formyl-[35S]Met-tRNAfMet (A) and Nac-[14C]-Val-tRNAVal (B) to WT (•), S9Δ3 (▴), and S13Δ5 (▪) 30S subunits. Increasing concentrations of 30S subunits were incubated with 200 nM tRNA in the presence of sm291 mRNA and assayed by filter binding as described in Materials and Methods.
Table 2. In vivo and in vitro effects of S9 and S13 deletions: 30S-tRNA binding affinities.
| f-[35S]Met-tRNAfMet, KD (× 10-9 M) | Nac-[14C]Val-tRNAVal, KD (× 10-9 M) | |
|---|---|---|
| WT | 11 ± 3 | 22 ± 8 |
| S9Δ26 | 36 ± 14 | |
| S13Δ36 | 62 ± 32 | |
| S9Δ3 | 18 ± 10 | 7,800 ± 680 |
| S13Δ5 | 24 ± 9 | 45 ± 23 |
| S9Δ3/S13Δ5 | 34 ± 8 |
Discussion
These studies show that E. coli cells are able to grow, with only modest decreases in growth rate, when all potential protein–tRNA interactions in the 30S P site are abolished by deletion of the C-terminal tails of proteins S9 and S13. Cells bearing the S9Δ3/S13Δ5 double deletion must carry out protein synthesis with ribosomes bearing an “all-RNA” 30S P site, in which all interactions between the ribosome and the ASL of tRNA are made by 16S rRNA. Cells containing larger deletions of the S9 and S13 tails (S9Δ26 and S13Δ36) or even cells with complete deletions of S9 were also able to grow, albeit with more impaired growth rates. Indeed, at least 17 different ribosomal proteins have been deleted without loss of cell viability, as shown earlier by Dabbs (14). More recently, Zengel et al. (23) have shown that deletion of the loops of proteins L4 and L24, which extend into the polypeptide exit channel, and were proposed to be important for ribosomal assembly and functional contacts with the nascent polypeptide chain (24), have no detectable effect on ribosomal assembly or on incorporation of 50S subunits into polysomes. These findings provide evidence to support the idea that presentday ribosomes evolved from preribosomal particles that contained only RNA (1–3). According to this view, ribosomal proteins were recruited later, as evolutionary refinements, to enhance the speed and accuracy of translation, increase the efficiency of ribosome assembly, and mediate such cellular processes as membrane trafficking and translational regulation.
The phenotypes conferred by the S9 and S13 tail deletions, both in vivo and in vitro, are clearly distinguishable. Whereas the S13 deletion mutants exhibit slowed growth rates at all temperatures, the S9Δ3 deletion produces a cold-sensitive phenotype, which is suppressed when combined with the S13Δ5 deletion. Cold sensitivity can be enhanced by destabilization of an early step of a multistep sequential process (25). In the case of the S9Δ3 mutation, cold sensitivity might arise from destabilization of some step in the binding of tRNA (for example, initiator tRNA) to the 30S P site. Loss of the S9 tail interaction could distort the positioning of the tRNA anticodon in a way that is compensated by deletion of the S13 tail.
The distinctly different in vitro properties of the S9 and S13 deletions are particularly interesting. Whereas the P site of S13Δ5 subunits shows a generally decreased affinity for tRNA, the S9Δ3 subunits selectively lose their affinity for a subset of tRNAs (Fig. 3 and Table 2). Because only the ASL interacts with the 30S P site, the structural basis for this selectivity must lie within this limited 17-nt region. Comparison of the structures of the eight different tRNA species that were assayed (Fig. 5) shows a striking correlation. The subset of tRNAs whose binding to S9Δ3 subunits is largely unaffected all contain two G-C base pairs at the second and third stem positions (nucleotides 29, 30, 40, and 41) from the anticodon loop, whereas none of the tRNAs whose binding is abolished has a G-C pair at the third position. These correspond to two of the three G-C pairs that are found in all initiator tRNAs, and which RajBhandary and Chow (26) have shown are critical for discrimination of initiator tRNA from elongator tRNAs. A possible explanation for the behavior of S9Δ3 subunits is that the 30S subunit is capable of specific recognition of tRNAs containing these G-C pairs, in addition to a nonspecific P-site affinity for tRNA that is facilitated by interaction of the backbone of the anticodon loop with the tail of protein S9. Deletion of the tail of S9 would result in loss of the nonspecific affinity, leaving the specific affinity for tRNAs that have initiator-like anticodon stems. Selectivity is still observed in the S9Δ3/S13Δ5 double deletion, indicating that the discrimination must be a function of 16S rRNA.
Fig. 5.

ASL sequences of tRNAs used in this study. Arrowheads indicate base pairs 29–41 and 30–40, which are G-C pairs in all nonmitochondrial initiator tRNAs (27).
The question arises as to why these deletion strains are only minimally defective in growth if they are so defective in binding some tRNAs. A likely explanation is that when 30S subunits associate with 50S subunits to form 70S ribosomes the binding defects of the 30S subunit are masked (Fig. 3), most likely because of the extensive additional interactions made between the 50S subunit and P-site tRNA (6). Thus, the impairments in 30S subunit function caused by the deletion mutations may have their greatest effect on translational initiation, the only physiological step in which tRNA binds to the 30S P site in the absence of the 50S subunit.
Acknowledgments
We thank H. Asahara, L. Lancaster, S. Takyar, R. Hickerson, K. Lieberman, M. Laurberg, L. Nguyenle, I. Ali, J. Nix, and K. Triman for helpful discussions; H. Asahara for a generous gift of tRNAThr and his expertise in calculating tRNA binding constants; and A. Baucom for generating Fig. 1. This work was supported by National Institutes of Health Grant GM17129, National Science Foundation Grant MCB-212689, and a grant to the Center for Molecular Biology of RNA from the W. M. Keck Foundation.
Abbreviation: ASL, anticodon stem-loop.
References
- 1.Noller, H. F. (1991) Annu. Rev. Biochem. 60, 191–227. [DOI] [PubMed] [Google Scholar]
- 2.Crick, F. H. C. (1968) J. Mol. Biol. 38, 367–379. [DOI] [PubMed] [Google Scholar]
- 3.Noller, H. F. (1999) in The RNA World, eds. Gestland, R. F., Cech, T. R. & Atkins, J. F. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 197–219.
- 4.Ban, N., Nissen, P., Hansen, J., Moore, P. B. & Steitz, T. A. (2000) Science 289, 905–920. [DOI] [PubMed] [Google Scholar]
- 5.Wimberly, B. T., Brodersen, D. E., Clemons, W. M., Jr., Morgan, R. J.-W., Carter, A. P., Vonrhein, C., Hartsch, T. & Ramakrishnan, V. (2000) Nature 407, 327–339. [DOI] [PubMed] [Google Scholar]
- 6.Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H. D. & Noller, H. F. (2001) Science 292, 883–896. [DOI] [PubMed] [Google Scholar]
- 7.Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F. & Yonath, A. (2001) Cell 107, 679–688. [DOI] [PubMed] [Google Scholar]
- 8.Nomura, M., Mizushima, S., Ozaki, M., Traub, P. & Lowry, C. V. (1969) in Cold Spring Harbor Symposia on Quantitative Biology, eds. Frisch, L. & Hershey, H. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 49–61. [DOI] [PubMed]
- 9.Traub, P., Hosokawa, K., Craven, G. R. & Nomura, M. (1967) Proc. Natl. Acad. Sci. USA 58, 2430–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sander, G., Marsh, R. C. & Parmeggiani, A. (1973) FEBS Lett. 33, 132–134. [DOI] [PubMed] [Google Scholar]
- 11.Marsh, R. C. & Parmeggiani, A. (1973) Proc. Natl. Acad. Sci. USA 77, 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dabbs, E. R. (1978) J. Bacteriol. 136, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dabbs, E. R., Hasenbank, R., Kastner, B., Rak, K.-H., Wartusch, B. & Stöffler, G. (1983) Mol. Gen. Genet. 192, 301–308. [DOI] [PubMed] [Google Scholar]
- 14.Dabbs, E. R. (1991) Biochimie 73, 639–645. [DOI] [PubMed] [Google Scholar]
- 15.Link, A. J., Phillips, D. & Church, G. M. (1997) J. Bacteriol. 179, 6228–6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fredrick, K., Dunny, G. M. & Noller, H. F. (2000) J. Mol. Biol. 298, 379–394. [DOI] [PubMed] [Google Scholar]
- 17.Miller, J. H. (1992) A Short Course in Bacterial Genetics, (Cold Spring Harbor Lab. Press, Plainview, NY).
- 18.Fredrick, K. & Noller, H. F. (2002) Mol. Cell 9, 1125–1131. [DOI] [PubMed] [Google Scholar]
- 19.Zamir, A., Miskin, R. & Elson, D. (1971) J. Mol. Biol. 60, 347–364. [DOI] [PubMed] [Google Scholar]
- 20.Moazed, D. & Noller, H. F. (1989) Cell 57, 585–597. [DOI] [PubMed] [Google Scholar]
- 21.Pleiss, J. A. & Uhlenbeck, O. C. (2001) J. Mol. Biol. 301, 895–905. [DOI] [PubMed] [Google Scholar]
- 22.Hartz, D., McPheeters, D. S. & Gold, L. (1989) Genes Dev. 3, 1899–1912. [DOI] [PubMed] [Google Scholar]
- 23.Zengel, J. M., Jerauld, A., Walker, A., Wahl, M. C. & Lindahl, L. (2003) RNA 9, 1188–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakatogawa, H. & Ito, K. (2002) Cell 108, 629–636. [DOI] [PubMed] [Google Scholar]
- 25.Dammel, C. & Noller, H. F. (1993) Genes Dev. 7, 660–670. [DOI] [PubMed] [Google Scholar]
- 26.RajBhandary, U. L. & Chow, C. M. (1995) in tRNA: Structure, Biosynthesis, and Function, eds. Söll, D. & RajBhandary, U. L. (Am. Soc. Microbiol., Washington, DC), pp. 511–528.
- 27.Gauss, D. H., Grüter, F. & Sprinzl, M. (1979) Nucleic Acids Res. 6, 520–537. [PMC free article] [PubMed] [Google Scholar]