

Abstract
We have determined the structure of adeno-associated virus type 2 (AAV2) Rep40 to 2.1-Å resolution with ADP bound at the active site. The complex crystallizes as a monomer with one ADP molecule positioned in an unexpectedly open binding site. The nucleotide-binding pocket consists of the P-loop residues interacting with the phosphates and a loop (nucleoside-binding loop) that emanates from the last strand of the central β-sheet and interacts with the sugar and base. As a result of the open nature of the binding site, one face of the adenine ring is completely exposed to the solvent, and consequently the number of protein–nucleotide contacts is scarce as compared with other P-loop nucleotide phosphohydrolases. The conformation of the ADP molecule in its binding site bears a resemblance to those found in only three other families of P-loop ATPases: the ATP-binding cassette transporter family, the bacterial RecA proteins, and the type II topoisomerase family. In all these cases, oligomerization is required to attain a competent nucleotide-binding pocket. We propose that this characteristic is native to superfamily 3 helicases and allows for an additional mechanism of regulation by these multifunctional proteins. Furthermore, it explains the strong tendency by members of this family such as simian virus 40 TAg to oligomerize after binding ATP.
Within recent years, there has been increased interest in the use of the human parvovirus adeno-associated virus (AAV) as a vector for human gene therapy. Many preclinical as well as clinical studies highlight the extraordinary potential of this virus as a delivery vehicle for long-term gene transfer. It is important to note that no adverse effects have yet been reported as a result of AAV-mediated gene transfer, making this virus likely to be the most promising gene-therapy tool (1). Possibly one of the most intriguing aspects of the AAV life cycle is that virus DNA integration occurs in a site-specific manner into the long arm of human chromosome 19 (2–5). It has been proposed that interactions of a viral protein (Rep) with short sequence motifs within the integration target sequence (AAVS1) represent the initiating steps of the underlying unique mechanism (6). The products of a single ORF (REP) are orchestrating all aspects of the AAV life cycle, including site-specific integration, replication, and DNA packaging. The use of two different promoters (p5 and p19) and a splice site results in the production of four nonstructural (Rep) proteins with overlapping amino acid sequences (Rep78, Rep68, Rep52, and Rep40) (reviewed in ref. 7). Biochemical activities of Rep are consistent with their role in the AAV life cycle and are separated into three domains. The N terminus possesses a DNA-binding and site-specific endonuclease activity (8–13). This domain is present in the larger Rep proteins (Rep78 and Rep68) and is responsible for origin interactions including DNA binding and nicking of origin DNA (in both AAV and AAVS1) and covalent attachment of Rep to the 5′ end of the nicked DNA (14, 15). The central core domain (shared by all Rep isoforms) represents the motor domain with motifs required for ATPase and helicase activity as well as a nuclear import signal (12, 14, 16–24). The C-terminal zinc-finger domain has been implied in a number of as-yet little-defined protein–protein interactions (25–28). All Rep proteins share the central motor domain that is represented by the smallest protein, Rep40.
Helicases are molecular motor proteins that couple the energy of nucleotide hydrolysis to unidirectional movement along nucleic acids [3′ to 5′ in the case of Rep (29)], removing nucleic acid-associated proteins or threading them through various pores. Helicases also are involved in many aspects of the cellular machinery, including DNA replication, repair, recombination, transcription, translation, RNA splicing, editing, transport and degradation, bacterial conjugation, and viral packaging (30–35). The AAV type 2 (AAV2) Rep40 protein belongs to helicase superfamily 3 (SF3), one of five major groups of helicases classified by Gorbalenya and Kooning (36). The signature motif for this group consists of a stretch of ≈100 aa encompassing the Walker A and B, the B′ box, and the sensor 1 motifs. These helicases are found mainly in the genomes of small DNA and RNA viruses. The structures of several members of each of the other helicase families have been solved, and studies on these proteins have given insight into potential mechanisms by which NTP hydrolysis is used to accomplish DNA unwinding (37–39). It was only recently that the structures of two SF3 helicases were solved. Our work (AAV Rep40) (23) as well as the structure of simian virus 40 large T antigen (40) have revealed that these helicases belong to the AAA+ families with a RecA-like nucleotide phosphohydrolase core. To reflect the fact that these SF3 proteins are somewhat different structurally from other AAA+ members, we introduced the term viral AAA+. To gain insight into the mechanism underlying the transmission of chemical energy through ATP hydrolysis to DNA unwinding and packaging by the viral AAA+ proteins, we solved the structure of Rep40 complexed to ADP. The ADP is bound in an unusually open binding site with one face of the adenine ring completely exposed to the solvent in a conformation that is not seen in other AAA+ proteins and helicase families. We propose that oligomerization is required to form a complete and catalytically competent nucleotide-binding site. As a corollary, it is reasonable to hypothesize that the binding of ATP acts as a regulatory mechanism to dictate the oligomeric state of the SF3 helicases.
Materials and Methods
Expression, Purification, and Crystallization. The Rep40 protein, residues 225–490, was cloned, expressed, and purified as described (23). A final concentration of 15.5 mg/ml of the protein (Bradford assay) was used to obtain crystals with the hanging-drop vapordiffusion technique. Initial crystallization trials were done with ATP, 5′-adenylyl γ-thiotriphosphate, and 5′-adenylyl imidodiphosphate by using a nucleotide concentration ranging from 1 to 10 mM. Crystals were grown in a solution of 0.2 M sodium acetate, 6% polyethylene glycol 8000 and 0.1 M Tris (pH 8.5), 10 mM nucleotide, and MgCl2 at 4°C. The crystals grew to a maximum size of 0.25 × 0.04 × 0.03 mm over a period of 72–96 h and were cryoprotected in a solution containing 15% glycerol and 30% polyethylene glycol 8000. Crystals diffracted to 2.1 Å and belonged to the P65 space group with unit-cell dimensions of a = b = 71.92 Å, c = 96.72 Å, α = β = 90°, and γ = 120°.
Data Collection, Phasing, and Refinement. Data for the nucleotide-containing crystals were collected at the National Synchrotron Light Source (beamline X12C, Brookhaven National Laboratories, Upton, NY) and processed by using the hkl2000 package (41). The structure was determined by molecular replacement in cns (42) by using the Rep40 monomer (chain B; PDB ID code 1U0J) as the search model. The final model yielded an R factor of 19.2% (Rfree, 23.7%) and contained 1 ADP and 213 water molecules after several rounds of refinement in cns and rebuilding in o (43) (see Table 1). There was poor electron density for the β-hairpin 1 loop between residues 402 and 407 as well as the side chains of five residues throughout the molecule. All the residues lie within the allowed regions of the Ramachandran plot, with 88.9% in the most favored regions. All the figures were created by using the software pymol (44).
Table 1. Data collection and refinement statistics.
| Rep40—ADP | |
|---|---|
| Data collection | |
| Wavelength, Å | 1.1 |
| Resolution, Å | 2.1 |
| No. of reflections measured | 77,853 |
| No. of unique reflections | 15,662 |
| % Completeness | 97.9 (93.7) |
| Rmeas,*† % | 6.9 (41.7) |
| Rmerge,*‡ % | 6.2 (36.7) |
| Mean I/σ* | 17.6 (4.3) |
| Refinement statistics | |
| Resolution range, Å | 20-2.1 |
| Reflections, F > 2σ (F) | 14,926 |
| Rcrys,§ % | 19.2 |
| Rfree,¶ % | 23.7 |
| Nonhydrogen atoms | |
| Protein | 2,044 |
| Water | 213 |
| ADP | 27 |
| rms deviations | |
| Bonds, Å | 0.0175 |
| Angles, ° | 2.54 |
| Average B factor, Å2 | 33.77 |
Values in parentheses are for the outermost shell.
These values were calculated with the program novel_r (69).
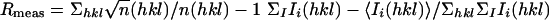 .
.
Rmerge = Σ |I — 〈I〉|/|Σ|, where I is the integrated intensity of a given reflection.
Rcrys = Σ |Fo| — |Fc|/Σ |F|.
Rfree was calculated by using 10% of data excluded from the refinement.
Results
Overall Structure. Although the protein was cocrystallized with both ATP and 5′-adenylyl γ-thiotriphosphate, a detailed inspection of the electron-density map showed only density for ADP. The monomeric Rep40–ADP complex is shown in Fig. 1a. The bimodular protein is composed of an N-terminal helical domain (α1–α4) and the C-terminal ATPase domain, a modified version of the AAA+ domain that we refer to as viral AAA+. This domain is composed of a central five-stranded β-sheet (β1–β5) flanked by four helices on one side (α6–α8 and α11) and two on the other (α9 and α10). The four conserved helicase motifs that characterize this family are located in a region of ≈100 amino acids going from strands β1to β4. The Walker A motif (residues 334–341) forms the loop following β1 and that connects to α8. The Walker B motif covers the end of β3 and a small part of the loop leading to α9. Conserved motif C encompasses the region across residues 416–420 (strand β4) and includes the sensor 1 residue N421. Motif B′ is found only in SF3 helicases and spans residues 391–404. Part of this motif forms a β-hairpin that protrudes into the solvent and may be involved in DNA binding. Five residues from this region are missing, suggesting that this region is very dynamic. As expected, the nucleotide sits in the region around the P-loop, and we can see clear density for an ADP molecule in the 2Fo – Fc simulated annealing omit map (Fig. 1b).
Fig. 1.

The Rep40–ADP complex. (a) The AAV2 Rep40 molecule (slate), complexed to ADP at 2.1 Å. The nucleotide sits in an unexpectedly open binding site formed by the P-loop residues and the NB-loop. The β-hairpin 1 loop (βa–βb) is disordered, with electron density for five consecutive residues, 402–407, missing. There are two secondary elements: a βe strand that is part of a three-stranded β-sheet together with strands βc–βd and a 310-helix h2 that is between β2 and β3. (b) A 2Fo – Fc simulated annealing omit map showing the electron density for the bound ADP at 1.5 σ.
Nucleotide-Binding Site. The structure of the Rep40–ADP complex shows the nucleotide in a surprisingly open binding site (Fig. 2a). The ADP molecule sits as expected, with the phosphate groups embedded in the groove formed by the P-loop residues. Residues K340 and T341 make hydrogen-bond contacts with the β-phosphate as observed in other P-loop ATPases. Additional protein–phosphate interactions are provided by the main-chain amide groups of T337, G339, and N342 (Fig. 2b). The electrostatic potential shows an electropositive region surrounding the phosphate groups that is large enough to accommodate the γ-phosphate group of an ATP molecule (Fig. 2c). The ribose torsion angle (γ) around the exocyclic C4′—C5′ bond is in a gauche-gauche (gg) conformation in contrast to the more typical trans-gauche conformation observed in all the structures of AAA+ proteins and helicase–nucleotide complexes solved to date. As a result, in a superposition of these two nucleotide conformations, the ribose ring in our complex appears to be rotated by almost 180° around the diphosphate bond (Fig. 2d). The ribose ring is in a C3′-endo conformation with the 2′ oxygen making a hydrogen bond with the main-chain carbonyl group of D455. G459 makes a water-mediated bond with the 2′ oxygen, whereas the O4′ interacts with the ND1 group of N342. The adenine base is in the anti conformation and points away from the protein core. Surprisingly, there is a lack of stacking interactions with the adenine ring, which is in marked contrast to most nucleotide-binding proteins, in which the adenine ring is sandwiched by hydrophobic residues that make stacking interactions with aromatic and/or aliphatic residues on both faces of the planar ring. In the Rep40–ADP complex, one face of the adenine is completely exposed to the solvent, whereas the other face sits on a relatively nonpolar region made by the main-chain atoms of G459 and K460 and the aliphatic portion of the K460 side chain. Additionally, the neutral residue N342 stacks below the adenine ring and is held in place by hydrogen-bond contacts with the carbonyl group of G459 and the amide group of V461. Thus, the loop connecting β5 to α11, which we call the nucleoside-binding (NB) loop, in addition to making direct interactions with the ribose and adenine acts as a “wall” that limits the rotational freedom of the nucleotide and restricts it to a gg conformation. Any other conformation, such as the trans-gauche seen in all AAA+ protein–nucleotide complex structures, would result in steric clashes with the NB-loop (Fig. 2d).
Fig. 2.

Rep40–ADP interactions. (a) The nucleotide binding pocket in Rep40. (b) Stereoview of the active site. The dotted lines represent hydrogen bonds; green spheres represent water molecules. Residues T337, T338, G339, K340, T341, and N342 are located in the P-loop, and residues D457, G459, K460, and V461 are located in the NB-loop. (c) Electrostatic surface potential of the Rep40–ADP molecule. Blue and red represent regions of positive and negative potential, respectively, as calculated in grasp (68). (d) Superposition of the ADP molecule in a trans-gauche conformation from the structure of NSF-D2 (cyan). Rep40 is shown as a surface representation (salmon).
Conformational Changes. Superposition of the apo and ADP-bound structures of Rep40 reveals no gross conformational changes after nucleotide binding (Fig. 3a). Comparison of all but seven residues from the loop of β-hairpin 1 gives an rms deviation of 1.53 Å over 260 Cα atoms. Most of the differences arise from the intrinsic mobility of the N-terminal domain. As a result, a least-squares alignment of only the viral AAA+ domains reduces the rms deviation to only 0.79 Å over 207 Cα atoms. To estimate the local effect of nucleotide binding between the two structures, Fig. 3b shows the Cα displacement versus residue number. There are two well defined regions with deviations >2 Å. The first region (residues 455–459) locates in the NB-loop. Most of the differences here arise from the “molding” of the loop around the adenosine group likely caused by van der Waals and hydrogen-bond contacts with the sugar and base groups of the nucleotide. Local changes in P-loop residues P335 and A336 can be attributed to the expansion of the P-loop to accommodate the phosphate groups after ADP binding as has been observed in other P-loop nucleotide phosphohydrolases. Another region with small conformational changes involves the sensor 1 residue N421 and includes all the residues of the second β-hairpin (βc–βd). Small differences can also be seen in the position of strand β4, which shows a small shift in the direction of strand β1 (Fig. 3a).
Fig. 3.

Comparison of the Rep40 apo and ADP-bound structures. (a) Superposition of the AAV2 Rep40 apo (red) and ADP-bound (cyan) structures. Small differences are observed in response to ADP binding. (b) Plot of the average difference distance (Å) versus residue number for the two superimposed molecules. Four regions of differences are seen: the P-loop between β1 and α8, the quasihelical loop connecting β2 and β3, the β-hairpin 2 (βc–βd) loop, and the NB-loop. Differences in the β-hairpin 1 (βa–βb) loop may be caused by the lack of crystal contacts in the ADP-bound form of the protein.
We can now see electron density accounting for the side-chain N421 that could not be seen previously in the apo structures. The side chain is stabilized by a hydrogen bond to a water molecule that is also contacted by K340. We believe that the small shift toward strand β1 will be accentuated after ATP binding and will put N421 within hydrogen-bond distance to the γ-phosphate. This residue is thought to detect the difference between the ATP- and ADP-bound state of the protein and transmit this difference through conformational changes to the nearby DNA-binding site as originally described for RecA (45). Indeed, the DNA-dependant ATPase activity is the landmark of all helicases, and this effect is mediated through the sensor 1 residue (46, 47), which in Rep40 is allosterically connected to the DNA-binding site located in β-hairpin 1. Surprisingly, the Walker B residues (E378 and E379) are still too far from the catalytic site (4 Å) as in the apo structure. The fact that Rep40 possesses ATPase activity (22, 23) suggests that oligomerization is sufficient to promote the required shifts in the Walker B residues to form a competent active site. DNA binding may help induce oligomerization, which could be responsible in part for the DNA-stimulated effect on Rep40 ATPase activity.
Discussion
The interaction of ADP with Rep40 illustrates the use of different modes of nucleotide recognition by P-loop nucleotide phosphohydrolases. On the one hand, the interactions of the Walker A residues with the phosphate moiety of the nucleotide is conserved in all of the P-loop containing nucleotide phosphohydrolases, reflecting the structural and functional conservation of this motif across several families of proteins (48, 49). The interaction of the protein with the ribose and base, on the other hand, shows the plasticity in the use of diverse motifs in nucleoside recognition (50, 51). In all the structures solved to date of AAA+ proteins with a bound nucleotide and in the structures of nucleotide complexes of SF1, SF2, and hexameric helicases, there is a unique preference for the trans-gauche conformation of the ribose torsion angle γ around the C4′—C5′ bond. This conformation positions the adenine ring inside a binding pocket composed of mostly aromatic and aliphatic residues that make stacking interactions with both faces of the ring. Furthermore, in these structures the entire nucleotide is buried in a deep cleft, and most groups with the potential to make hydrogen bonds are satisfied. In the case of AAA+ proteins, the conserved C-terminal helical domain II sits on “top” of the adenine ring, forming a tight adenine-binding pocket together with regions of the N-terminal domain I (52). In Rep40, the lack of an equivalent domain II leaves the nucleotide partially exposed to the solvent with the adenine ring pointing away from the protein core where the number of direct protein–nucleotide interactions is sparse. Indeed, the number of direct interactions between Rep40 and ADP is only one third of those made by a typical AAA+ protein such as NSF (53).
A search through the protein data bank for P-loop ATPases with the bound nucleotide in a gg conformation resulted in proteins belonging to only three other families: RecA proteins (45, 54), the ATP-binding cassette (ABC) transporter family of proteins (55–57), and the type II topoisomerase family (58–60). A structural feature shared by all these proteins is the presence of a “steric wall” that is selective of the gg conformation to avoid steric clashes (Fig. 4). However, this conformation produces an energetically unfavorable, solvent-exposed adenine in the monomeric state of these proteins. Stabilization of the nucleotide bound is then achieved through formation of an oligomeric interface, at which the gg conformation increases both the number of potential hydrogenbonding groups available and the molecular surface area of the ADP molecule accessible to a neighboring subunit.
Fig. 4.

Comparison of nucleotide-binding pockets of Rep40, RecA, TAP-1, and NSF-D2. Proteins are shown as a surface representation and colored according to curvature.
SF3 helicases such as Rep68/78 and simian virus 40 TAg share with RecA and ABC transporters their strong tendency to oligomerize after ATP binding (61, 62). In the case of RecA, it readily oligomerizes to form filaments without nucleotide or DNA, but the filaments formed are not competent in ATP hydrolysis (63). A recent model that is based on electron-microscopy reconstruction of RecA-DNA-ATP filaments suggests the formation of a competent ATP-binding site between adjacent subunits (64). In the case of the ABC transporters, ATP binding induces the formation of a tight nucleotide sandwich dimer, the oligomerization interface of which is made up by the strong interaction of the ABC LSGGQ signature motif from one subunit and the ATP bound to the second subunit (65, 66).
An incomplete nucleotide-binding site in the Rep40 monomer explains several biochemical results such as the fact that Rep40 binds nucleotides poorly, as indirectly shown by the extremely high Km value of ≈1 mM for ATP hydrolysis (22); its ability to use other nucleotides such as CTP and GTP, albeit at lower efficiency than ATP (18, 22); and by the fact that we could only obtain cocrystals by using high nucleotide concentrations (10 mM) and at the temperature of 4°C. We previously suggested that the presence of an arginine finger implies the requirement of Rep40 oligomerization for ATP hydrolysis (23). A closer inspection of this putative interface shows several extra residues with the potential to make interaction with the nucleotide (Fig. 5). Of particular interest are K327 and K391, which are conserved in most SF3 family members. The latter has been mutated in the context of Rep68 and was shown to be defective in ATP hydrolysis and helicase activity, thus supporting the hypothesis of its active role in ATP binding and/or catalysis (10). Most of the residues that are predicted to form this oligomeric interface are part of the conserved motif B′. This motif, present only in SF3 helicases, is poised to play multiple roles during the helicase reaction. Some residues will be part of the oligomeric interface interacting directly with the nucleotide, others will be involved in the coupling of DNA binding to ATP hydrolysis, and a third set of residues such as K404 and K406 are directly involved in mediating DNA interactions (M. Yoon-Robarts, A.K.A., C.R.E., and R.M.L., unpublished work).
Fig. 5.

Interface between adjacent monomers of a Rep40 molecule modeled on the structure of the simian virus 40 TAg hexamer. Residues of one subunit (turquoise) point toward the ADP moiety of the adjacent subunit (purple). K327, K391, and R444 are highly conserved residues in the SF3 helicases. E388, K391, S397, and K398 (for which electron density is missing in the ADP structure) all precede the putative DNA-binding loop (69).
The inability to isolate a stable ATP-bound Rep40 oligomer either by gel filtration or during our crystallization attempts suggests that the ATP-induced oligomerization of Rep40 may be a transient event that may require other factors for its stabilization. This behavior parallels that of ABC transporter proteins, with which initial attempts to biochemically characterize and obtain the structure of an ABC-ATPase domain bound to ATP in the dimeric state were unsuccessful, and only versions of the protein that included the transmembrane domain or mutants that were shown to stabilize the dimer resulted in the structure of the dimeric species bound to ATP (65, 66). The larger Rep proteins (Rep78/Rep68) contain an additional domain (the origin-interaction domain) that has been shown to promote oligomerization (67). The absence of this domain in the smaller Rep proteins explains the monomeric character of these proteins in solution. However, the active role that Rep40 plays in DNA packaging reinforces the notion of a factor or factors that would promote oligomerization (e.g., hexamerization) of Rep40/Rep52 during DNA translocation through the capsid. Whether this factor is the capsid itself or some other cellular factor remains to be answered.
We believed that the conclusions drawn from the Rep40–ADP structure can be extended to the large Rep68/78 proteins, in which there is a precedent for nucleotide-induced oligomerization (61). At the same time, the nucleotide-induced oligomerization of simian virus 40 TAg suggests that this may be a general feature for the SF3 family members.
Acknowledgments
We thank the staff at beamlines X25 and X12C (National Synchrotron Light Source, Brookhaven National Laboratories, Upton, NY) for facilitating x-ray data collection. R.M.L. is supported by National Institutes of Health Grant R01 GM62234, and C.R.E. is supported by National Institutes of Health Grant R01 AI41706.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: AAV, adeno-associated virus; SF3, superfamily 3; NB, nucleoside-binding; gg, gauche-gauche; ABC, ATP-binding cassette.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1U0J).
References
- 1.Samulski, R. J. (2003) Ernst Schering Res. Found. Workshop 43, 25–40. [DOI] [PubMed] [Google Scholar]
- 2.Kotin, R. M., Siniscalco, M., Samulski, R. J., Zhu, X. D., Hunter, L., Laughlin, C. A., McLaughlin, S., Muzyczka, N., Rocchi, M. & Berns, K. I. (1990) Proc. Natl. Acad. Sci. USA 87, 2211–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kotin, R. M., Linden, R. M. & Berns, K. I. (1992) EMBO J. 11, 5071–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samulski, R. J., Zhu, X., Xiao, X., Brook, J. D., Housman, D. E., Epstein, N. & Hunter, L. A. (1991) EMBO J. 10, 3941–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutheil, N., Shi, F., Dupressoir, T. & Linden, R. M. (2000) Proc. Natl. Acad. Sci. USA 97, 4862–4866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Linden, R. M., Winocour, E. & Berns, K. I. (1996) Proc. Natl. Acad. Sci. USA 93, 7966–7972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linden, R. M. & Berns, K. I. (2000) Contrib. Microbiol. 4, 68–84. [DOI] [PubMed] [Google Scholar]
- 8.Hickman, A. B., Ronning, D. R., Kotin, R. M. & Dyda, F. (2002) Mol. Cell 10, 327–337. [DOI] [PubMed] [Google Scholar]
- 9.Hickman, A. B., Ronning, D. R., Perez, Z. N., Kotin, R. M. & Dyda, F. (2004) Mol. Cell 13, 403–414. [DOI] [PubMed] [Google Scholar]
- 10.Davis, M. D., Wu, J. & Owens, R. A. (2000) J. Virol. 74, 2936–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cathomen, T., Collete, D. & Weitzman, M. D. (2000) J. Virol. 74, 2372–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon, M., Smith, D. H., Ward, P., Medrano, F. J., Aggarwal, A. K. & Linden, R. M. (2001) J. Virol. 75, 3230–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoon-Robarts, M. & Linden, R. M. (2003) J. Biol. Chem. 278, 4912–4918. [DOI] [PubMed] [Google Scholar]
- 14.Im, D. S. & Muzyczka, N. (1990) Cell 61, 447–457. [DOI] [PubMed] [Google Scholar]
- 15.Snyder, R. O., Im, D. S., Ni, T., Xiao, X., Samulski, R. J. & Muzyczka, N. (1993) J. Virol. 67, 6096–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou, X., Zolotukhin, I., Im, D. S. & Muzyczka, N. (1999) J. Virol. 73, 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarty, D. M., Ni, T. H. & Muzyczka, N. (1992) J. Virol. 66, 4050–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Im, D. S. & Muzyczka, N. (1992) J. Virol. 66, 1119–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walker, S. L., Wonderling, R. S. & Owens, R. A. (1997) J. Virol. 71, 6996–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gavin, D. K., Young, S. M., Jr., Xiao, W., Temple, B., Abernathy, C. R., Pereira, D. J., Muzyczka, N. & Samulski, R. J. (1999) J. Virol. 73, 9433–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King, J. A., Dubielzig, R., Grimm, D. & Kleinschmidt, J. A. (2001) EMBO J. 20, 3282–3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collaco, R. F., Kalman-Maltese, V., Smith, A. D., Dignam, J. D. & Trempe, J. P. (2003) J. Biol. Chem. 278, 34011–34017. [DOI] [PubMed] [Google Scholar]
- 23.James, J. A., Escalante, C. R., Yoon-Robarts, M., Edwards, T. A., Linden, R. M. & Aggarwal, A. K. (2003) Structure (London) 11, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 24.Yang, Q., Kadam, A. & Trempe, J. P. (1992) J. Virol. 66, 6058–6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Pasquale, G. & Stacey, S. N. (1998) J. Virol. 72, 7916–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiorini, J. A., Zimmermann, B., Yang, L., Smith, R. H., Ahearn, A., Herberg, F. & Kotin, R. M. (1998) Mol. Cell. Biol. 18, 5921–5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmidt, M., Afione, S. & Kotin, R. M. (2000) J. Virol. 74, 9441–9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hermonat, P. L., Santin, A. D. & Batchu, R. B. (1996) Cancer Res. 56, 5299–5304. [PubMed] [Google Scholar]
- 29.Smith, R. H. & Kotin, R. M. (1998) J. Virol. 72, 4874–4881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hickson, I. D. (2003) Nat. Rev. Cancer 3, 169–178. [DOI] [PubMed] [Google Scholar]
- 31.Nakayama, H. (2002) Oncogene 21, 9008–9021. [DOI] [PubMed] [Google Scholar]
- 32.Theis, K., Skorvaga, M., Machius, M., Nakagawa, N., Van Houten, B. & Kisker, C. (2000) Mutat. Res. 460, 277–300. [DOI] [PubMed] [Google Scholar]
- 33.Luking, A., Stahl, U. & Schmidt, U. (1998) Crit. Rev. Biochem. Mol. Biol. 33, 259–296. [DOI] [PubMed] [Google Scholar]
- 34.de la Cruz, J., Kressler, D. & Linder, P. (1999) Trends Biochem. Sci. 24, 192–198. [DOI] [PubMed] [Google Scholar]
- 35.Matson, S. W. (1991) Prog. Nucleic Acid Res. Mol. Biol. 40, 289–326. [DOI] [PubMed] [Google Scholar]
- 36.Gorbalenya, A. E. & Kooning, E. V. (1993) Curr. Opin. Struct. Biol. 3, 419–429. [Google Scholar]
- 37.Korolev, S., Hsieh, J., Gauss, G. H., Lohman, T. M. & Waksman, G. (1997) Cell 90, 635–647. [DOI] [PubMed] [Google Scholar]
- 38.Singleton, M. R., Sawaya, M. R., Ellenberger, T. & Wigley, D. B. (2000) Cell 101, 589–600. [DOI] [PubMed] [Google Scholar]
- 39.Soultanas, P., Dillingham, M. S., Velankar, S. S. & Wigley, D. B. (1999) J. Mol. Biol. 290, 137–148. [DOI] [PubMed] [Google Scholar]
- 40.Li, D., Zhao, R., Lilyestrom, W., Gai, D., Zhang, R., DeCaprio, J. A., Fanning, E., Jochimiak, A., Szakonyi, G. & Chen, X. S. (2003) Nature 423, 512–518. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 42.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 43.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard. (1991) Acta Crystallogr. A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- 44.DeLano, W. L. (2002) pymol (Delano Scientific, San Carlos, CA), Version 0.95.
- 45.Story, R. M. & Steitz, T. A. (1992) Nature 355, 374–376. [DOI] [PubMed] [Google Scholar]
- 46.Dillingham, M. S., Soultanas, P. & Wigley, D. B. (1999) Nucleic Acids Res. 27, 3310–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelley, J. A. & Knight, K. L. (1997) J. Biol. Chem. 272, 25778–25782. [DOI] [PubMed] [Google Scholar]
- 48.Leipe, D. D., Wolf, Y. I., Kooning, E. V. & Aravind, L. (2002) J. Mol. Biol. 317, 41–72. [DOI] [PubMed] [Google Scholar]
- 49.Smirnova, I. N., Kasho, V. N. & Faller, L. D. (1998) FEBS Lett. 431, 309–314. [DOI] [PubMed] [Google Scholar]
- 50.Babor, M., Sobolev, V. & Edelman, M. (2002) J. Mol. Biol. 323, 523–532. [DOI] [PubMed] [Google Scholar]
- 51.Moodie, S. L., Mitchell, J. B. & Thornton, J. M. (1996) J. Mol. Biol. 263, 486–500. [DOI] [PubMed] [Google Scholar]
- 52.Ogura, T. & Wilkinson, A. J. (2001) Genes Cells 6, 575–597. [DOI] [PubMed] [Google Scholar]
- 53.Lenzen, C. U., Steinmann, D., Whiteheart, S. W. & Weis, W. I. (1998) Cell 94, 525–536. [DOI] [PubMed] [Google Scholar]
- 54.Datta, S., Ganesh, N., Chandra, N. R., Muniyappa, K. & Vijayan, M. (2003) Proteins 50, 474–485. [DOI] [PubMed] [Google Scholar]
- 55.Gaudet, R. & Wiley, D. C. (2001) EMBO J. 20, 4964–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karpowich, N., Martsinkevich, O., Millen, L., Yuan, Y. R., Dai, P. L., MacVey, K., Thomas, P. J. & Hunt, J. F. (2001) Structure (London) 9, 571–586. [DOI] [PubMed] [Google Scholar]
- 57.Yuan, Y. R., Blecker, S., Martsinkevich, O., Millen, L., Thomas, P. J. & Hunt, J. F. (2001) J. Biol. Chem. 276, 32313–32321. [DOI] [PubMed] [Google Scholar]
- 58.Berger, J. M., Gamblin, S. J., Harrison, S. C. & Wang, J. C. (1996) Nature 379, 225–232. [DOI] [PubMed] [Google Scholar]
- 59.Wigley, D. B., Davies, G. J., Dodson, E. J., Maxwell, A. & Dodson, G. (1991) Nature 351, 624–629. [DOI] [PubMed] [Google Scholar]
- 60.Brino, L., Urzhumtsev, A., Mousli, M., Broner, C., Mitschler, A., Oudet, P. & Moras, D. (2000) J. Biol. Chem. 275, 9468–9475. [DOI] [PubMed] [Google Scholar]
- 61.Smith, R. H., Spano, A. J. & Kotin, R. M. (1997) J. Virol. 71, 4461–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dean, F. B., Borowiec, J. A., Eki, T. & Hurwitz, J. (1992) J. Biol. Chem. 267, 14129–14137. [PubMed] [Google Scholar]
- 63.Takahashi, M. & Norden, B. (1994) Adv. Biophys. 30, 1–35. [DOI] [PubMed] [Google Scholar]
- 64.VanLoock, M. S., Yu, X., Yang, S., Lai, A. L., Low, C., Campbell, M. J. & Egelman, E. H. (2003) Structure (London) 11, 187–196. [DOI] [PubMed] [Google Scholar]
- 65.Smith, P. C., Karpowich, N., Millen, L., Moody, J. E., Rosen, J., Thomas, P. J. & Hunt, J. F. (2002) Mol. Cell 10, 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Locher, K. P., Lee, A. T. & Rees, D. C. (2002) Science 296, 1091–1098. [DOI] [PubMed] [Google Scholar]
- 67.Davis, M. D., Wonderling, R. S., Walker, S. L. & Owens, R. A. (1999) J. Virol. 73, 2084–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins 11, 281–296. [DOI] [PubMed] [Google Scholar]
- 69.Diederichs, K. & Karplus, A. (1997) Nat. Struct. Biol. 4, 269–275. [DOI] [PubMed] [Google Scholar]