

Abstract
Acid-induced unfolding of apomyoglobin (apoMb) proceeds in a multistate process involving at least one equilibrium intermediate (I) at pH 4.2. The structure of the I form has been investigated thoroughly, with significant effort devoted to identifying potentially stabilizing native contacts. Here, we test whether rigid side-chain packing interactions like those in holomyoglobin persist at a buried position, Met-131, within the low-pH apoMb intermediate. We have measured the urea-induced unfolding transitions of overpacking, underpacking, and polar substitutions of Met-131 to determine the effect on the stability of the native and intermediate states of apoMb. Whereas underpacking substitutions should destabilize the I form irrespective of the degree of native side-chain-packing interactions, we anticipate that overpacking replacements might show opposite effects in a tightly packed environment, compared with a region lacking native side-chain packing interactions. We observe that, whereas underpacking and polar substitutions destabilize the I form, overpacking substitutions are stabilizing, implying that I is structurally plastic. We also report a strong correlation between the I state unfolding free energies and side-chain transfer free energies from water to octanol. Our results suggest that, whereas side-chain hydrophobicity is important for the stability of the I form, specific side-chain packing interactions are not.
The folding of apomyoglobin (apoMb, myoglobin with the heme group removed) has been studied extensively. Like holomyoglobin (holoMb), native apoMb is compact and contains a well-ordered hydrophobic core (1). However, apoMb has a more dynamic structure than holoMb, showing a slight decrease in helix content and protection from hydrogen exchange (2–4). Multidimensional NMR studies suggest that the F helix, which contacts the heme in holoMb, is structurally disordered and that the N terminus of the G helix and the C terminus of the H helix are frayed (2, 5, 6).
Acid-induced unfolding of apoMb is multistate, involving at least one equilibrium intermediate (I) at pH 4.2 (1, 7). The I form is structurally compact and contains ≈35% helical content (1, 2). Hydrogen exchange results suggest that the A, G, and H helix regions are protected and are thus likely to be structured (2, 8, 9).
Attempts to assess the tertiary interactions and their contributions to the stability of the I form of apoMb have lead to somewhat contradictory interpretations. Hughson et al. (10) used site-directed mutagenesis to target residues in the A–H and G–H interfaces and monitored the effects of substitution by using acid-induced denaturation. Their results suggested that the I form is more tolerant of mutation than the native state (N) and is stabilized by weak, nonspecific hydrophobic interactions, rather than specific, native-like packing interactions (10). Interpreted in this way, the I form of apoMb resembles a molten globule intermediate (11–13). Later, Kay and Baldwin (14) used urea denaturation to assess stability changes in the I form resulting from point substitution and interpreted a handful of substitution-induced destabilizations in I to be evidence for specific native side-chain packing. A subsequent study of underpacking variants at buried positions revealed a similar level of destabilization of the I form (15).
One of the hallmarks of specific side-chain packing interactions is that both overpacking and underpacking substitutions should be destabilizing (16). Thus, overpacking substitutions provide a means to discriminate between stabilization by specific native-like packing interactions and nonspecific hydrophobic stabilization, because substitution with larger, nonpolar side chains should destabilize tightly packed native-like regions of structure but should stabilize hydrophobic regions that lack tight native packing. In contrast, underpacking substitutions destabilize both specific packing and nonspecific hydrophobic stabilization; thus, underpacking substitutions alone cannot be used to resolve these two different sources of stability.
The studies supporting the presence of native packing interactions in the I form have only focused on underpacking substitutions (14, 15). The observed destabilization of the I form in underpacking variants could either result from disruption of native-like side-chain packing interactions, as has been proposed (14), or from a loss of the nonpolar surface area that contributes to stability through loose nonspecific hydrophobic interactions (10).
Here, we have measured the urea-induced unfolding transitions of overpacking, underpacking, and polar substitutions at a buried residue, Met-131, to determine the effect of different types of substitutions on the stability of the N and I forms. Based on the structure of holoMb, Met-131 seems a good choice for substitution, because it connects the A, G, and H helices through direct van der Waals contacts (14). In the native form of apoMb, the side chain of Met-131 has nuclear Overhauser effects to the A and G helices consistent with the holoMb structure (3). Further, nuclear Overhauser effect-based structure determination suggests a holoMb-like fold in this region, in which Met-131 is docked, along with more N-terminal residues of the H helix, against the A and G helices (4, 6). Position 131 was one of the three sites at which an underpacking substitution was found to decrease the stability of I (14). The structural origin of previously observed destabilizations resulting for underpacking substitutions can be resolved by examining the effects of overpacking substitutions at this interhelical contact site (17). If specific side-chain packing interactions are retained in the I form at position 131, substitution with large aromatic side chains, such as Phe and Trp, will destabilize the I form. However, if specific side-chain packing is absent, and instead burial of hydrophobic surface area is the primary determinant of the stability of I, these substitutions should increase the stability of the I form.
Materials and Methods
Mutagenesis. Substitutions were made in the synthetic myoglobin gene encoded by pMb413b (10, 18) by using either the QuikChange Mutagenesis Kit (Stratagene) or cassette mutagenesis (10) and were verified by dideoxy sequencing.
Protein Expression and Purification. Proteins were expressed in Escherichia coli strain TB-1, in 1-liter LB cultures. For the M131G and M131Q variants, expression was reproducibly low. Better levels of expression were obtained when these variants were grown in smaller volumes; thus, the M131G and M131Q variants were grown in 10-ml cultures. Typically, 150 of these 10-ml cultures were combined for protein purification. Purification was as described (19), except that ammonium sulfate precipitation was omitted. Instead, cleared lysates were dialyzed overnight against 10 mM imidazole hydrochloride (pH 6.5) before passage over sequential DE52 and CM52 ion-exchange columns. Purified proteins were exchanged into distilled water and were stored at –80°C. Mb was converted to apoMb for stability measurements by using the 2-butanone method of Teale (20). apoMb concentration was determined by using the method of Edelhoch (21). All apoMbs were stored at 0–4°C and used within 36 h of preparation.
Acid-Induced Denaturation. Acid-induced unfolding of apoMb was monitored by CD spectroscopy at 222 nm by using an Aviv 62-DS Spectropolarimeter (Aviv Associates, Lakewood, NJ). Buffers at different pH values were prepared by combining 10 mM sodium acetate with 10 mM acetic acid; to obtain pH values of <3, a small amount of HCl was added to 10 mM acetic acid. apoMb was then added to a concentration of 2 μM. Samples were equilibrated for 5 min before data acquisition at 4°C. After titration, the pH of each sample was measured at room temperature. To obtain numerical estimates of pH midpoints for unfolding, acid-induced unfolding transitions were fitted by a simple proton-binding model by using nonlin for macintosh (22). Although this model, which treats the N–I and I–U transitions with two separate phenomenological Hill expressions, provides reasonable pH midpoints for the two transitions, it is not meant to provide a mechanistic description of acid-induced unfolding.
Urea-Induced Denaturation. Urea (ICN) was treated for 1 h with AG-301-X8 (D) resin (Bio-Rad), filtered, and buffered at either pH 7.8 or 4.2. The urea concentration was determined by refractometry (23).
I–U Transitions. Urea-induced unfolding of the I form was monitored by CD and tryptophan fluorescence. Both signals were monitored with an Aviv 62-DS spectropolarimeter. CD at 222 nm was measured as described above. Trp fluorescence was monitored by using a 320-nm cutoff filter in front of a perpendicular photomultiplier, after excitation at 280 nm. A Hamilton Microlab 500 titrator was used to titrate denatured protein (2 μM protein/7–8 M urea/4 mM sodium citrate, pH 4.2) into a 1-cm path-length cuvette containing a 2 μM concentration of the I form of apoMb (4 mM sodium citrate, pH 4.2). After equilibration for 300 s at 4°C, the CD and fluorescence signals were recorded.
N–U Transitions. CD-monitored unfolding transitions of the N form (10 mM Hepes, pH 7.8) were performed as described for the I form. Because the cutoff-filtered fluorescence change accompanying the N–U transition is modest, we measured fluorescence-detected unfolding transitions with an Aviv ATF-105 spectrofluorimeter so that emission at a specific wavelength could be monitored. Fluorescence emission at 320 nm was recorded after excitation at 288 nm. For both the CD- and fluorescence-monitored urea denaturations of the N form, the titration system was flushed with denatured protein solution in a mock titration to avoid what appeared to be a slight adsorption of the protein to the titration lines.
Data Analysis. The urea-induced unfolding transitions measured for the I form were fitted by a two-state model as described by Santoro and Bolen (24) using the nonlinear least-squares analysis tool of kaleidegraph 3.6 (Synergy Software, Reading, PA). Pre- and posttransition baselines were treated as linear functions of denaturant concentration. Pretransition baselines at pH 4.2 are short and significantly sloped, resulting in part from the fact that at low pH apoMb populates two intermediate forms, Ia and Ib. During urea-induced unfolding, Ib is converted to Ia between 0 and 1 M urea; Ia then unfolds cooperatively (7). Thus, the unfolding transition at pH 4.2 largely reports on the major Ia-to-U unfolding transition. To reduce the error in determining the free energy of unfolding (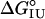 ) caused by the poor native baselines and the presence of Ib in the baseline region, the m-value was fixed at the average value measured in CD- and fluorescence-monitored denaturations of WT apoMb I (m = 1.52 ± 0.04), as was done previously (14). This procedure is justified in part by the observation that when m-values are adjusted during the fit, the optimized m-values for the I–U transitions of all variants are close to that of WT apoMb (not shown). By using this procedure, unfolding free energy estimates are determined largely by the midpoint of the unfolding transition (Cm). In addition to being fitted separately by the linear extrapolation model, the CD- and fluorescence-monitored I–U transitions were analyzed simultaneously by global fitting. CD and fluorescence data were scaled from zero to one so that the two types of curves have equal influence on the global fitting procedure.
) caused by the poor native baselines and the presence of Ib in the baseline region, the m-value was fixed at the average value measured in CD- and fluorescence-monitored denaturations of WT apoMb I (m = 1.52 ± 0.04), as was done previously (14). This procedure is justified in part by the observation that when m-values are adjusted during the fit, the optimized m-values for the I–U transitions of all variants are close to that of WT apoMb (not shown). By using this procedure, unfolding free energy estimates are determined largely by the midpoint of the unfolding transition (Cm). In addition to being fitted separately by the linear extrapolation model, the CD- and fluorescence-monitored I–U transitions were analyzed simultaneously by global fitting. CD and fluorescence data were scaled from zero to one so that the two types of curves have equal influence on the global fitting procedure.
The urea-induced unfolding transitions measured for the apoMb N (pH 7.8) form were also fitted by a two-state model individually and globally as described above. However, the apparent fitted m-values varied significantly, both between unfolding transitions of different variants monitored with the same probe and also between fluorescence- and CD-monitored unfolding transitions of the same variants, indicating the presence of a substantial and variable population of intermediate conformations. Thus, m-values were not restricted to the WT values but were adjusted during fitting. Of the numerical parameters resulting from fitting of the pH 7.8 unfolding transitions, the Cm values are most likely to provide a rough estimate of stability. The m-values are likely to provide qualitative information on the buildup of intermediates in these transitions, but should not be interpreted thermodynamically.
Results
Effects of Position 131 Substitutions on the Stability of I. To determine the effect of substitutions at position 131 on the stability of the I form, we monitored the urea-induced unfolding transitions of the position 131 variants by CD spectroscopy (Fig. 1). Nearly all the substitutions at position 131 produce a measurable change in the stability of the I form. This change can be seen in the CD-monitored urea-induced unfolding transitions, where the midpoints are shifted, relative to that for WT apoMb I (Fig. 1 A). This shift is more clearly represented by converting the unfolding transitions (Fig. 1 A) into the fraction of apoMb in the I form (Fig. 1B). This conversion facilitates comparison of the midpoints of the curves by normalizing the endpoints of the denaturation curves† and by compensating for sloping baselines. Some substitutions at position 131 appear to stabilize the intermediate form of apoMb, based on an increase in the midpoint of the urea-induced unfolding transition relative to WT apoMb I; this trend is apparent both in the primary unfolding data (Fig. 1 A) and in the population-based curves (Fig. 1B). We have also measured the urea-induced unfolding transitions of the position 131 variants in the I form by monitoring the fluorescence signal arising from two tryptophan residues in the A helix (Table 1 and Fig. 4, which is published as supporting information on the PNAS web site). The fluorescence-detected transitions show measurable differences in the midpoints similar to those observed by CD.
Fig. 1.

Representative urea-induced unfolding transitions of the I form (pH 4.2) of apoMb Met-131 variants. (A) Raw data. (B) Normalized data with native and denatured baselines fixed at 1 and 0, respectively. Solid lines indicate fits by a two-state model. •, Wild type; ▪, M131W; ▵, M131V; ♦, M131F; ○, M131A; □, M131G; ⋄, M131Q. Conditions: 2 μM protein/4 mM sodium citrate (pH 4.2), 4°C.
Table 1. Urea-induced unfolding parameters for the I form of the apoMb Met-131 variants.
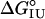 , kcal·mol-1 , kcal·mol-1
|
Cm, M
|
χ2R
|
|||||
|---|---|---|---|---|---|---|---|
| CD | Fluorescence | Global | CD | Fluorescence | Global | Global | |
| WT | 2.77 ± 0.05 | 2.44 ± 0.03 | 2.46 ± 0.03 | 1.82 ± 0.03 | 1.61 ± 0.02 | 1.62 ± 0.02 | 1.61 × 10-4 |
| M131W | 2.98 ± 0.13 | 2.80 ± 0.17 | 2.92 ± 0.08 | 1.96 ± 0.09 | 1.84 ± 0.11 | 1.92 ± 0.09 | 2.55 × 10-4 |
| M131F | 3.03 ± 0.05 | 2.69 ± 0.04 | 2.72 ± 0.05 | 1.99 ± 0.04 | 1.77 ± 0.03 | 1.79 ± 0.03 | 1.87 × 10-4 |
| M131V | 2.45 ± 0.06 | 2.20 ± 0.03 | 2.24 ± 0.03 | 1.61 ± 0.04 | 1.45 ± 0.02 | 1.47 ± 0.02 | 1.17 × 10-4 |
| M131A | 1.91 ± 0.02 | 1.72 ± 0.06 | 1.74 ± 0.05 | 1.25 ± 0.01 | 1.13 ± 0.04 | 1.14 ± 0.03 | 3.74 × 10-4 |
| M131G | 1.79 ± 0.19 | 1.40 ± 0.14 | 1.44 ± 0.12 | 1.18 ± 0.13 | 0.92 ± 0.09 | 0.95 ± 0.08 | 3.16 × 10-4 |
| M131Q | 1.90 ± 0.04 | 1.65 ± 0.03 | 1.63 ± 0.04 | 1.25 ± 0.03 | 1.09 ± 0.02 | 1.07 ± 0.03 | 2.74 × 10-4 |
Parameters were estimated by fitting to the linear model, with m-values fixed at the average WT value. Reported values are the mean of n ≥ 3 independent experiments, with the exception of the fluorescence data reported for M131V where n = 2. Uncertainties represent the standard error on the mean. χ2R is sum of the residuals squared divided by the degrees of freedom. Conditions: 2 μM protein/4 mM sodium citrate (pH 4.2), 4°C.
Comparison of CD- and fluorescence-monitored unfolding data is a standard test for two-state unfolding. Coincidence of unfolding transitions monitored by these two independent structural probes is considered to support the two-state approximation. For WT and variant forms of apoMb I, urea-induced unfolding transitions obtained by CD and fluorescence are coincident, with the exception of M131G. [M131G has short native baselines, apparently resulting from significant destabilization of the apoMb I form upon substitution with glycine (Fig. 4).] Likewise, Cm values obtained by CD and fluorescence for WT apoMb and variants in the I form are the same, within error (as are other fitted thermodynamic parameters; Table 1).
The strong similarity between fluorescence- and CD-monitored urea-induced unfolding transitions, and the close agreement between fitted parameters from the two types of curves is consistent with a simple two-state unfolding model for apoMb I at pH 4.2. Adherence to a two-state model is also supported by the observation that global fitting of a two-state model to CD- and fluorescence-monitored unfolding transitions results in a good fit for all variants, as indicated by uniformly low 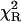 values (Table 1). A two-state mechanism is also supported by the narrow range of m-values for the pH 4.2 urea-induced unfolding transitions of the position 131 variants, when this parameter is allowed to float during fitting. Thus, parameters resulting from fitting a two-state model to urea-induced unfolding transitions at pH 4.2 are likely to accurately reflect the thermodynamic stability of WT and position 131 variant forms of apoMb I.
values (Table 1). A two-state mechanism is also supported by the narrow range of m-values for the pH 4.2 urea-induced unfolding transitions of the position 131 variants, when this parameter is allowed to float during fitting. Thus, parameters resulting from fitting a two-state model to urea-induced unfolding transitions at pH 4.2 are likely to accurately reflect the thermodynamic stability of WT and position 131 variant forms of apoMb I.
Fitted unfolding free energies reveal significant decreases in stability of apoMb I as a result of underpacking substitution. This stability decrease could either result from loss of native-like side-chain packing interactions, or from the loss of buried hydrophobic surface area. Within the nonpolar packing substitutions (Val, Ala, and Gly), loss in stability of the I form correlates with the amount of hydrophobic side-chain volume lost in the replacement. Thus, M131V is the least destabilized underpacking variant ( ),‡ M131A is intermediately destabilized (
),‡ M131A is intermediately destabilized (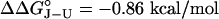 ), and M131G is most destabilized (
), and M131G is most destabilized (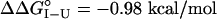 ) (Fig. 1 and Table 1). Substitution at position 131 with the polar residue Gln also destabilizes the I form (
) (Fig. 1 and Table 1). Substitution at position 131 with the polar residue Gln also destabilizes the I form (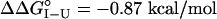 ), consistent either with disruption of specific native side-chain packing or with the burial of a polar group in a nonpolar environment. To resolve whether the destabilization of apoMb I observed for the underpacking substitutions stems from a loss of specific side-chain packing interactions or simply a reduction in nonpolar surface area, we examined the effects of overpacking substitutions of Phe and Trp at position 131 on stability. In contrast to the underpacking substitutions, overpacking substitutions stabilize apoMb I by ≈0.3 kcal/mol (Fig. 1 and Table 1). We expect these bulky aromatic side chains to be too large to substitute for a Met in a well-packed environment without significant structural rearrangement, which is likely to be destabilizing. The observation that these substitutions actually increase stability suggests that the environment surrounding the position 131 side chain in apoMb I is plastic (i.e., not tightly packed) and can rearrange without large energetic penalties.
), consistent either with disruption of specific native side-chain packing or with the burial of a polar group in a nonpolar environment. To resolve whether the destabilization of apoMb I observed for the underpacking substitutions stems from a loss of specific side-chain packing interactions or simply a reduction in nonpolar surface area, we examined the effects of overpacking substitutions of Phe and Trp at position 131 on stability. In contrast to the underpacking substitutions, overpacking substitutions stabilize apoMb I by ≈0.3 kcal/mol (Fig. 1 and Table 1). We expect these bulky aromatic side chains to be too large to substitute for a Met in a well-packed environment without significant structural rearrangement, which is likely to be destabilizing. The observation that these substitutions actually increase stability suggests that the environment surrounding the position 131 side chain in apoMb I is plastic (i.e., not tightly packed) and can rearrange without large energetic penalties.
Although the substitution-induced changes in unfolding free energy of apoMb I, as determined by urea denaturation at pH 4.2, are modest (in some cases, smaller than thermal energy, RT; R is the universal gas constant and T is absolute temperature), these changes exceed the error margins on unfolding free energies. Furthermore, these changes are mirrored in the low-pH midpoints of acid-induced unfolding transitions for the position 131 variants (Fig. 5, which is published as supporting information on the PNAS web site). In particular, nonpolar overpacking substitutions decrease the low pH midpoint, suggesting an increase in stability of the I form, whereas underpacking substitutions increase the low pH midpoint, consistent with decreased stability (Table 3, which is published as supporting information on the PNAS web site). Although pH midpoints are a less reliable way to assess the stability of the I form of apoMb than urea-induced unfolding (14), the agreement of these two independent quantities supports the interpretation that specific side-chain packing interactions are absent from apoMb I at position 131.
Effects of Position 131 Substitution on the Unfolding of N. To determine the consequences of substitution of position 131 in a conformational state that has more structure overall, we examined the effect of substitutions on native apoMb (N). We have measured urea-induced unfolding transitions of the overpacking, underpacking, and polar variants using CD and fluorescence spectroscopy at pH 7.8, where the N form is expected to be the predominant conformation in the absence of urea.
As with the unfolding transitions of the I form at pH 4.2, all the substitutions at position 131 significantly altered the pH 7.8 unfolding transition (Fig. 2). However, in contrast to the low-pH urea unfolding transitions, where both CD and fluorescence unfolding could be described by using a simple two-state (I-to-U) model, several features of the pH 7.8 unfolding transitions are not compatible with a simple two-state (N-to-U) model. First, for several variants (M131A, M131G, and M131Q), multiple unfolding transitions are observed in fluorescence-detected urea-induced unfolding transitions (Fig. 6, which is published as supporting information on the PNAS web site). Multiple unfolding transitions have been observed previously in fluorescence-monitored high-pH urea-induced unfolding curves of apoMb variants, including M131A (14). In addition, the folded baseline for the M131W pH 7.8 urea-induced unfolding curve has a large positive slope and may also reflect partial formation of partly folded intermediates in the transition region. Furthermore, a large variation occurs in slopes of the urea-induced unfolding transitions of the position 131 variants (Fig. 2). The pH 7.8 transitions of most variants are shallower than that for WT apoMb by as much as 3-fold (see fitted m-values, Table 2). Whereas modest changes in the steepness of denaturant-induced unfolding m-values are sometimes interpreted as changes in exposed surface area in the denatured state (26), these large decreases seem more likely to reflect the population of partly folded conformations in the transition zone, especially given the relative insensitivity of m-values associated with the I-to-U transitions at pH 4.2 to these same substitutions (Table 1).
Fig. 2.

Representative urea-induced unfolding transitions of the N form (pH 7.8) of apoMb Met-131 variants. (A) Raw data. (B) Normalized data with native and denatured baselines fixed at 1 and 0, respectively. Solid lines indicate fits by a two-state model. •, Wild type; ▪, M131W; ▵, M131V; ♦, M131F; ○, M131A; □, M131G; ⋄, M131Q. Conditions: 2 μM protein/10 mM Hepes (pH 7.8), 4°C.
Table 2. Urea-induced unfolding parameters for the N form of the apoMb Met-131 variants.
| Cm, M
|
m-value, kcal·mol-1·M-1
|
χ2r
|
|||
|---|---|---|---|---|---|
| CD | Fluorescence | CD | Fluorescence | Global | |
| WT | 3.63 ± 0.05 | 3.82 ± 0.05 | 1.77 ± 0.04 | 1.78 ± 0.03 | 9.54×10-4 |
| M131W | 3.52 ± 0.04 | 3.98 ± 0.01 | 1.15 ± 0.07 | 1.98 ± 0.03 | 6.53×10-4 |
| M131F | 3.73 ± 0.05 | 3.78 ± 0.01 | 1.52 ± 0.08 | 1.67 ± 0.02 | 1.22×10-4 |
| M131V | 2.93 ± 0.05 | 3.79 ± 0.08 | 1.15 ± 0.04 | 1.41 ± 0.007 | 1.17×10-3 |
| M131A | 2.02 ± 0.06 | 3.39 ± 0.05 | 1.07 ± 0.04 | 1.33 ± 0.09 | 1.00×10-3 |
| M131G* | 0.8 ± 0.3 | ND | 0.65 ± 0.01 | ND | ND |
| M131Q* | 1.0 ± 0.2 | ND | 0.63 ± 0.04 | ND | ND |
Parameters were estimated by fitting a two-state model, treating m-values as adjustable parameters. Errors and χ2r are described in Table 1. Conditions: 2 μM protein/10 mM Hepes (pH 7.8), 4°C.
For M131G and M131Q, fitting of the fluorescence curves was not attempted, as the unfolding transitions were multiphasic. ND, not determined.
Another feature of the pH 7.8 urea-induced unfolding transitions that is inconsistent with a two-state unfolding mechanism is the substantial difference between CD- and fluorescence-monitored unfolding transitions at pH 7.8. This is clearest for M131W and M131V (Fig. 6), where the CD-monitored transitions are centered at a lower urea concentration than the fluorescence-monitored transition. Given that the low pH apoMb I form has decreased CD intensity but greater fluorescence intensity than the N form, these data are consistent with formation of an intermediate with similar spectroscopic features in the pH 7.8 unfolding transitions of M131W and M131V. In addition, global fitting of a two-state model to the CD- and fluorescence-monitored pH 7.8 transitions produces substantially different fitted parameters, large systematic residuals, and high  values (Table 2), again indicating that urea-induced unfolding of the position 131 variants at pH 7.8 involves more than two distinct states.
values (Table 2), again indicating that urea-induced unfolding of the position 131 variants at pH 7.8 involves more than two distinct states.
Discussion
Although it is clear that the I form of apoMb contains substantial α-helical structure, and the distribution of helical structure has been mapped (2, 5), the type of tertiary interactions present is less clear. In particular, the degree to which specific side-chain packing like that seen in native holoMb is retained in the I form of apoMb remains an open question.
The use of single-residue substitutions to resolve whether one interaction or another is responsible for protein stability requires that substitutions have opposite effects on the two types of interactions. To resolve the contributions of rigid side-chain packing and hydrophobicity to stability, substitutions must either disrupt rigid side-chain packing while increasing hydrophobicity or stabilize native side-chain packing while decreasing hydrophobicity. Further, evaluation of the degree to which different types of interactions stabilize a protein structure can be facilitated by analysis of multiple substitutions at a single position. If a single type of interaction is the dominant factor in determining stability, a strong correlation should be obtained between stability and the chemical feature related to that interaction, whereas other chemical features should be uncorrelated. In contrast, if multiple types of interactions contribute to stability, no single chemical feature should show a strong correlation to stability.
Here, we have attempted to gain insight into the degree to which different interactions (specific side-chain packing, hydrophobicity, and α-helical structure) stabilize the I form of apoMb by comparing stability changes of a diverse collection of substitutions at position 131, a position involved in extensive hydrophobic packing interactions at the contact region between the A, G, and H helices of holoMb. Comparing the relative stabilities of different variants with each other and with the basic chemical features of the substituting side chains suggests that native-like side-chain packing does not stabilize the I form of apoMb. Instead, burial of nonpolar surface area in the vicinity of position 131 seems to be the dominant contributor to I form stability, suggesting that this region of the I form resembles a molten globule conformation (10).
Origins of Stability in the I Form of apoMb. The increase in stability of apoMb I variants containing overpacking substitutions at position 131 suggests the absence of rigid side-chain packing interactions in the vicinity of the substitutions. If such interactions are absent, the increase in stability of apoMb I overpacking variants may be related to the increased nonpolar surface area introduced by the substitutions. To examine the relationship between apoMb I stability and nonpolar surface area of position 131 variants, we plotted 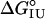 values versus free energies of transfer from water to octanol (
values versus free energies of transfer from water to octanol (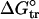 ) for substituting side chains (27). A striking correlation between these two quantities is observed, with r = 0.98 (Fig. 3A), strongly suggesting that side-chain hydrophobicity is a dominant component of stability for the I form of apoMb.
) for substituting side chains (27). A striking correlation between these two quantities is observed, with r = 0.98 (Fig. 3A), strongly suggesting that side-chain hydrophobicity is a dominant component of stability for the I form of apoMb.
Fig. 3.

Correlation between apoMb I-to-U unfolding free energy changes ( , from global fits) for position 131 variants with octanol–water transfer energies (A) (21) and free energies of helix formation in alanine-based peptides (B) (25).
, from global fits) for position 131 variants with octanol–water transfer energies (A) (21) and free energies of helix formation in alanine-based peptides (B) (25).
Another potential source of stability differences among the position 131 variants is differences in helix propensity. Several studies have shown that the stability of the I form can be modulated by changes in helix propensity in the A and G helices (28–30). Consistent with this idea, M131G is significantly less stable than M131A, as would be expected from differences in the helix propensities of these two residues (31). However, 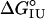 values for the entire collection of position 131 variants do not appear to be correlated (r = 0.14; Fig. 3B). This lack of correlation may indicate that the H helix is retained in the U form of apoMb, consistent with the observation that peptides corresponding to the H helix show significant helix formation in isolation (10, 32). Previous substitution studies that show a correlation between helix propensity and stability in apoMb I have targeted surface residues of the A and G helices, for which peptide models show little or no helical structure (10, 32, 33). Regardless of whether the lack of correlation between helix propensities and
values for the entire collection of position 131 variants do not appear to be correlated (r = 0.14; Fig. 3B). This lack of correlation may indicate that the H helix is retained in the U form of apoMb, consistent with the observation that peptides corresponding to the H helix show significant helix formation in isolation (10, 32). Previous substitution studies that show a correlation between helix propensity and stability in apoMb I have targeted surface residues of the A and G helices, for which peptide models show little or no helical structure (10, 32, 33). Regardless of whether the lack of correlation between helix propensities and 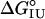 values for position 131 variants results from helix formation in the U form of apoMb, the strong correlation with octanol transfer free energies suggests that burial of nonpolar surface at position 131 makes a major contribution to the relative stability of the I form. Previous studies demonstrating a correlation between helix propensity and apoMb I stability focused on sites that are exposed in native holoMb (28–30); thus, the effects of side-chain hydrophobicity are not likely to have contributed to stability changes.
values for position 131 variants results from helix formation in the U form of apoMb, the strong correlation with octanol transfer free energies suggests that burial of nonpolar surface at position 131 makes a major contribution to the relative stability of the I form. Previous studies demonstrating a correlation between helix propensity and apoMb I stability focused on sites that are exposed in native holoMb (28–30); thus, the effects of side-chain hydrophobicity are not likely to have contributed to stability changes.
Origins of Stability in the N Form of apoMb. Although the primary goal of this study is to investigate the tertiary structure and origins of stability in the I form of apoMb, we have also attempted to examine the effects of substitution at position 131 on the unfolding transitions of the native form of apoMb at pH 7.8, which has more overall structure than the low-pH equilibrium intermediate. In principle, determination of the effects of substitution on the N form of apoMb provides a control in which the effects of substitution on stability in a structured environment can be assessed. However, most of the position 131 variants display multistate unfolding transitions, complicating analysis of stability changes.
Previous studies have resolved high-pH unfolding transitions of apoMb variants by globally fitting a multistate model to the CD- and fluorescence-monitored urea unfolding curves (14). However, application of multistate fitting to the position 131 variants studied here did not yield unique spectroscopic and thermodynamic parameter values. Lacking a mechanistic model with which to quantify the effects of substitution at position 131 on the N form of apoMb, we present a qualitative analysis of the perturbations to the stability of N by comparing Cm values for pH 7.8 urea-induced unfolding curves (Table 2) and also pH midpoints for the N–I reaction determined from acid-induced unfolding (Table 3). Because fluorescence curves are more sensitive to apoMb I as its fluorescence is enhanced, CD-monitored transitions at pH 7.8 are more likely to capture the unfolding of the apoMb N form, whereas the fluorescence-monitored unfolding transitions are more likely be influenced by the I form. This expectation is supported by comparison of Cm values from pH 7.8 urea-induced unfolding transitions with pH midpoints for the N–I transition from acid-induced unfolding (Table 3); Cm values determined by CD show a higher degree of correlation with and sensitivity to pH midpoints (r = 0.97, slope = 1.74) than those determined by fluorescence (r = 0.79, slope = 0.45).
For variants that show a single urea-induced unfolding transition at pH 7.8, rankings of Cm values for CD- and fluorescence-monitored transitions from high to low urea concentration are similar:
 |
This ranking is also similar to that obtained by ranking pH midpoints for the N–I transition from low to high pH. [For M131G, the native form is so severely destabilized that it does not appear to be populated even at pH 7.8 (Fig. 5).] Underpacking substitutions produce the largest shift in pH 7.8 Cm values, whereas overpacking substitutions have little effect on Cm values. For underpacking substitutions that show a single fluorescence-detected transition, decreases in the fitted Cm values for the pH 7.8 unfolding transitions are significantly greater than for the pH 4.2 transitions, especially for the CD-monitored transitions (compare Figs. 1 and 2). For M131Q and M131G, which show multistage fluorescence-monitored unfolding at pH 7.8, the decrease in the Cm corresponding to the first transition is also greater than the corresponding Cm change at pH 4.2, as are CD-detected Cm changes.
In contrast with underpacking substitutions, Cm values at pH 7.8 are relatively insensitive to overpacking substitutions. Moreover, pH midpoints for the N–I transition are modestly decreased relative to WT, suggesting that, like the I form, the N form of apoMb might actually be stabilized by overpacking. Given the expectation that, based on the structure of holoMb, the region surrounding Met-131 is tightly packed, stabilization of the apoMb N form by hydrophobic overpacking substitutions is somewhat of a surprise. One explanation for this apparent insensitivity is that these substitutions destabilize the N form by disrupting native packing, but offset this destabilization by increasing nonpolar surface area burial. However, the apparent tolerance of the N form of apoMb to overpacking substitutions suggests that, in the region of Met-131, even the native form may exhibit a substantial amount of structural plasticity at the level of side-chain packing (34). Although NMR studies indicate that Met-131 is in close proximity to sites on the A and G helices in the N form of apoMb, little evidence exists of structure C-terminal to position 131 (4, 5, 35). If position 131 is at the junction between structural and disordered regions, defects in side-chain packing might be more easily accommodated. Clearly, native holoMb would be the ideal form for examining the effects of packing substitutions on stability, because it is this form in which rigid packing is directly evaluated crystallographically. However, we have been unable to obtain reversible unfolding curves for holoMb under any condition, even using the cyanomet form, which has been suggested to improve reversibility (15).
Comparison with Other Studies of Molten Globule Intermediates. The strong correlation between the stabilities of the position 131 variants of apoMb I with 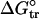 , a measure of side-chain hydrophobicity, suggests that hydrophobic interactions play a major role in determining the stability of this partly folded intermediate. The enhanced stability of apoMb I resulting from overpacking substitution is consistent with this interpretation but suggests that the specific side-chain packing, as seen in holoMb, is absent at this position and its surroundings. This interpretation is at odds with an earlier study (14), which interpreted mutagenesis results as indicative that specific residues make energetically important packing interactions. However, with the exception of A130L, which slightly stabilizes the I form, none of the substitutions in the earlier study resolve packing from hydrophobicity (14). This resolution is provided here with the two overpacking variants, M131W and M131F, the effects of which argue against specific side-chain packing interactions.
, a measure of side-chain hydrophobicity, suggests that hydrophobic interactions play a major role in determining the stability of this partly folded intermediate. The enhanced stability of apoMb I resulting from overpacking substitution is consistent with this interpretation but suggests that the specific side-chain packing, as seen in holoMb, is absent at this position and its surroundings. This interpretation is at odds with an earlier study (14), which interpreted mutagenesis results as indicative that specific residues make energetically important packing interactions. However, with the exception of A130L, which slightly stabilizes the I form, none of the substitutions in the earlier study resolve packing from hydrophobicity (14). This resolution is provided here with the two overpacking variants, M131W and M131F, the effects of which argue against specific side-chain packing interactions.
Specific native-like side-chain packing interactions have been identified in other equilibrium intermediates, including cytochrome c, α-lactalbumin, and staphylococcal nuclease (17, 36–38). Unlike the effects of the position 131 variants on the I form of apoMb, overpacking substitutions that increase hydrophobicity in cytochrome c, α-lactalbumin, and staphylococcal nuclease lead to destabilization, suggesting that rigid side-chain packing plays a role at some sites within these partly folded proteins. However, for α-lactalbumin, overpacking substitutions at some positions behave similarly to position 131 of apoMb, suggesting that some regions are stabilized by nonspecific hydrophobic interactions rather than specific packing interactions. As the present study examines only one site in apoMb, it remains possible that specific holoMb-like packing interactions stabilize some regions of the I form of apoMb (but not that of position 131). Resolution of this issue will require a substitution set that separates packing from hydrophobicity in such regions.
Supplementary Material
Acknowledgments
We thank Frederick Hughson and Juliette Lecomte for critical comments and suggestions and Bertrand Garcia-Moreno E. and David Draper for use of fluorimeters. This work was supported by American Heart Association Grant-in-Aid AHA 9930126N (to D.B.).
Abbreviations: apoMb, apomyoglobin; holoMb, holomyoglobin; N, I, and U, native, acid intermediate, and unfolded forms of apomyoglobin.
Footnotes
Endpoint normalization has a large effect for M131W, which shows a reproducibly larger (more negative) ellipticity in the I form than WT apoMb I. Whether this change results from increased structure in the peptide backbone or from a CD transition involving the substituting tryptophan side chain is not clear (25).
ΔΔG° is the apparent unfolding free energy difference between the WT protein and each variant: 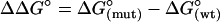 .
.
References
- 1.Griko, Y. V., Privalov, P. L., Venyaminov, S. Y. & Kutyshenko, V. P. (1988) J. Mol. Biol. 202, 127–138. [DOI] [PubMed] [Google Scholar]
- 2.Hughson, F. M., Wright, P. E. & Baldwin, R. L. (1990) Science 249, 1544–1548. [DOI] [PubMed] [Google Scholar]
- 3.Cocco, M. J. & Lecomte, J. T. J. (1990) Biochemistry 29, 11072–11078.2125502 [Google Scholar]
- 4.Lecomte, J. T. J., Kao, Y.-H. & Cocco, M. J. (1996) Proteins 22, 267–285. [DOI] [PubMed] [Google Scholar]
- 5.Eliezer, D. & Wright, P. E. (1996) J. Mol. Biol. 263, 531–538. [DOI] [PubMed] [Google Scholar]
- 6.Lecomte, J. T. J., Sukits, S. F., Bhattacharya, S. & Falzone, C. J. (1999) Protein Sci. 8, 1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jamin, M. & Baldwin, R. L. (1998) J. Mol. Biol. 276, 491–504. [DOI] [PubMed] [Google Scholar]
- 8.Eliezer, D., Chung, J., Dyson, H. J. & Wright, P. E. (2000) Biochemistry 39, 2894–2901. [DOI] [PubMed] [Google Scholar]
- 9.Nishimura, C., Dyson, H. J. & Wright, P. E. (2002) J. Mol. Biol. 322, 483–489. [DOI] [PubMed] [Google Scholar]
- 10.Hughson, F. M., Barrick, D. & Baldwin, R. L. (1991) Biochemistry 30, 4113–4118. [DOI] [PubMed] [Google Scholar]
- 11.Kuwajima, K. (1989) Proteins 6, 87–103. [DOI] [PubMed] [Google Scholar]
- 12.Ptitsyn, O. B. (1987) J. Protein Chem. 6, 273–293. [Google Scholar]
- 13.Ptitsyn, O. B., Pain, R. H., Semisotnov, G. V., Zerovnik, E. & Razgulyaev, O. I. (1990) FEBS Lett. 262, 20–24. [DOI] [PubMed] [Google Scholar]
- 14.Kay, M. S. & Baldwin, R. L. (1996) Nat. Struct. Biol. 3, 439–445. [DOI] [PubMed] [Google Scholar]
- 15.Kay, M. S., Ramos, C. H. I. & Baldwin, R. L. (1999) Proc. Natl. Acad. Sci. USA 96, 2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsumura, M., Becktel, W. J. & Matthews, B. W. (1988) Nature 334, 406–410. [DOI] [PubMed] [Google Scholar]
- 17.Wu, L. C. & Kim, P. S. (1998) J. Mol. Biol. 280, 175–182. [DOI] [PubMed] [Google Scholar]
- 18.Springer, B. A. & Sligar, S. G. (1987) Proc. Natl. Acad. Sci. USA 84, 8961–8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrick, D. & Dalquist, F. W. (2000) Proteins 39, 278–290. [DOI] [PubMed] [Google Scholar]
- 20.Teale, F. W. J. (1959) Biochim. Biophys. Acta 35, 543. [DOI] [PubMed] [Google Scholar]
- 21.Edelhoch, H. (1967) Biochemistry 6, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 22.Brenstein, R. J. (1991) nonlin for macintosh (Robelko Software, Carbondale, IL).
- 23.Pace, C. N. (1986) Methods Enzymol. 131, 266–280. [DOI] [PubMed] [Google Scholar]
- 24.Santoro, M. M. & Bolen, D. W. (1988) Biochemistry 27, 8063–8068. [DOI] [PubMed] [Google Scholar]
- 25.Chakrabartty, A., Kortemme, T., Padmanabhan, S. & Baldwin, R. L. (1993) Biochemistry 32, 5560–5565. [DOI] [PubMed] [Google Scholar]
- 26.Sondek, J. & Shortle, D. (1992) Proteins 13, 132–140. [DOI] [PubMed] [Google Scholar]
- 27.Fauchère, J. & Pliska, V. (1983) Eur. J. Med. Chem. 18, 369–375. [Google Scholar]
- 28.Luo, Y., Kay, M. S. & Baldwin, R. L. (1997) Nat. Struct. Biol. 4, 925–930. [DOI] [PubMed] [Google Scholar]
- 29.Luo, Y. & Baldwin, R. L. (1998) J. Mol. Biol. 279, 49–57. [DOI] [PubMed] [Google Scholar]
- 30.Luo, Y. & Baldwin, R. L. (2001) Biochemistry 40, 5283–5289. [DOI] [PubMed] [Google Scholar]
- 31.Chakrabartty, A., Kortemme, T. & Baldwin, R. L. (1994) Protein Sci. 3, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waltho, J. P., Feher, V. A., Merutka, G., Dyson, H. J. & Wright, P. E. (1993) Biochemistry 32, 6337–6347. [DOI] [PubMed] [Google Scholar]
- 33.Barrick, D. (1993) Ph.D. thesis (Stanford University, Stanford, CA).
- 34.Lin, L, Pinker, R. J., Forde, K., Rose, G. D. & Kallenbach, N. R. (1994) Nat. Struct. Biol. 1, 447–452. [DOI] [PubMed] [Google Scholar]
- 35.Cocco, M. J. & Lecomte J. T. J. (1994) Protein Sci. 3, 267–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marmorino, J. L. & Pielak, G. J. (1995) Biochemistry 34, 3140–3143. [DOI] [PubMed] [Google Scholar]
- 37.Marmorino, J. L., Lehti, M. & Pielak, G. J. (1998) J. Mol. Biol. 275, 379–388. [DOI] [PubMed] [Google Scholar]
- 38.Carra, J. H., Anderson, E. A. & Privalov, P. L. (1994) Protein Sci. 3, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}