

Abstract
Mucopolysaccharidosis type VII is a lysosomal storage disorder resulting from inherited deficiency of β-glucuronidase (GUS). Mucopolysaccharidosis type VII is characterized by glycosaminoglycan storage in most tissues, including brain. In these disorders, enzyme delivery across the blood-brain barrier (BBB) is the main obstacle to correction of lysosomal storage in the CNS. Prior studies suggested mouse brain is accessible to GUS in the first 2 weeks of life but not later. To explore a possible role for the mannose 6-phosphate/insulin-like growth factor II receptor in GUS transport across the BBB in neonatal mice, we compared brain uptake of phosphorylated GUS (P-GUS) and nonphosphorylated GUS (NP-GUS) in newborn and adult mice. 131I-P-GUS was transported across the BBB after i.v. injection in 2-day-old mice. The brain influx rate (Kin) of 131I-P-GUS in 2-day-old mice was 0.21 μl/g·min and decreased with age. By 7 weeks of age, transport of 131I-P-GUS was not significant. Capillary depletion revealed that 62% of the 131I-P-GUS in brain was in brain parenchyma in 2-day-old mice. In addition, uptake of 131I-P-GUS into brain was significantly reduced by coinjection of unlabeled P-GUS or M6P in a dose-dependent manner. In contrast, the Kin of 131I-NP-GUS (0.04 μl/g·min) was significantly lower than 131I-P-GUS in 2-day-old mice. Transcardiac brain perfusion confirmed that neither 131I-P-GUS nor 131I-NP-GUS crossed the BBB in adult mice. These results indicate that 131I-P-GUS transport into brain parenchyma in early postnatal life is mediated by the mannose 6-phosphate/insulin-like growth factor II receptor. This receptor-mediated transport is not observed in adult mice.
Keywords: β-glucuronidase, mannose 6-phosphate/insulin-like growth factor II receptor, central nervous system, lysosomal storage disease, phosphorylated β-glucuronidase
The enzyme β-glucuronidase (β-d-glucuronoside glucuronosohydrolase; International Union of Biochemistry and Molecular Biology no. EC 3.2.1.31) (GUS) is a tetrameric glycoprotein acid hydrolase localized primarily in lysosomes and found in virtually all mammalian cells (1, 2). GUS acts in lysosomes as an exoglycosidase to cleave glucuronic acid residues from the nonreducing termini of glycosaminoglycans (GAGs). Mucopolysaccharidosis type VII (MPS VII; Sly syndrome) is an autosomal recessive lysosomal storage disorder resulting from an inherited deficiency of GUS. MPS VII patients have mental retardation, hepatosplenomegaly, multiple skeletal anomalies referred to as dysostosis multiplex, coarse facial features, corneal clouding, hearing impairment, increased urinary GAGs, widespread lysosomal storage of GAGs in most tissues, including the CNS, and premature death (3, 4). Enzyme replacement therapy with murine or human GUS in MPS VII mice reduces visceral lysosomal storage, normalizes the pathological phenotype, and prolongs lifespan (5-7). It also improves abnormal storage in brain if treatment with GUS is begun before 2 weeks of age (8). Why GUS treatment reverses brain disease only when begun before age 2 weeks is unclear, but it must cross the blood-brain barrier (BBB) to do so.
The BBB is formed by a continuous layer of endothelial cells that regulate the passage of solutes between the CNS and the blood. It restricts the entry of serum proteins into the CNS and also controls the passage of regulatory proteins and various essential substrates in both the brain-to-blood and blood-to-brain directions (9). The microvasculature in the brain is so dense that no neuron or glial cell is >20 μm from the nearest capillary (10). Thus, every neuron is virtually perfused by its own microvessel. For this reason, a circulating substance that crosses the brain microvascular wall is widely distributed and immediately available throughout the brain. An enzyme as large as GUS (>300 kDa) would likely cross the BBB only by receptor-mediated transcytosis.
The mannose 6-phosphate (M6P)/insulin-like growth factor II (IGF-II) receptor (M6P/IGF2R) is a multifunctional cell surface receptor that binds lysosomal enzymes that contain the M6P marker, resulting in their sorting to the lysosome. The receptor also has a distinct binding site for IGF-II and binds this mitogenic growth factor at the cell surface, leading to its internalization and degradation in the lysosome (11-14). This study was undertaken to examine the role of the M6P/IGF2R in delivery of lysosomal enzymes to brain by comparing the passage of phosphorylated and nonphosphorylated forms of human GUS across the BBB in neonatal and adult mice.
Materials and Methods
Recombinant Human GUS Production. Phosphorylated human GUS (P-GUS) was produced in overexpressing, M6P/IGF2R-deficient mouse L cells.¶ The enzyme was purified from conditioned media by anti-human GUS monoclonal antibody affinity column chromatography. P-GUS was eluted with 3.5 M MgCl2 and then desalted over Bio-Gel P6 sizing resin.
Nonphosphorylated human GUS (NP-GUS) was produced in insect cells by using the baculovirus system as described for mouse GUS in ref. 15. NP-GUS was purified from media from virus-infected Sf21 insect cells by anti-human GUS monoclonal antibody Affi-Gel 10 affinity column chromatography by the same procedure used for P-GUS. The concentrations of both the P-GUS and NP-GUS were adjusted to 2.5 × 105 units per ml (1 unit = 1 nmol of substrate cleaved per h) and both purified enzymes were stored at -70°C. M6P-specific uptake of the P-GUS and NP-GUS purified enzymes by human fibroblasts was 185 and 0 units per mg/h, respectively (data not shown).
Radioactive Labeling. P-GUS and NP-GUS were radioactively labeled by the iodobead method (Pierce) with 131I-Na (Amersham Pharmacia). Labeled, active enzyme was separated from free iodine on a Sephadex G-10 column. Albumin was labeled with 125I-Na (Amersham Pharmacia) by the chloramine-T method and purified on a column of Sephadex G-10. Each agent was freshly prepared on the day of experiment.
Animals. Male CD-1 mice from our in-house colony were studied at 2 days, 5 days, 1 week, 2 weeks, and 7 weeks of age. The mice had free access to food and water and were maintained on a 12-h dark/light cycle in a room with controlled temperature (24 ± 1°C) and humidity (55 ± 5%). Studies were approved by the local Animal Care and Use Committee and done in a facility approved by the Association for Assessment and Accreditation of Laboratory Animal Care.
Drug Administration and Experimental Procedures. Mice anesthetized with isoflurane received an i.v. injection of 131I-P-GUS or 131I-NP-GUS with 125I-albumin (550,000 cpm of each) into the superficial temporal vein (neonates) or jugular vein (adults). At 1, 2, 3, 4, 5, 7.5, and 10 min after the injection, mice were killed under light anesthesia with isoflurane, and the blood, brain, heart, lung, liver, spleen, and kidneys were collected immediately. Serum from mouse blood was isolated by centrifugation. To test the specificity of the uptake and tissue distribution of 131I-P-GUS, unlabeled P-GUS (1, 10, and 30 μg), M6P (0.02, 0.2, and 2 μmol), or IGF-II (1.35 nmol) was included in the i.v. injection.
Multiple Time Regression Analysis. This method (16, 17) was used to calculate the blood-to-brain unidirectional influx rate (Kin) of radiolabeled compounds into the brain. The brain/serum ratios were plotted against exposure time estimated from the following equation:
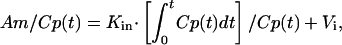 |
1 |
where Am and Cp(t) are the cpm/g of brain and the cpm/μl of serum at time t, respectively. Kin was measured as the slope for the linear portion of the relation between the brain/serum ratios and respective exposure times. The exposure time was calculated as the area under the serum concentration time curve (the integral part of above equation, where dt is the differential of time) divided by the serum concentration at time t. The y-intercept of the line represents the initial distribution volume (Vi) in the brain at t = 0.
The percentage of the injected dose taken up per gram of brain at time t (%Inj/g) was calculated from the following equation:
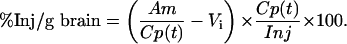 |
2 |
Capillary Depletion. To determine whether 131I-P-GUS crosses the full width of the BBB, capillary depletion was performed (18). The brain was removed 10 min after i.v. injection of 131I-P-GUS and 125I-albumin and emulsified in a glass homogenizer (8-10 strokes) at 4°C in a 9-fold volume of physiological buffer (10 mM Hepes/141 mM NaCl/4 mM KCl/2.8 mM CaCl2/1 mM MgSO4/1 mM NaH2PO4/10 mM d-glucose adjusted to pH 7.4). Dextran solution was added to the homogenate to a final concentration of 26%. An aliquot was centrifuged at 5,400 × g for 15 min at 4°C in a swinging bucket rotor. The pellet containing the brain microvessels and the supernatant containing the brain parenchyma were carefully separated. Results were expressed as capillary/serum and parenchyma/serum ratios, both corrected for vascular space contamination by subtracting the respective ratios for 125I-albumin.
Transcardiac Brain Perfusion (19). Adult male CD-1 mice were anesthetized i.p. with urethane, and, for each mouse, the heart was exposed, both jugulars were severed, and the descending aorta was ligated. A 26-gauge butterfly needle was inserted into the left ventricle of the heart and the perfusate, containing 131I-P-GUS or 131I-NP-GUS with 125I-albumin, was infused at a rate of 2 ml/min for 1-10 min. This rate of perfusion quickly fills the vascular space in the brain without disrupting the BBB (20). After perfusion, the brain was removed and weighed. The level of radioactivity was determined in a gamma counter. Brain/perfusate ratios were calculated by dividing the radioactivity in a gram of brain by the radioactivity in a microliter of perfusate. 131I-P-GUS, 131I-NP-GUS, and 125I-albumin were diluted in prewarmed physiological buffer [7.19 g/liter NaCl/0.3 g/liter KCl/0.28 g/liter CaCl2/2.1 g/liter NaHCO3/0.16 g/liter KH2PO4/0.17 g/liter anhydrous MgCl2/0.99 g/liter d-glucose/1% (wt/vol) BSA] at the amounts of 200,000 cpm/ml for each agent. The perfusate was freshly prepared each day.
In Vivo Stability of 131I-P-GUS in Serum and Brain. The stability of 131I-P-GUS in serum and brain was examined by HPLC 10 min after i.v. injection of 131I-P-GUS at 5,000,000 cpm in 2-day-old mice. Serum (10 μl) was diluted with 90 μl of mobile phase. Mouse brain was homogenized in 200 μl of mobile phase and centrifuged at 20,000 × g for 15 min, and the supernatant was collected. Each sample (100 μl) was injected onto an HPLC (pump model 2350, Teledyne ISCO, Lincoln, NE; BioSep-SEC-S 2000 size exclusion column, 7.8 ϕ × 300 mm, Phenomenex, Torrance, CA). Degradation of 131I-P-GUS during the extraction process in brain was determined by the addition of 131I-P-GUS into brain homogenate. The mobile phase consisted of 25 mM sodium phosphate buffer (pH 6.8). Fractions were collected at 1-min intervals for 60 min at a flow rate of 1.0 ml/min, and the radioactivity in each fraction was detected with a gamma counter.
Statistical Analysis. Means are presented with their standard errors and compared by one-way ANOVA followed by Dunnett's test or by unpaired t test with Welch's correction by using the prism 4.0 program (GraphPad, San Diego).
Results
The Distribution of 131I-P-GUS After i.v. Injection. The concentration-time profile of 131I-P-GUS and 125I-albumin in the serum fraction up to 10 min after the i.v. injection is shown in Fig. 1. The serum concentration of 131I-P-GUS declined linearly with time after i.v. injection into 2-day-, 5-day-, 1-week-, 2-week-, and 7-week-old mice. In contrast, the levels of 125I-albumin in serum were sustained throughout the experiment, regardless of age. The entry of 131I-P-GUS into brain was examined in 2-day-old and 7-week-old mice (Fig. 2A). The percent of 131I-P-GUS taken up by the brain, as calculated from Eq. 2, showed a rapid increase, peaking at 0.95 ± 0.16% injected dose per gram of brain 10 min after i.v. injection in 2-day-old mice. In contrast, there was negligible uptake of 131I-P-GUS in the brain in 7-week-old mice (0.008 ± 0.006% Inj/g). Despite this difference in M6P-mediated uptake with age, the plasma clearance was not markedly affected by age, because most of the enzyme was cleared by mannose receptors in the liver.
Fig. 1.

Time courses of radioactivity in serum after i.v. coinjection of 131I-P-GUS (A) and 125I-albumin (B) (550,000 cpm of each) in 2-day-old (•), 5-day-old (▴), 1-week-old (▪), 2-week-old (▾), and 7-week-old (♦) mice. n = 3-8 mice per point.
Fig. 2.

The percent (A) of 131I-P-GUS taken up by the brain and multiple-time regression analyses (B) of 131I-P-GUS and 125I-albumin after i.v. injection in 2-day-old and 7-week-old mice. 131I-P-GUS and 125I-albumin (550,000 cpm of each) were coinjected. n = 5-7 mice per point.
Fig. 9, which is published as supporting information on the PNAS web site, shows typical HPLC chromatograms of radioactivity for serum and brain extract 10 min after the i.v. injection of 131I-P-GUS in 2-day-old mice. 131I-P-GUS and 131I-Na eluted at 6 min and 11 min as single peaks, respectively. Intact 131I-P-GUS in the serum and brain extract accounted for 95.1 ± 0.7% and 73.1 ± 11.1% of the total radioactivity, respectively. The degradation of 131I-P-GUS in brain during the extraction process was 9.1 ± 1.1% of the total radioactivity, suggesting that 82.2% of 131I-P-GUS in the brain was still intact at 10 min after i.v. injection in 2-day-old mice.
Blood-to-Brain Kin. The relation between the brain/serum ratios of 131I-P-GUS and 125I-albumin and their exposure times is shown in Fig. 2B. The brain/serum ratio of 131I-P-GUS increased with the exposure time in 2-day-old mice, giving a Kin of 0.21 ± 0.04 μl/g·min. However, the Kin for 125I-albumin was too low to measure. The Vi values, estimated from the y-intercept, in 2-day-old mice were 5.67 ± 0.33 (131I-P-GUS) and 4.40 ± 0.32 μl/g (125I-albumin). In 7-week-old mice, there was no significant uptake of either.
The Kin and Vi were determined for 131I-P-GUS and 125I-albumin in 2-day-, 5-day-, 1-week-, 2-week-, and 7-week-old mice. As shown in Fig. 3, the Kin value of 131I-P-GUS was the highest in 2-day-old mice. The Kin gradually decreased with increasing age and was not measurable at the age of 7 weeks (Fig. 3A). The Vi values of 131I-P-GUS did not vary with age. The Kin values for 125I-albumin in 2-day-, 1-week-, and 7-week-old mice were below the levels of detection. In contrast, the Vi values of 125I-albumin increased with age, consistent with increased vascularization (10). The Vi value of 125I-albumin in 7-week-old mice was consistent with prior reports (21, 22).
Fig. 3.

Kin (A) and Vi (B) values of 131I-P-GUS and 125I-albumin for brain after i.v. coinjection at various ages. n = 3-8 mice per bar. Asterisks indicate a significant difference in comparison with 2-day-old mice: *, P < 0.05; **, P < 0.01; ***, P < 0.001. ND, not detected (no blood-to-brain entry).
Capillary depletion was conducted 10 min after i.v. coinjection of 131I-P-GUS and 125I-albumin in 2-day-old and 7-week-old mice (Fig. 4). About 62.1% of the 131I-P-GUS taken up by brain entered the brain parenchyma fraction (1.39 ± 0.21 μl/g), demonstrating transfer across the capillary wall, and ≈37.9% remained in the capillary fraction (0.85 ± 0.11 μl/g) in 2-day-old mice. In 7-week-old mice, the uptake of 131I-P-GUS was not observed in either fraction.
Fig. 4.

Distribution volume of 131I-P-GUS in brain parenchyma and capillary fraction 10 min after i.v. injection in 2-day-old and 7-week-old mice. n = 4-8 mice per mean fraction. Asterisk indicates a significant difference (P < 0.05) between the parenchyma and capillary fraction. ND, not detected.
Coinjection of unlabeled P-GUS (1, 10, or 30 μg) with 131I-P-GUS in 2-day-old mice produced a dose-dependent reduction in 131I-P-GUS uptake (Fig. 5A). The reduction was significant with the coinjection of both 10 or 30 μg of unlabeled P-GUS. The brain/serum ratio at 30 μg was equal to the vascular space within the brain as estimated by 125I-albumin. This result suggests that there was a saturable transport process for 131I-P-GUS into the brain in 2-day-old mice that was totally inhibited by 30 μg of unlabeled P-GUS.
Fig. 5.

Effects of unlabeled P-GUS (A), M6P, and IGF-II (B) on brain/serum ratios of 131I-P-GUS 10 min after i.v. injection in 2-day-old mice. Mice received 131I-P-GUS (550,000 cpm) i.v. with unlabeled P-GUS (1, 10, and 30 μg), M6P (0.02, 0.2, and 2 μmol), or IGF-II (1.35 nmol) and were killed 10 min later. The dashed line indicates the vascular space as measured with 125I-albumin in the 2-day-old mice shown in Fig. 3. n = 3-12 mice per mean. Asterisks indicate a significant difference from the control value: *, P < 0.05; ***, P < 0.001.
Coinjection of M6P or IGF-II on the uptake of 131I-P-GUS by brain was also tested in 2-day-old mice (Fig. 5B). The difference in slope of the disappearance curve for radiolabeled P-GUS and NP-GUS is very small because only a small amount of the plasma clearance reflects M6P receptor-mediated clearance. However, there was a significant dose-dependent decrease in brain uptake of 131I-P-GUS with the coinjection of M6P at doses of 0.2 and 2 μmol (P < 0.001). IGF-II (1.35 nmol) produced a reduction (78.8% of control) in 131I-P-GUS uptake by brain that was not statistically significant. Unlabeled P-GUS, M6P, or IGF-II produced no statistically significant effect on brain uptake of 125I-albumin in 2-day-old mice (data not shown).
To confirm the role of M6P in transport of 131I-P-GUS into the brain, 131I-P-GUS was compared to NP-GUS, which lacks M6P. Fig. 6A illustrates the levels of 131I-P-GUS and 131I-NP-GUS in the serum after i.v. injection of these agents in 2-day-old mice. The serum levels of both agents rose rapidly after injection, thereafter decreasing with time. However, the level of 131I-NP-GUS in serum was higher than that of 131I-P-GUS during the course of experiment. The relation between the brain/serum ratios of 131I-P-GUS and 131I-NP-GUS and their exposure times is shown in Fig. 6B. The Kin for 131I-NP-GUS (0.04 ± 0.37 μl/g·min) was significantly (P < 0.05) lower than that for 131I-P-GUS (0.31 ± 0.06 μl/g·min) in 2-day-old mice. In fact, although the Kin value of 131I-P-GUS significantly (P < 0.001) deviated from zero, the Kin value of 131I-NP-GUS was statistically not different from zero in 2-day-old mice; that is, no influx into brain could be measured for 131I-NP-GUS. In addition, there was a significant (P < 0.05) difference between the Vi values for 131I-P-GUS (7.23 ± 0.74 μl/g) and those for 131I-NP-GUS (4.52 ± 0.37 μl/g).
Fig. 6.

Time courses of radioactivity in serum (A) and multiple-time regression analyses (B) of 131I-P-GUS and 131I-NP-GUS 1-10 min after i.v. injection in 2-day-old mice. 131I-P-GUS (•) or 131I-NP-GUS (○) (550,000 cpm of each) was injected. n = 4 mice per point.
Transcardiac brain perfusion of 131I-P-GUS and 131I-NP-GUS corrected for 125I-albumin was conducted in 7-week-old mice (Fig. 7). The Kin values for 131I-P-GUS and 131I-NP-GUS in 7-week-old mice were not significant. The Vi values for 131I-P-GUS and 131I-NP-GUS were 7.70 ± 0.56 and 8.67 ± 0.65, respectively, not significantly different.
Fig. 7.

Time courses of the brain/perfusate ratios of 131I-P-GUS (•) and 131I-NP-GUS (○) corrected for 125I-albumin with transcardiac brain perfusion in 7-week-old mice. Neither 131I-P-GUS nor 131I-NP-GUS had a measurable uptake by brain. n = 3-4 mice per point.
Distribution of 131I-P-GUS and 131I-NP-GUS in Peripheral Tissues. Uptake of 131I-NP-GUS by brain, heart, lung, and kidney in 2-day-old mice was significantly less than the uptake of 131I-P-GUS (Fig. 8). However, there was no significant difference between the distribution of 131I-P-GUS and 131I-NP-GUS in the liver and spleen in 2-day-old mice. We also compared the effects of age on the uptakes of 131I-P-GUS and 125I-albumin in peripheral tissues (Table 1, which is published as supporting information on the PNAS web site). The uptake of 131I-P-GUS was significantly greater than that of 125I-albumin at day 2 in all tissues (6.2-fold for heart and 13-fold for liver). With aging, the difference between 131I-P-GUS uptake and that of 125I-albumin decreased in all tissues except spleen.
Fig. 8.

Comparison of the distributions of 131I-P-GUS and 131I-NP-GUS in various tissues 10 min after i.v. injection in 2-day-old mice. 131I-P-GUS or 131I-NP-GUS (550,000 cpm of each) was injected. The distribution volumes of the brain (A), heart (B), lung (C), liver (D), spleen (E), and kidney (F) were normalized by the percent of 131I-P-GUS in each tissue. n = 4-5 mice per mean. Asterisks indicate a significant difference from the control value: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We also compared the effects of M6P and IGF-II on the distribution of 131I-P-GUS and 125I-albumin in peripheral tissues 10 min after injection into 2-day-old mice (Table 2, which is published as supporting information on the PNAS web site). M6P significantly inhibited the uptake of 131I-P-GUS in all tissues except spleen in a dose-dependent manner. IGF-II had little effect at the single dose (1.35 nmol) examined. These data support the notion that uptake of 131I-P-GUS is mediated by the M6P receptor, which is developmentally regulated and down-regulated over the first 7 weeks of life in heart, lung, liver, and kidney, as well as in brain. 125I-albumin uptake was unchanged by M6P or IGF-II, except for spleen.
Discussion
Enzyme replacement therapy has been important for the treatment of lysosomal storage disorders (23). Although both CNS and peripheral tissues respond to i.v. replacement therapy in neonates, only peripheral tissues respond in adults (8). Inability to deliver enzyme across the BBB after the newborn period remains a major obstacle to the treatment of CNS lysosomal storage diseases. Until now, the reason why the CNS of neonates, but not adults, responds to enzyme replacement therapy was to our knowledge unknown. Here we report that the BBB of neonatal mice possesses a transport system for phosphorylated lysosomal enzymes, that this transporter is the M6P/IGF2R, and that transport is progressively lost with aging. In fact, by adulthood, the BBB has totally lost this transport capacity.
When 131I-P-GUS was injected intravenously into neonatal and adult mice at various ages, the 131I-P-GUS was rapidly taken up by brain in the neonate but not in the adult. Almost 1% of the i.v. dose of 131I-P-GUS was taken up per gram of brain by 10 min in 2-day-old mice. This uptake rate is very high for a protein of this size (312 kDa) (24, 25), exceeding that of morphine by >50-fold (26) and of interleukin-1 by at least 10-fold (27). The Kin value of 131I-P-GUS and the Vi value of 131I-P-GUS were both larger than those of 125I-albumin, clearly demonstrating specific uptake by brain. Capillary depletion confirmed that the 131I-P-GUS had entered the brain parenchymal space. HPLC analysis showed that the radioactivity in blood and brain during the course of study represented intact 131I-P-GUS. Consistent with the literature, we found the BBB of the neonate was no more permeable to 125I-albumin than the adult BBB (10, 28, 29). Thus, 131I-P-GUS did not enter the brain because of a leaky BBB. The BBB uptake of 131I-P-GUS steadily declined with aging until 7 weeks of age, when uptake was not measurable. It is this ability of the neonate, but not the adult, to transport the enzyme GUS across the BBB that explains why replacement therapy was effective in treating CNS manifestations of disease only if started before 2 weeks of age in murine mucopolysaccharidosis type VII (6).
Although the BBB of the neonate was no more permeable to 125I-albumin than the adult BBB, the albumin space was larger in the adult because of increases in the density, length, and capillary surface area of brain microvessels. These changes occur primarily between 1 and 3 weeks of age (10).
The M6P/IGF2R is known to participate in the endocytosis of lysosomal enzymes from the cell surface (30). We speculated that M6P/IGF2R could be the BBB transporter for P-GUS because M6P/IGF2R is important in the uptake of P-GUS by peripheral tissues (15) and it had been demonstrated on bovine and rodent brain capillaries (31, 32). In this study, we found that both P-GUS and M6P inhibited the transport of 131I-P-GUS across the BBB in 2-day-old mice in a dose-dependent manner. IGF-II, which can sterically inhibit binding of GUS to the M6P/IGF2R (33), also tended to reduce the uptake of 131I-P-GUS in the brain. 131I-NP-GUS, which does not contain M6P on its oligosaccharide chains, was not transported across the BBB. These observations support the view that the M6P/IGF2R mediates the observed transport of 131I-P-GUS across the BBB in early neonatal life.
We ruled out the possibility that the loss of transport of 131I-P-GUS across the adult BBB is caused by some blood-borne factor. A circulating substance capable of binding or rapidly degrading GUS enzymatically or an excess of endogenous inhibitor (e.g., GUS, M6P, or IGF-II) could have explained why the adult was no longer able to transport 131I-P-GUS across the BBB (34). However, the brain perfusion method we used replaces the blood with perfusion buffer and negates the possible effects of circulating substances that might impair BBB function (35, 36) or that might interfere with the interaction of 131I-P-GUS with the BBB. With brain perfusion, 131I-P-GUS still did not cross the adult BBB. Therefore, it is a change in the BBB itself, and not circulatory factors, that accounts for the loss in brain uptake with age in the mouse.
Our findings agree with prior studies showing that M6P/IGF2R is developmentally regulated and declines in most tissues of the rat and mouse during the postnatal period (37-41). We found that 131I-P-GUS was taken up by multiple tissues in excess of 125I-albumin or of 131I-NP-GUS, that this uptake was inhibited by M6P, and that 131I-P-GUS uptake declined with aging (see supporting information).
In conclusion, the present study indicates that the neonate transports 131I-P-GUS across the BBB into brain parenchyma by a saturable transport system mediated by M6P/IGF2R. This transport is developmentally regulated in early postnatal life and is lost by 7 weeks of age. The presence of this transport system in the newborn can explain the effectiveness of neonatal i.v. enzyme therapy in correcting lysosomal storage in the CNS, and its loss can explain why the adult CNS is resistant to treatment. Understanding why this transporter is lost with aging and how one might induce its expression in the adult BBB could have great therapeutic importance.
Supplementary Material
Acknowledgments
This study was supported by Veterans Affairs Merit Reviews R01 NS41863 and R01 AA12743 (to W.A.B.) and by National Institutes of Health Grant R01 GM34182 (to W.S.S.).
Abbreviations: BBB, blood-brain barrier; GUS, β-glucuronidase; P-GUS, phosphorylated human GUS; NP-GUS, nonphosphorylated human GUS; M6P, mannose 6-phosphate; IGF-II, insulin-like growth factor II; M6P/IGF2R, mannose 6-phosphate/IGF-II receptor.
Footnotes
Grubb, J. H., Kyle, J. W., Cody, L. B. & Sly, W. S. (1993) FASEB J. 7, 1225a.
References
- 1.Musa, B. U., Doe, R. P. & Seal, U. S. (1965) J. Biol. Chem. 240, 2811-2816. [PubMed] [Google Scholar]
- 2.Paigen, K. (1989) Prog. Nucleic Acid Res. Mol. Biol. 37, 155-205. [DOI] [PubMed] [Google Scholar]
- 3.Sly, W. S., Quinton, B. A., McAlister, W. H. & Rimoin, D. L. (1973) J. Pediatr. 82, 249-257. [DOI] [PubMed] [Google Scholar]
- 4.Vogler, C., Levy, B., Kyle, J. W., Sly, W. S., Williamson, J. & Whyte, M. P. (1994) Mod. Pathol. 7, 132-137. [PubMed] [Google Scholar]
- 5.Sands, M. S., Vogler, C., Kyle, J. W., Grubb, J. H., Levy, B., Galvin, N., Sly, W. S. & Birkenmeier, E. H. (1994) J. Clin. Invest. 93, 2324-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogler, C., Sands, M. S., Levy, B., Galvin, N., Birkenmeier, E. H. & Sly, W. S. (1996) Pediatr. Res. 39, 1050-1054. [DOI] [PubMed] [Google Scholar]
- 7.Sands, M. S., Vogler, C., Torrey, A., Levy, B., Gwynn, B., Grubb, J., Sly, W. S. & Birkenmeier, E. H. (1997) J. Clin. Invest. 99, 1596-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vogler, C., Levy, B., Galvin, N. J., Thorpe, C., Sands, M. S., Barker, J. E., Baty, J., Birkenmeier, E. H. & Sly, W. S. (1999) Pediatr. Res. 45, 838-844. [DOI] [PubMed] [Google Scholar]
- 9.Davson, H. & Segal, M. B. (1996) in Physiology of the CSF and Blood-Brain Barriers (CRC, Boca Raton, FL), pp. 303-485.
- 10.Bar, T. (1980) Adv. Anat. Embryol. Cell Biol. 59, 1-62. [DOI] [PubMed] [Google Scholar]
- 11.Oka, Y., Rozek, L. M. & Czech, M. P. (1985) J. Biol. Chem. 260, 9435-9442. [PubMed] [Google Scholar]
- 12.Oka, Y. & Czech, M. P. (1986) J. Biol. Chem. 261, 9090-9093. [PubMed] [Google Scholar]
- 13.Nielsen, F. C., Wang, E. & Gammeltoft, S. (1991) J. Neurochem. 56, 12-21. [DOI] [PubMed] [Google Scholar]
- 14.Auletta, M., Nielsen, F. C. & Gammeltoft, S. (1992) J. Neurosci. Res. 31, 14-20. [DOI] [PubMed] [Google Scholar]
- 15.Sands, M. S., Vogler, C. A., Ohlemiller, K. K., Roberts, M. S., Grubb, J. H., Levy, B. & Sly, W. S. (2001) J. Biol. Chem. 276, 43160-43165. [DOI] [PubMed] [Google Scholar]
- 16.Blasberg, R. G., Fenstermacher, J. D. & Patlak, C. S. (1983) J. Cereb. Blood Flow Metab. 3, 8-32. [DOI] [PubMed] [Google Scholar]
- 17.Urayama, A., Yamada, S., Ohmori, Y., Deguchi, Y., Uchida, S. & Kimura, R. (2003) Drug Metab. Pharmacokin. 18, 310-318. [DOI] [PubMed] [Google Scholar]
- 18.Triguero, D., Buciak, J. & Pardridge, W. M. (1990) J. Neurochem. 54, 1882-1888. [DOI] [PubMed] [Google Scholar]
- 19.Banks, W. A., Clever, C. M. & Farrell, C. L. (2000) Am. J. Physiol. 278, E1158-E1165. [DOI] [PubMed] [Google Scholar]
- 20.Shayo, M., McLay, R. N., Kastin, A. J. & Banks, W. A. (1997) Life Sci. 60, L115-L118. [DOI] [PubMed] [Google Scholar]
- 21.Banks, W. A. & Broadwell, R. D. (1994) J. Neurochem. 62, 2404-2419. [DOI] [PubMed] [Google Scholar]
- 22.Banks, W. A., Kastin, A. J. & Ehrensing, C. A. (1994) J. Physiol. (London) 479, 257-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grabowski, G. A. & Hopkin, R. J. (2003) Annu. Rev. Genomics Hum. Genet. 4, 403-436. [DOI] [PubMed] [Google Scholar]
- 24.Brot, F. E., Bell, C. E., Jr., & Sly, W. S. (1978) Biochemistry 17, 385-391. [DOI] [PubMed] [Google Scholar]
- 25.Gupta, G. S. & Singh, G. P. (1983) Biochim. Biophys. Acta 748, 398-404. [DOI] [PubMed] [Google Scholar]
- 26.Banks, W. A. & Kastin, A. J. (1994) Peptides 15, 23-29. [DOI] [PubMed] [Google Scholar]
- 27.Banks, W. A., Ortiz, L., Plotkin, S. R. & Kastin, A. J. (1991) J. Pharmacol. Exp. Ther. 259, 988-996. [PubMed] [Google Scholar]
- 28.Johanson, C. E. (1989) in Implications of the Blood-Brain Barrier and Its Manipulation, ed. Neuwelt, E. A. (Plenum, New York).
- 29.Davson, H. & Segal, M. B. (1996) in Physiology of the CSF and Blood-Brain Barriers (CRC Press, Boca Raton, FL), pp. 607-662.
- 30.Stein, M., Zijderhand-Bleekemolen, J. E., Geuze, H., Hasilik, A. & von Figura, K. (1987) EMBO J. 6, 2677-2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenfeld, R. G., Pham, H., Keller, B. T., Borchardt, R. T. & Pardridge, W. M. (1987) Biochem. Biophys. Res. Commun. 149, 159-166. [DOI] [PubMed] [Google Scholar]
- 32.Reinhardt, R. R. & Bondy, C. A. (1994) Endocrinology 135, 1753-1761. [DOI] [PubMed] [Google Scholar]
- 33.Nolan, C. M., Kyle, J. W., Watanabe, H. & Sly, W. S. (1990) Cell Regul. 1, 197-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banks, W. A. & Kastin, A. J. (1993) in Proceedings of the International Symposium on Blood Binding and Drug Transfer, eds. Tillement, J. P., Eckert, H., Albengres, E., Barre, J., Bauman, P. & Lemaire, M. (Fort et Clair, Paris), pp. 223-242.
- 35.Takasato, Y., Rapoport, S. I. & Smith, Q. R. (1984) Am. J. Physiol. 247, H484-H493. [DOI] [PubMed] [Google Scholar]
- 36.Smith, Q. R., Momma, S., Aoyagi, M. & Rapoport, S. I. (1987) J. Neurochem. 49, 1651-1658. [DOI] [PubMed] [Google Scholar]
- 37.Lee, S. J. & Nathans, D. (1988) J. Biol. Chem. 263, 3521-3527. [PubMed] [Google Scholar]
- 38.Sklar, M. M., Kiess, W., Thomas, C. L. & Nissley, S. P. (1989) J. Biol. Chem. 264, 16733-16738. [PubMed] [Google Scholar]
- 39.Alexandrides, T., Moses, A. C. & Smith, R. J. (1989) Endocrinology 124, 1064-1076. [DOI] [PubMed] [Google Scholar]
- 40.Senior, P. V., Byrne, S., Brammar, W. J. & Beck, F. (1990) Development (Cambridge, U.K.) 109, 67-73. [DOI] [PubMed] [Google Scholar]
- 41.Ballesteros, M., Scott, C. D. & Baxter, R. C. (1990) Biochem. Biophys. Res. Commun. 172, 775-779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}