

Abstract
Series of N-substituted carbazole analogues bearing an indole ring were synthesized as anti-methicillin-resistant Staphylococcus aureus (MRSA) agents from a molecular hybridization approach. The representative compound 19 showed an MIC = 1 μg/mL against a panel of MRSA clinical isolates as it possessed comparable in vitro activities to that of vancomycin. Moreover, compound 19 also exhibited MIC = 1 μg/mL activities against a recent identified Z172 MRSA strain (vancomycin-intermediate and daptomycin-nonsusceptible phenotype) and the vancomycin-resistant Enterococcus faecalis (VRE) strain. In a mouse model with lethal infection of MRSA (4N216), a 75% survival rate was observed after a single dose of compound 19 was intravenously administered at 20 mg/kg. In light of their equipotent activities against different MRSA isolates and VRE strain, the data underscore the importance of designed hybrid series for the development of new N-substituted carbazoles as potential anti-MRSA agents.
Keywords: Methicillin-resistant Staphylococcus aureus, vancomycin, carbazole derivatives, molecular hybridization, resistant bacteria
The great discovery and deployment of antibiotics had succumbed to the steady emergence of multidrug-resistant bacterial strains and new microbial pathogens, which placed tremendous pressure on the race to develop novel antimicrobial agents.1−5Staphylococcus aureus is the leading Gram-positive pathogen in clinical setting responsible for a wide spectrum of diseases ranging from mild skin infections to severe life threatening pneumonia and bacteremia.6−10 Methicillin was introduced in early 1960s but methicillin-resistant S. aureus (MRSA) strains emerged soon after. The prevalence of MRSA increased steadily starting in the 1980s, and MRSA has become resistant to many marketed antibiotics due to the extensive selective pressure from utility of these drugs in the hospital setting.2,6 Decreased efforts in the development of other classes of antibiotics further complicated the limited treatment options.2,3,7
MRSA arises when methicillin-susceptible S. aureus (MSSA) acquires a large mobile genetic element called staphylococcal cassette chromosome mec (SCCmec) through horizontal gene transfer.11,12 Incorporation of the SCCmec leads to the expression of an altered penicillin-binding protein PBP2′ (PBP2a), which has >100-fold lower binding affinity for nearly all available β-lactam antibiotics, rendering them ineffective against MRSA.1 Glycopeptide-based antibiotics, especially vancomycin, have been the main drug of choice for treating severe MRSA infections.13,14 However, a decrease in efficacy of vancomycin has attracted great attention due to the emergence of heterogeneous vancomycin-intermediate S. aureus (hVISA) that was associated with increasing treatment failures.15 In addition, reports of vancomycin-resistant S. aureus (VRSA) are alarming due to the presence of transferable vancomycin resistance plasmids among the infection strains.16 In the clinical setting, coinfection of MRSA and vancomycin-resistant enterococci (VRE) after surgical procedures has gradually but surely reduced the effectiveness of glycopeptide-based antibiotics over the past decade, leading to yet another challenge in the fight against bacterial infection.17,18 Moreover, the increasing prevalence of MRSA in hospitals and emergence of community-associated MRSA (CA-MRSA) strains have become a leading cause of serious complications in patient treatments.19−21 It has been noted that the over millions of hospitalizations associated with these infections yearly would place a tremendous economic burden on many healthcare systems worldwide.22,23
With the shift in MRSA susceptibility and an increase in CA-MRSA infections,19−21,24 there is an urgent medical need for the development of new antimicrobial agents.14 To bolster the dwindling antibiotic arsenal, our research group has conducted an in-house screening program, leading to the identification of a novel hit (1) possessing a N-substituted bromoindole scaffold with a moderate anti-MRSA (ATCC 43300) activity at MIC = 8 μg/mL (Figure 1, Table S1).
Figure 1.

Strategy to generate novel antibiotic agents based on molecular hybridization.
Interestingly, a series of marine natural products known as bromomethylthioindoles were isolated from red algae Laurencia brongniartii by Y. Kamei et al. and identified to possess potent anti-MRSA activities (Figure 1).25 The regioselective synthesis of these alkylthio-and arylthioindoles was recently reported by Suzuki et al.26 Meanwhile, it was noted that naturally occurring carbazoles (Figure 1) possessed moderate antibacterial activities against both Gram-positive and Gram-negative bacteria.27−29 In particular, numerous series of N-substituted carbazole derivatives were synthesized and have been reported to exhibit diverse biological activities, such as antimicrobial, antitumor, antioxidatives, and anti-inflammatory properties.30−32 Moreover, carbazole-containing drug, carvedilol, was approved for the treatment of mild to severe congestive heart failure and of high blood pressure.33 Yet, reported N-substituted carbazoles or different carbazole cores only exhibited moderate activities against MSSA or MRSA with the MIC ranging from 4 to 16 μg/mL.29,34 In view of the above-reported bioactivities, it was foreseen to amalgamate the two anti-MRSA pharmacophores in one molecular unit to generate a new scaffold for anti-MRSA evaluation. In particular, we envisioned that N-alkylation of carbazole with halogenated indole analogues would provide a platform for generation of anti-MRSA derivatives. We have designed a facile synthetic route to generate a series of hybrids, several of which have been shown potent activities against MSSA, MRSA, and VRE. Herein, we wish to disclose our findings, including (1) a simple and efficient synthetic route to obtain potent N-substituted carbazole derivatives based on molecular hybridization; (2) the structure–activity relationship analysis; (3) a broad spectrum of antimicrobial activities against clinical MRSA and VRSA isolates, and (4) an in vivo study of compound 19 in the MRSA (4N216) systemic infection mouse model.
To validate whether the in-house hit is a false positive, hit compound 1 and its analogue 2 were resynthesized according to a two-step synthetic sequence in Scheme 1, involving N-substitution with epibromohydrin under basic conditions (78%) followed by coupling with an aryl amine under refluxing ethanol (80%).
Scheme 1. Synthesis of N-Substituted Indole Derivatives 1 and 2.

Reagents and conditions: (a) KOH, DMF, 0 °C, 30 min then epibromohydrin, rt, 16 h, 78%; (b) aryl amine, EtOH, reflux, 80%.
In general, carbazole-based derivatives with a β-hydroxy amine moiety were readily prepared through a two-step synthetic sequence as shown in Scheme 2. Accordingly, carbazole-based derivatives 3–24, containing dichloro-, dibromo-, and monobromo substituents at C-3 and C-6 positions of the carbazole ring, were first deprotonated with potassium hydroxide (1.4 equiv), and the resulting anion was allowed to react with epibromohydrin (2.5 equiv) to provide epoxide intermediates B–E in 78–81% yields. These intermediates were further coupled with various aryl amines to furnish the corresponding products 3–24, respectively, in 68–75% yields.
Scheme 2. Synthesis of N-Substituted Carbazole Derivatives 3–24 with a β-Hydroxyl Amine.

Reagents and conditions: (a) KOH, DMF, 0 °C, 30 min then epibromohydrin, rt, 16 h, 78–81%; (b) aryl amines, EtOH, reflux, 68–75%.
For comparison purposes, compounds 25 and 26 without containing a β-hydroxyl amine group were also synthesized according to Scheme 3. 9H-Carbazole substrate, containing dichloro substituents at C-3 and C-6 positions, was first treated with potassium hydroxide and 2-(3-bromopropoxy)-tetrahydro-2H-pyran followed by acidic hydrolysis and Dess–Martin periodinane oxidation, giving rise to aldehyde F in 72% yields over three steps. Intermediate F was further reacted with 2-(6-chloro-1H-indol-3-yl)ethan-1-amine or 2-(6-fluoro-1H-indol-3-yl)ethan-1-amine followed by reduction with NaBH4 to afford compounds 25 and 26, respectively, in 80 and 82% yields through a reductive amidation process.
Scheme 3. Synthesis of N-Substituted Carbazole Derivatives 25–26 without a β-Hydroxy Amine.

Reagents and conditions: (a) KOH, 2-(3-bromopropoxy)-tetrahydro-2H-pyran, DMF, 0 °C to rt, 12 h; (b) cat. PTSA, MeOH, rt, 12 h; (c) Dess–Martin periodinane (DMP), CH2Cl2, 0 °C to rt, 1 h, 72% over three steps; (d) indole amines, MeOH/THF (1:1), 0 °C to rt, 1 h then NaBH4, 0 °C to rt, 12 h, 80–82%.
However, to investigate whether stereoisomers arising at the β-hydroxylamine position within these derivatives affect the anti-MRSA activities, we further synthesized enantiomeric pairs (+)-13 and (−)-14 as well as (+)-20 and (−)-21 to compare the MIC values with their respective racemic counterparts (±)-12 and (±)-19. The synthesis was achieved by using commercially available (−)-glycidol ([α]D20 −15; neat) and (+)-glycidol ([α]D20 +15; neat) as enantioenriched source and through a three-step synthetic sequence reported in the literature,35 including tosylation of the hydroxyl group and 9H-carbozale-N alkylation, followed by opening the epoxide ring with an indole amine.
Derivatives hybridized with carbazole and indole units were first evaluated for bacterial growth inhibitions using standard techniques against a methicillin-susceptible S. aureus (MSSA) strain ATCC 29213 and a methicillin-resistant S. aureus (MRSA) strain ATCC 43300 (Table S1 in Supporting Information). From initial screening of our in-house compounds, the discovery of N-substituted carbazoles with anti-MRSA activities had prompted us to synthesize a series of compounds (Schemes 1–3) and investigate their structure–activity relationships as listed in Table S1. Hit 1, an upper N-substituted bromoindole connecting to a lower 2-(3-fluoro-phenyl)ethylamine fragment with a 2-hydroxypropyl linker, was resynthesized and exhibited both anti-MSSA and anti-MRSA activities at a moderate concentration of MIC = 16–32 and 8 μg/mL, respectively. Along with hit 1, the corresponding 3-halogenated 2 was also prepared and showed an improvement in activity against both MSSA and MRSA by 2- to 8- fold, respectively. These results had prompted us to further modify these hits to obtain a novel series of N-substituted carbazole derivatives incorporating an indole functionality. Concurrently, structurally related N-substituted carbazole derivatives, referred to as DCAP analogues, were reported to be antibacterial against a panel of clinically isolated BSL-2 pathogenic strains.36 Inspired by the structural similarity, we then symmetrized the upper N-substituted bromoindole to the N-substituted dibromo-carbazole ring to afford compound 3, resulting in a remarkable 8-fold increase in both anti-MSSA and anti-MRSA activities relative to hit 1. Encouraged by these findings, we started to systematically modify the peripheral functional groups of the carbazole ring with particular emphasis on the NH-substitution fixed with a 2-hydroxypropyl linker. Carbazole derivatives 4–24 (Table S1) with structural modifications extending from the tail of the 2-hydroxypropyl linker were then synthesized according to Schemes 1–3. As illustrated with compounds 3 and 4, substitution of a hydroxyl or halogen group on the terminal phenyl ring was first examined. As a result, activities (MIC = 1–2 μg/mL) against tested ATCC 29213 and 43300 strains were retained. However, increasing the hydrophilicity met with little success as seen in compound 5 with a dramatic decrease in anti-MSSA and anti-MRSA activity by 8-fold (MIC = 16 μg/mL). Replacing the phenyl unit with an imidazole ring at the tail portion gave rise to compound 6 showing less activity (MIC = 4 μg/mL) against tested strains. We then prepared compounds 7–11 to address the importance of the size and length of the lower projected functionalities. With the triazole group lacking the carbon extension from the 2-hydroxypropyl moiety in compound 7, both anti-MSSA and anti-MRSA activities were significantly reduced to MIC = 16 μg/mL. Despite incorporating a larger functional group at the terminal portion as indicated in compounds 8 and 9 (MIC > 32 μg/mL), both anti-MSSA and anti-MRSA activities could not be restored. This observation appeared to reveal that flexibility and the extension of the lower accessory groups could influence the anti-MSSA and anti-MRSA activities as seen with compound 10 (MIC = 2 μg/mL). To test this hypothesis, we readily introduced different aliphatic chain length at the lower portion of the indole ring. The choice of the indole unit was inspired by compound 10 and the bromomethylthioindoles isolated from red algae that possessed potent anti-MRSA activities.25,26 Consistent with the regiochemistry of the isolated bromomethylthioindoles, we connected the carbazole moiety via the C-3 position on the indole ring while maintaining a halogen group on the phenyl ring. Compound 11 thus obtained had a substantial (>16 fold) improvement in both anti-MSSA and anti-MRSA activities as compared to compounds 8 and 9, suggesting that the flexibility of the functional group extended from 2-hydroxypropyl linker might allow for a favorable conformation fitting into the target protein. However, there was no substantial difference in activities between the methylene (−CH2−) and ethylene (−CH2–CH2−) carbon extensions as seen with compounds 10 (MIC = 2 μg/mL) and 11 (MIC = 1 μg/mL). When comparing compound 12 with 11, varying the halogen substituent and its position on the indole ring had limited effects on their activities against tested strains. Compound 15 containing a weak electron-donating group on the indole ring accounted for a marginal 2-fold decrease in potency. More importantly, racemic 12 and its enantiomers (S)-13 and (R)-14 showed equipotent activities against tested strains at a MIC value of 1 μg/mL, suggesting that inhibitory activities are little influenced by the stereochemistry of the secondary hydroxyl group in the linker. A similar phenomenon was observed for racemic 19 and its enantiomers (S)-20 and (R)-21 with MIC values around 0.5–1 μg/mL. These results imply that antimicrobial activities of this series of compounds might arise from binding to bacterial membranes rather than inhibiting an enzyme or protein. To examine the significance of the halogen atom around the carbazole ring, we found that the dibromo decoration was advantageous. As compared to compound 16 (MIC = 4 μg/mL) without any halogen substitution, compound 11 (MIC = 1 μg/mL) bearing a bromo atom at C-3 and C-6 positions exhibited a 4-fold increase in potency, suggesting that an electron-withdrawing group(s) on the carbazole ring could be a structural requirement for showing anti-MRSA and anti-MSSA activity. Indeed, irrespective of the halogen substituent on the indole ring, compounds 17 and 18, merely containing a bromo atom on the carbazole ring, possessed anti-MRSA activities as potent as its dibromo counterpart 11 (MIC = 1 μg/mL). Further structural modifications were carried out to remove the hydroxyl moiety of the linker. As a result, the inhibitory activities of compounds 25 and 26 were retained as compared to their hydroxyl analogue 19 (MIC = 1 μg/mL), verifying that the hydroxyl group was structurally redundant.
The antibacterial spectra of potent carbazole derivatives, such as compounds 11, 12, 19, 22, and 24, were further evaluated against a panel of clinical isolates of MSSA and MRSA, results of which were compiled in Table 1. M056 is a MSSA clinical isolate and the above five compounds have exhibited comparable potent antibacterial activities to that of vancomycin. Moreover, these compounds also possessed a broad spectrum of inhibitory activities against multiple clinically isolated MRSA strains, including M013, N043, C217, Y069, L166, Z234, and 4N216, at MIC values as low as 0.5–1 μg/mL. In contrast to the indole-containing N-substituted carbazoles (11, 12, 19), compound 24 showed a higher MIC value ranging from 1 to 4 μg/mL against majority of the clinical MRSA strains tested. Structural comparison between compounds 19 and 24 suggests that the indole moiety, with the corresponding linker length, plays a vital role in the growth inhibition of the clinical MRSA strains. Interestingly, shortening of the linker length in compound 22 has restored the potent anti-MRSA activities. Taken together, these data strongly supported the structural hybridization with enhanced anti-MRSA activities and provided a new lead structural motif.
Table 1. Antibacterial Activity of Compounds 11, 12, 19, 22, and 24 against Clinical MSSA and MRSA Strains.

| minimum
inhibitory concentration (μg/mL) |
||||||||
|---|---|---|---|---|---|---|---|---|
| MSSAa | MRSAb |
|||||||
| compd | M056 | M013 | N043 | C217 | Y069 | L166 | Z234 | 4N216 |
| 11 | 1–2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 12 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 19 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 0.5 |
| 22 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| 24 | 1 | 4 | 4 | 4 | 4 | 4 | 4 | 0.5–1 |
| vancomycin | 2 | ≤0.5 | 1 | 1 | 1 | 2 | 1 | 1 |
Clinical MSSA strain M056.
Clinical MRSA strains M013, N043, C217, Y069, L166, Z234, and 4N216.
Recently, the emergence of heterogeneous vancomycin-intermediate S. aureus (hVISA) has been reported from healthcare system around the world. VISA was associated with increasing treatment failures.15 Alternatively, daptomycin has been employed for the management of serious infections caused by MRSA with reduced susceptibility to vancomycin, such as VISA.37 In addition, MRSA and VRE have emerged as the two most common Gram-positive multidrug-resistant pathogens associated with many health care facilities. Z172, a hospital-associated MRSA strain in Taiwan, exhibited a vancomycin-intermediate and daptomycin-nonsusceptible phenotype.38 Even though the occurrence of MRSA strains with reduced susceptibility to vancomycin and daptomycin along with VRE strains resistant to daptomycin are still not prevalent, their rising appearances in the hospital settings have necessitated the development of new class of anti-MRSA agents in combating these bacteria. In particular, compounds 19 exhibited potent inhibitory activities (MIC = 1 μg/mL) against Z172 and VRE (Table 2). Indeed, compound 19 showed an increase of 4–16-fold in the inhibitory activities in comparison to those of vancomycin (Table 2). Of note, the carbazole-based compounds 12 and 19 are about 4–18 times more potent than the recently reported coumarin-based inhibitors against VRE (ATCC 51299).39 To assess the antibacterial spectrum across other genera, several Gram-negative bacteria, including Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter baumannii, were further selected for evaluation. Nevertheless, we observed a significant loss of activities against various Gram-negative bacterial strains as mentioned above. Thus, the carbazole-based series are tentatively designated as Gram-positive-specific inhibitors. Interestingly, comparing compound 11 to 12, substitution of halogens on the indole ring provided a 4-fold increase in inhibitory activities against E. coli and A. baumannii. These results suggested that carbazole-based inhibitors might hit a different target(s) compared to vancomycin as compounds 12 and 19 exerted potent activities against both VRE (ATCC 51299) and a vancomycin-intermediate and daptomycin-nonsusceptible MRSA strain (Z172).
Table 2. Antibacterial Activity of Compounds 11, 12, 19, 22, and 24 against Z172, VRE and Several Gram Negative Bacteria.
| minimum
inhibitory concentration (μg/mL) |
||||||
|---|---|---|---|---|---|---|
| compd | Z172a | ATCC 51299 (VRE) | ATCC 25922 (E. coli) | ATCC 27853 (P. aeruginosa) | ATCC 700603 (K. pneumoniae) | ATCC 19606 (A. baumannii) |
| 11 | 2 | 2 | 8–16 | >64 | >64 | 16 |
| 12 | 2 | 1 | >64 | >64 | >64 | >64 |
| 19 | 1 | 1 | 4 | >64 | >64 | 8 |
| 22 | 1 | 2 | >64 | >64 | >64 | >64 |
| 24 | 4 | 2 | >64 | >64 | >64 | >64 |
| vancomycin | 4 | 8–16 | N.A. | N.A. | N.A. | N.A. |
Z172 is a hospital-associated MRSA strain in Taiwan that exhibits vancomycin-intermediate and daptomycin-nonsusceptible phenotypes.
Of the synthesized carbazole-based inhibitors, compound 19 has a broader spectrum of antimicrobial activities particularly for resistant strains VRE (ATCC 51299) and MRSA (Z172); thus, its pharmacokinetic studies were conducted prior to in vivo efficacy studies. As compiled in Table 3, compound 19 (10 mg/kg, iv) displays a longer half-life (t1/2 = 10.3 vs 0.6 h) and larger blood exposure (AUC = 20546 vs 13715 ng/mL) than vancomycin (10 mg/kg, iv), which served as a positive control in animal studies.
Table 3. Pharmacokinetic Studies of Compound 19 and Vancomycin in Male ICR Micea.
| compd | route | dose (mg/kg) | Cmax (ng/mL) | T1/2 (h) | CL (mL/min/kg) | Vss (L/kg) | AUC(0–24h) (ng/mL·h) |
|---|---|---|---|---|---|---|---|
| 19 | i.v. | 10 | 29667 | 10.3 | 6.9 | 4.9 | 20546 |
| vancomycin | i.v. | 10 | 58500 | 0.6 | 11.7 | 0.3 | 13715 |
Values indicate mean (n = 3, per time point). Abbreviations: half-life (T1/2); total body clearance (CL); volume of distribution at steady-state (Vss); area under the curve (AUC). Cmax was taken at t = 20 min.
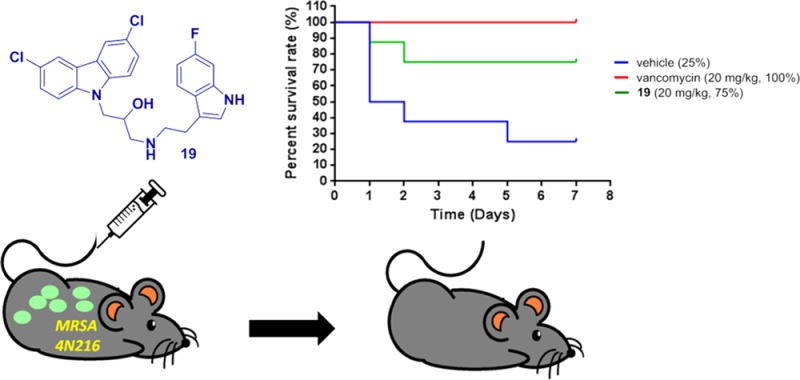
To evaluate the in vivo tolerability of the carbazole series, compounds 11 or 19 were intravenously administered at a dose of 100 mg/kg into ICR mice. Body weight was monitored for 2 weeks after the injection, and survival with increased body weight was observed in all animals with the treatment (Figure S1 in Supporting Information). These results, together with the pharmacokinetic properties provide strong evidence that these novel anti-MRSA carbazole series are well-tolerated in vivo. Encouraged by these promising data, compound 19 was then subjected to a MRSA (4N216) mouse systemic infection model. Mice were infected via intraperitoneal injection (ip) at 3 × 106 CFU/0.2 mL of MRSA strain 4N216 per mouse. Survival rate was calculated at 7 days postinfection. As depicted in Figure 2, following a single intravenous injection at a dose of 20 mg/kg (five times less than the amount used in the in vivo toxicity experiment) at 30 min postinfection, compound 19 could neutralize MRSA (4N216) infection in an effective manner, resulting in a 75% survival rate compared to vancomycin (100% survival rate) as a reference under the same treatment conditions. These results indicate that the newly developed hybrid series might have great potential to become a new generation of antimicrobial agents by further structural modifications especially using 19 as a template.
Figure 2.

Survival rate of 19 (20 mg/kg, iv) in a MRSA (methicillin-resistant Staphylococcus aureus) mouse systemic infection model. Five-week-old ICR female mice were infected via intraperitoneal injection (ip) at 3 × 106 CFU/0.2 mL of MRSA strain 4N216 per mouse (n = 8 mice/group). Survival rate was calculated at 7 days postinfection. Arrow denotes time of dosing at 30 min postinfection.
We have designed and synthesized a series of novel anti-MRSA agents through a molecular hybridization approach by combining a carbazole scaffold with a halogenated indole moiety. Among them, the representative compound 19 not only exhibited potent in vitro activities against various MSSA and MRSA clinical isolates, and VRE (ATCC 51299) strain with a MIC value as low as 0.5–1 μg/mL, but also maintained a 75% survival rate in a MRSA (4N216) mouse systemic infection model at a dose of 20 mg/kg following intravenous administration. Originally inspired by marine natural products possessing similar in vitro anti-MRSA activities to the marketed vancomycin, current findings provide a good starting point for further lead optimization. Although the precise mechanism of the newly developed hybrids remains unclear, however, they might possess a novel mechanism of action different from any known classes of antibiotics in light of their equipotent activities (MIC = 1 μg/mL) against a broad spectrum of tested MSSA, MRSA, and VRE strains.
Acknowledgments
We are grateful to the National Health Research Institutes and Ministry of Science and Technology of the Republic of China (MOST 103-2113-M-400-002-MY3) for financial support
Glossary
ABBREVIATIONS
- MRSA
Methicillin-resistant Staphylococcus aureus
- VRE
vancomycin-resistant enterococci
- MSSA
methicillin-susceptible S. aureus
- MIC
minimal inhibitory concentration
- SAR
structure–activity relationships
- AUC
area under the curve
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00289.
Synthetic procedures, spectral data, protocols of bioassay, and 1H and 13C NMR spectra of key compounds 19, 25 and 26; structure–activity relationships of compounds 1–26 against ATCC 29213 and ATCC 43300 strains; acute toxicity studies of compounds 11 and 19 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Walsh C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. 10.1038/35021219. [DOI] [PubMed] [Google Scholar]
- Davies J. Bacteria on the rampage. Nature 1996, 383, 219–220. 10.1038/383219a0. [DOI] [PubMed] [Google Scholar]
- Neu H. C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. 10.1126/science.257.5073.1064. [DOI] [PubMed] [Google Scholar]
- Piddock L. J. V. The crisis of no new antibiotics-what is the way forward?. Lancet Infect. Dis. 2012, 12, 249–253. 10.1016/S1473-3099(11)70316-4. [DOI] [PubMed] [Google Scholar]
- Levy S. B.; Marshall B. Antibacterial resistance worldwide: causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- Chambers H. F.; Deleo F. R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann H.; Aires-De-Sousa M.; Boyce J.; Tiemersma E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 2006, 368, 874–885. 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- Klevens R. M.; Morrison M. A.; Nadle J.; Petit S.; Gershman K.; Ray S.; Harrison L. H.; Lynfield R.; Dumyati G.; Townes J. M.; Craig A. S.; Zell E. R.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Fridkin S. K. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007, 298, 1763–1771. 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- Hersh A. L.; Chambers H. F.; Maselli J. H.; Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch. Intern. Med. 2008, 168, 1585–1591. 10.1001/archinte.168.14.1585. [DOI] [PubMed] [Google Scholar]
- Hope R.; Livermore D. M.; Brick G.; Lillie M.; Reynolds R. Non-susceptibility trends among staphylococci from bacteraemias in the UK and Ireland, 2001–06. J. Antimicrob. Chemother. 2008, 62, ii65–ii74. 10.1093/jac/dkn353. [DOI] [PubMed] [Google Scholar]
- Deurenberg R. H.; Stobberingh E. E. The evolution of Staphylococcus aureus. Infect., Genet. Evol. 2008, 8, 747–763. 10.1016/j.meegid.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Ito T.; Katayama Y.; Asada K.; Mori N.; Tsutsumimoto K.; Tiensasitorn C.; Hiramatsu K. Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1323–1336. 10.1128/AAC.45.5.1323-1336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhanel G. G.; Trapp S.; Gin A. S.; DeCorby M.; Lagacé-Wiens P. R.; Rubinstein E.; Hoban D. J.; Karlowsky J. A. Dalbavancin and telavancin: novel lipoglycopeptides for the treatment of Gram-positive infections. Expert Rev. Anti-Infect. Ther. 2008, 6, 67–81. 10.1586/14787210.6.1.67. [DOI] [PubMed] [Google Scholar]
- Bassetti M.; Merelli M.; Temperoni C.; Astilean A. New antibiotics for bad bugs: where are we?. Ann. Clin. Microbiol. Antimicrob. 2013, 12, 22–36. 10.1186/1476-0711-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel L. M.; Clewell D. B.; Gill S. R.; Clark N. C.; McDougal L. K.; Flannagan S. E.; Kolonay J. F.; Shetty J.; Killgore G. E.; Tenover F. C. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science 2003, 302, 1569–1571. 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- Furuno J. P.; Perencevich E. N.; Johnson J. A.; Wright M. O.; McGregor J. C.; Morris J. G.; Strauss S. M.; Roghman M. C.; Nemoy L. L.; Standiford H. C.; Hebden J. N.; Harris A. D. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci co-colonization. Emerging Infect. Dis. 2005, 11, 1539–1544. 10.3201/eid1110.050508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfee D. P. Methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci, and other Gram-positive in healthcare. Curr. Opin. Infect. Dis. 2012, 25, 385–394. 10.1097/QCO.0b013e3283553441. [DOI] [PubMed] [Google Scholar]
- Meyer E.; Ziegler R.; Mattner F.; Schwab F.; Gastmeier P.; Martin M. Increase of patients co-colonised or co-infected with methicillin-resistant Staphylococcus aureus, vancomycin-resistant Enterococcus faecium or extended-spectrum beta-lactamase-producing. Infection 2011, 39, 501–506. 10.1007/s15010-011-0154-0. [DOI] [PubMed] [Google Scholar]
- Huson M. A.; Kalkman R.; Remppis J.; Beyeme J. O.; Kraef C.; Schaumburg F.; Alabi A. S.; Grobusch M. P. Methicillin-resistant Staphylococcus aureus as a cause of invasive infections in Central Africa: a case report and review of the literature. Infection 2014, 42, 451–457. 10.1007/s15010-014-0589-1. [DOI] [PubMed] [Google Scholar]
- Muileboom J.; Hamilton M.; Parent K.; Makahnouk D.; Kirlew M.; Saginur R.; Lam F.; Kelly L. Community-associated methicillin-resistant Staphylococcus aureus in northwest Ontario: a five-year report of incidence and antibiotic resistance. Can. J. Infect. Dis. Med. Microbiol. 2013, 24, e42–e44. 10.1155/2013/169409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridkin S. K.; Hageman J. C.; Morrison M.; Sanza L. T.; Como-Sabetti K.; Jernigan J. A.; Harriman K.; Harrison L. H.; Lynfield R.; Farley M. M.; Surveillance A. B. C. Methicillin-resistant Staphylococcus aureus disease in three communities. N. Engl. J. Med. 2005, 352, 1436–1444. 10.1056/NEJMoa043252. [DOI] [PubMed] [Google Scholar]
- Moran G. J.; Krishnadasan A.; Gorwitz R. J.; Fosheim G. E.; McDougal L. K.; Carey R. B.; Talan D. A. Methicillin-resistant S. aureus infections among patients in the emergency department. N. Engl. J. Med. 2006, 355, 666–674. 10.1056/NEJMoa055356. [DOI] [PubMed] [Google Scholar]
- Calfee D. P. Crisis in hospital-acquired, healthcare-associated infections. Annu. Rev. Med. 2012, 63, 359–371. 10.1146/annurev-med-081210-144458. [DOI] [PubMed] [Google Scholar]
- Moran G. J.; Amii R. N.; Abrahamian F. M.; Talan D. A. Methicillin-resistant Staphylococcus aureus in community-acquired skin infections. Emerging Infect. Dis. 2005, 11, 928–930. 10.3201/eid1106.040641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horikawa M.; Noro T.; Kamei Y. In vitro anti-methicillin-resistant Staphylococcus aureus activity found in extracts of marine algae indigenous to the coastline of Japan. J. Antibiot. 1999, 52, 186–189. 10.7164/antibiotics.52.186. [DOI] [PubMed] [Google Scholar]
- Hamashima T.; Mori Y.; Sawada K.; Kasahara Y.; Murayama D.; Kamei Y.; Okuno H.; Yokoyama Y.; Suzuki H. A practical regioselective synthesis of alkylthio- or arylthioindoles without the use of smelly compounds such as thiols. Chem. Pharm. Bull. 2013, 61, 292–303. 10.1248/cpb.c12-00882. [DOI] [PubMed] [Google Scholar]
- Maneerat W.; Ritthiwigrom T.; Cheenpracha S.; Laphookhieo S. Carbazole alkaloids and coumarins from Clausena lansium roots. Phytochem. Lett. 2012, 5, 26–28. 10.1016/j.phytol.2011.08.013. [DOI] [Google Scholar]
- Maneerat W.; Phakhodee W.; Ritthiwigrom T.; Cheenpracha S.; Promgool T.; Yossathera K.; Deachathai S.; Laphookhieo S. Antibacterial carbazole alkaloids from Clausena harmandiana twigs. Fitoterapia 2012, 83, 1110–1114. 10.1016/j.fitote.2012.04.026. [DOI] [PubMed] [Google Scholar]
- Zhang F. F.; Gan L. L.; Zhou C. H. Synthesis, antibacterial and antifungal activities of some carbazole derivatives. Bioorg. Med. Chem. Lett. 2010, 20, 1881–1884. 10.1016/j.bmcl.2010.01.159. [DOI] [PubMed] [Google Scholar]
- Bashir M.; Bano A.; Ijaz A. S.; Chaudhary B. A. Recent developments and biological activities of N-substituted carbazole derivatives: A review. Molecules 2015, 20, 13496–13517. 10.3390/molecules200813496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saturnino C.; Palladino C.; Napoli M.; Sinicropi M. S.; Botta A.; Sala M.; Carcereri de Prati A.; Novellino E.; Suzuki H. Synthesis and biological evaluation of new N-alkylcarbazole derivatives as STAT3 inhibitors: Preliminary study. Eur. J. Med. Chem. 2013, 60, 112–119. 10.1016/j.ejmech.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Głuszyńska A. Biological potential of Carbazole derivatives. Eur. J. Med. Chem. 2015, 94, 405–426. 10.1016/j.ejmech.2015.02.059. [DOI] [PubMed] [Google Scholar]
- Ruffolo R. R. Jr.; Gellai M.; Hieble J. P.; Willette R. N.; Nichols A. J. The pharmacology of carvedilol. Eur. J. Clin. Pharmacol. 1990, 38, S82–S88. 10.1007/BF01409471. [DOI] [PubMed] [Google Scholar]
- Gu W.; Qiao C.; Wang S. F.; Hao Y.; Miao T. T. Synthesis and biological evaluation of novel N-substituted 1H-dibenzo[a,c]carbazole derivatives of dehydroabietic acid as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2014, 24, 328–331. 10.1016/j.bmcl.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Naidoo J.; De Jesus-Cortes H.; Huntington P.; Estill S.; Morlock L. K.; Starwalt R.; Mangano T. J.; Williams N. S.; Pieper A. A.; Ready J. M. Discovery of a neuroprotective chemical, (S)-N-(3-(3,6-Dibromo-9H-carbazol-9-yl)-2-fluoropropyl)-6-methoxypyridin-2-amine [(−)-P7C3-S243], with improved druglike properties. J. Med. Chem. 2014, 57, 3746–3754. 10.1021/jm401919s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley K. A.; Heinrich V. A.; Hershfield J. R.; Demons S. T.; Weibel D. B. Membrane-targeting DCAP analogues with broad-spectrum antibiotic activity against pathogenic bacteria. ACS Med. Chem. Lett. 2015, 6, 466–471. 10.1021/acsmedchemlett.5b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.; Bayer A.; Cosgrove S. E.; Daum R. S.; Fridkin S. K.; Gorwitz R. J.; Kaplan S. L.; Karchmer A. W.; Levine D. P.; Murray B. E. J.; Rybak M.; Talan D. A.; Chambers H. F. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children: executive summary. Clin. Infect. Dis. 2011, 52, 285–292. 10.1093/cid/cir034. [DOI] [PubMed] [Google Scholar]
- Chen F. J.; Lauderdale T. L.; Wang L. S.; Huang I. W. Complete genome sequence of Staphylococcus aureus Z172, a vancomycin-Intermediate and daptomycin-nonsusceptible methicillin-resistant strain isolated in Taiwan. Genome Announc. 2013, 1, e01011. 10.1128/genomeA.01011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Pai R.; Di M.; Aiello D.; Barnes M. H.; Butler M. M.; Tashjian T. F.; Peet N. P.; Bowlin T. L.; Moir D. T. Coumarin-based inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicase: chemical optimization, biological evaluation, and antibacterial activities. J. Med. Chem. 2012, 55, 10896–10908. 10.1021/jm300922h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.