

Abstract
Self-assembled polymer/porous silicon nanocomposites overcome intracellular and systemic barriers for in vivo application of peptide nucleic acid (PNA) anti-microRNA therapeutics. Porous silicon (PSi) is leveraged as a biodegradable scaffold with high drug cargo loading capacity. Functionalization with a diblock polymer improves PSi nanoparticle colloidal stability, in vivo pharmacokinetics, and intracellular bioavailability through endosomal escape, enabling PNA to inhibit miR-122 in vivo.
Keywords: PNA, peptide nucleic acids, porous silicon, anti-miRNA, nanocomposite
Graphical abstract
Peptide nucleic acid (PNA) DNA mimics are promising drug candidates for modifying gene expression in diseased cells because their synthetic, neutrally-charged amide backbone imparts increased stability, nuclease resistance, and binding affinity for complimentary nucleic acid targets. [1, 2] One intriguing therapeutic application of PNA is for the competitive binding and inhibition of micro-RNAs (miRNAs), which are endogenous negative regulators of gene expression.[2–4, 5, 6] Inhibition of disease-associated miRNA is a potentially potent therapeutic strategy because a single miRNA can control the expression of hundreds of different genes.[7] PNA have been successfully delivered in vitro using approaches such as fusion with cell penetrating peptides or lipophilic moieties,[8] but the clinical translation of PNA anti-miRNA therapeutics is limited by physiologic delivery barriers, including poor cellular uptake, lack of endosomal escape, and rapid clearance following intravenous delivery (<5 min blood circulation half-life).[9] While the neutrally-charged PNA backbone can provide a functional advantage, it obviates the use of conventional nucleic acid delivery strategies that rely on charge, such as formulation with cationic polymers and lipids to form nanomedicines. To overcome these delivery barriers and formulation challenges, we have developed polymer/porous silicon nanocomposites with high cargo loading capacity independent of electrostatics, stability in the blood based on surface PEGylation, and endosomal escape functionality to facilitate PNA delivery to the cytoplasm where miRNA targets are located.[10]
Porous silicon is a biocompatible, biodegradable material with a large internal surface area (>100 m2 cm−3) for drug loading.[11] We previously demonstrated that packaging PNA into “naked” porous silicon nanoparticles (PSNPs) improves the cellular uptake and therapeutic activity of PNA in vitro.[3] After internalization, however, porous silicon cannot efficiently trigger release of PNA from endo-lysosomal vesicles into the cytoplasm,[12] and we have observed that PSNPs are not colloidally stable at high concentrations in saline solutions amenable to intravenous administration. One PSNP modification strategy to enable endosomal escape, developed by the Santos group, is to covalently attach a zwitterionic copolymer of polyethyleneimine (PEI) and poly(methyl vinyl ether-co-maleic acid) (PMVE-MA) to the porous silicon surface.[12] PEI, which is debated to mediate endosome escape via a proton sponge mechanism,[13] was shown to improve endosomal escape and therapeutic activity of the small molecule drug methotrexate in these studies, though not larger biologics. The PMVE-MA-based particles have a highly negative zeta potential (−31.7 ±1.7mV) which is associated with poor in vivo pharmacokinetics relative to surface charge neutral nanoparticles.[14] Furthermore, PMVE-MA activates the innate immune system through both toll like receptor and complement activation pathways and thus is not an ideal material for systemic therapeutic applications.[15] Nanoprecipitation-based coating of hydrophobic porous silicon nanoparticles with amphiphilic block-copolymers containing poly(histidine) has also been explored for pH-triggered release of small molecules. However, this approach has not been utilized to deliver intracellular-acting biologics and its potential endosomal-disrupting function has not been explored.[16]
Our group recently developed an endosomolytic polymer poly[(ethylene glycol) – block – (2-(dimethylamino)ethyl methacrylate – co – butyl methacrylate)] (PEGDB), and tuned it to have optimal endosomal escape properties for systemic delivery of negatively-charged nucleic acids.[17] PEGDB consists of a hydrophilic PEG block and a second block comprising 50:50 mol% of cationic and pH-responsive 2-(dimethylamino)ethyl methacrylate (DMAEMA; D) and hydrophobic butyl methacrylate (BMA; B). The resulting polymer is finely tuned to disrupt membranes in acidic environments representative of early (pH 6.8) and late endosomes (pH 6.2) but not at physiologic pH (pH 7.4).[17] Neutrally charged PNA cannot be electrostatically loaded into PEGDB polyplexes. Here, we explored oxidized, PNA-loaded PSNPs with a highly negative surface charge as “scaffolds” for PEGDB electrostatic surface coating. We demonstrate that this approach yields surface charge neutral composite NPs with enhanced stability, improved pharmacokinetics, and increased cytosolic delivery and bioactivity of PNA relative to PSNPs without a polymeric coating.
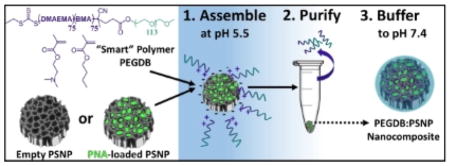
Scheme 1 depicts the strategy for PSNP-polymer nanocomposite formation. PSNPs were fabricated by ultrasonic fracture of multi-layer porous silicon films and then oxidized in H2O2 at 115ºC for 4 hours. The PEGDB diblock polymer was synthesized based on previous work by Nelson et al., and comprised a 5kDa PEG block and a BMA/DMAEMA copolymer block with approximately 50 mol% of each monomer (Supplementary Figure 1).[17] In deionized water at pH 5.5, the surfaces of oxidized PSNPs are negatively charged (ζ = −20 mV), while the tertiary amines on DMAEMA (pKa ~ 11) are predominantly protonated, making PEGDB positively charged and unimeric in solution. Composite particles were formed electrostatically by mixing the two components together in de-ionized H2O (pH 5.5) while stirring at room temperature for 30 min. Coated PSNPs were purified from excess polymer by centrifugally pelleting the PSNP-polymer composites and removing the supernatant containing free, unbound polymer. The particle pellet was then re-suspended in phosphate buffered saline at physiologic pH (pH 7.4). Exposure of the DB polymer block to physiologic pH makes it water-insoluble and triggers polymer self-aggregation,[17] potentially contributing to hydrophobic stabilization of the surface coating and stabilization of the drug loading into the PSNP interior pores. At physiologic pH, addition of PEGDB neutralizes negative PSNP surface charge (Scheme 1), which is important for systemic delivery applications. [14]
Scheme 1.

PSi-polymer nanocomposite fabrication. 1. Electrostatic assembly of PEGDB on the negatively charged PSNP surface. 2. Purification of nanocomposites by removal of free PEGDB in the supernatant following centrifugation. 3. Buffering the nanoparticle solution to physiologic pH increases polymer self-aggregation, potentially contributing to hydrophobic stabilization of the surface coating and drug loading into the PSNP interior pores. The bottom panel shows surface charge of oxidized PSNPs, PEGDB, and nanocomposites during assembly at pH 5.5, and after buffering pH to 7.4.
The ratio of PEGDB polymer to porous silicon was tuned to create a library of nanocomposites with varied degrees of polymer surface functionalization (Figure 1). Reactant weight ratios of 1:5, 1:1, 20:1, and 80:1 (PEGDB:PSNP) yield composite particles containing 12, 24, 50, and 60 wt% PEGDB, respectively, as determined by thermal gravimetric analysis (TGA) (Figure 1A). Dynamic light scattering (DLS) measurements reveal that as % PEGDB is increased, the PSNP ζ-potential at physiologic pH (7.4) increases and approaches charge neutrality (Figure 1B). The average hydrodynamic diameter of nanocomposites with up to 24 wt% PEGDB is the same as that of uncoated PSNPs (220nm) (Figure 1C). Scanning transmission electron microscopy-energy-dispersive X-ray spectrometry (STEM-EDS) analysis of nanocomposites confirms that nitrogen and sulfur signals generated by PEGDB co-localize with the Si matrix at the nano-scale, supporting our hypothesis that electrostatic interactions facilitate PEGDB assembly onto the PSNP surface (Figure 1D and Supplementary Figure 2). Spectroscopic evaluation of PSNP colloidal stability reveals that coating of PSNPs with PEGDB minimizes particle aggregation and precipitation in the presence of salt-containing physiologic buffered saline (Figure 1E–F). This increased stability can be attributed to PEG’s ability to sterically block surface adsorption of proteins and ions, consequently preventing particle aggregation.[18]
Figure 1.

PEGDB effectively coats PSNPs, neutralizing particle surface charge and imparting colloidal stability within physiologic buffered saline. Characterization of nanocomposite library by (A) thermal gravimetric analysis, (B) ζ-potential, and (C) hydrodynamic size measurements acquired in PBS at pH 7.4. (D) STEM-EDS elemental mapping of (top) an uncoated PSNP and (bottom) composite PSNPs coated with 24% PEGDB. The increased strength and dispersion of N and S signals in elemental maps of composite particles (bottom) indicates successful PEGDB coating of the PSNP matrix. High-angle annular dark-field (HAADF) images are shown in the left-most panels. Maps of Si, N, and S are indicated in red, teal, and yellow, respectively. Scale bar = 200 nm. (E) Particle aggregation and precipitation out of solution, quantified by monitoring PSNP absorption at 450nm over time, shows that fully coated 50% PEGDB nanocomposites have increased colloidal stability in PBS. (F) Photographs depict PSNP and composite colloidal stability in PBS after 60 min. Black arrow indicates precipitated PSNPs.
To assess the impact of PEGDB coating density on PSNP uptake and anti-miRNA activity, composites with the minimum amount of PEGDB necessary to fully shield the porous silicon surface (50 wt%, zeta −3.1 ± 4.0 mV), and composites which were only partially shielded by PEGDB (24 wt%, zeta −8.6 ± 3.0 mV) were selected for further in vitro characterization. Cell internalization and bioactivity was assessed for a PNA designed to inhibit miR-122, a liver-specific miRNA involved in cholesterol biosynthesis. Inhibition of miR-122 is a promising therapeutic approach for reducing viremia in patients infected with Hepatitis C, as well as lowering elevated cholesterol and triglyceride levels due to hypercholesterolemia.[19] In this study, nanocomposites were loaded with anti-miR122 PNA (NH2-ACA AAC ACC ATT GTC ACA CTC CA-cys-COOH) by physically adsorbing PNA within oxidized PSNPs, followed by coating with PEGDB as described above. The average PNA loading in the nanoparticle formulations was quantified by LCMS to be 34, 22, and 20 nmoles PNA per mg porous silicon (21, 14, and 12 wt% PNA) for uncoated, partially coated, and fully coated nanocomposites, respectively (Supplemental Figure 3). To our knowledge, this is the highest reported PNA loading in any nanoparticle system, and is 60× higher than what has been reported for anti-miRNA PNA loading in PLGA nanoparticles,[5] highlighting an important advantage of highly porous PSNPs that do not require emulsion fabrication/loading approaches.
Cellular uptake and miR-122 inhibition studies were performed in vitro using Huh7 human liver cancer cells (Figure 2). Cells were treated for 24 h in Dubelcco’s modified eagle medium (DMEM, Gibco Cell Culture, Carlsbad, CA) supplemented with 10% fetal bovine serum, at a 2μM dose of PNA. Following treatment, uptake of fluorescently-labeled PNA was quantified by flow cytometry and imaged by confocal microscopy. PNA encapsulated in uncoated PSNPs is ~50× more efficiently internalized than free PNA (Figure 2A). Additionally, PNA uptake decreases proportionately with increasing wt% PEGDB (Figure 2A and 2B). This is likely due to PEG shielding on the outer surface of the composite.[20] Importantly, the extent of cytosolic PNA delivery increased with increasing PEGDB content, as quantified by co-localization analysis of PNA and lysosomes stained with LysoTracker®, 24 h after treatment (Figure 2C). This enhanced cytosolic PNA delivery can be attributed to both full and partial PEGDB nanocomposites possessing pH-dependent, membrane disruptive activity in a relevant endo-lysosomal range, whereas uncoated PSNPs do not (Figure 2D).
Figure 2.

Coating of PSNPs with PEGDB decreases PNA uptake but increases both endosome escape potential and anti-miRNA activity relative to uncoated PSNPs in Huh7 human hepatocellular carcinoma cells. PEGDB functionalization decreases cellular uptake, as characterized by (A) flow cytometry and (B) confocal microscopy, 24 h after treatment with Alexa Fluor 488–labeled anti-miR122 PNA at a 2 μM PNA dose. Top scale bar = 100 μm, bottom scale bar = 10 μm. (C) PEGDB functionalization increases PNA cytosolic delivery, as shown by colocalization analysis of Alexa Fluor 488–labeled PNA with LysoTracker at 24 h after treatment with 2 μM PNA. Endosomal entrapment was quantified by calculating the Manders’ overlap coefficients for green and red pixels, shown at the right as means ± SEM (n ≥ 3 separate images). Increased cytosolic delivery observed for composite particles is due to (D) the pH dependent membrane disruptive function (grey arrow) of PEGDB, as determined by a hemolysis assay. Composites did not disrupt erythrocyte membranes at pH 7.4, but produced robust hemolysis at pH 6.2, which is representative of late endosomes. (E) A firefly luciferase assay reveals that all PSNP treatments are non-toxic at a 2 μM PNA dose, in contrast to the gold-standard of a 2’OMe modified RNA delivered using a commercial cationic transfection reagent (AMO+Fugene6). (F) Therapeutic anti-miR122 activity increases with increasing PEGDB polymer functionalization (based on renilla luciferase readout tied directly to miR-122 inhibition) 24 hours after treatment, when compared to free, unencapsulated PNA and the control, 2’OMe AMO. (p<0.05 when compared to *Free PNA or PSNP, **AMO+Fugene6, and ***Partial Comp).
Anti-miR-122 activity was quantified using Huh7 cells stably transfected with a Renilla luciferase sensor for endogenous miR-122.[21] Inhibition of miR-122 in these cells causes an increase in luciferase signal. Both anti-miRNA activity and cytotoxicity were benchmarked against the anti-miRNA oligonucleotide (AMO) agent used in development and validation of this luciferase reporter cell line: 2’OMe PS modified RNA delivered with the cationic commercial transfection reagent, FuGENE® 6. All PSNP treatments cause significantly less cytotoxicity than FuGENE® 6 (Figure 2E), which is too toxic (and colloidally instable) for in vivo translation. Although cell uptake was reduced (Figure 2A), anti-miR-122 activity was 6-fold and 10-fold greater than uncoated PSNPs for the partial and fully coated nanocomposites, respectively (Figure 2F). Furthermore, fully coated nanocomposite PSNPs demonstrate 2.3 fold higher miR-122 inhibition relative to the AMO standard. Taken together, these data suggest that fully coated composites are non-toxic and have potent anti-miRNA activity due to increased delivery of PNA to the cytosol, where miR-122 is located.
To evaluate whether nanocomposites improve the blood circulation half-life and miR inhibitory bioactivity of PNA in vivo, CD-1 mice (10 weeks of age, Charles River) were injected intravenously via the tail vein with 1 mg kg−1 cy5-labeled anti-miR122 PNA (free, loaded into uncoated PSNPS or fully coated nanocomposites) (Figure 3). Blood samples were collected 5, 10, 40, and 80 minutes after injection, and circulation half-life was determined based on the quantity of PNA in the plasma collected at each time point (Figure 3A). Uncoated PSNPs extended the circulation half-life of free PNA from <1 min to ~30min, and addition of the PEGDB coating to PSNPs more than doubled the half-life to nearly 70 min. As a result of increased circulation time, encapsulation of PNA anti-miR-122 in nanocomposites increased its bioavailability by 73×, as quantified by the area under the curve (AUC).
Figure 3.

PSNP-polymer nanocomposites increase PNA blood circulation half-life, bioavailability, and anti-miRNA activity in vivo. (A) Blood pharmacokinetics curves generated using cy5-labeled PNA show that PSNPs increase circulation half-life of PNA when delivered I.V. through the tail vein of mice at a 1 mg kg−1 dose (n=8 per group for 0–20 min, n=5 per group for 40–80 min). (B) In vivo biodistribution of cy5-PNA cargo and Si from the PSNP carriers was analyzed by fluorescent imaging and ICP-OES, respectively. PNA and Si organ distributions 160 min after injection show that PSNPs increase PNA accumulation in the liver and decrease uptake in the kidneys. (C) Quantification of bioavailability in blood, liver, and lungs demonstrates that PEGDB functionalization improves blood circulation stability and decreases particle lung accumulation. (D–H) In vivo miR-122 inhibition studies following injection of a 5 mg kg-1 dose of PNA, every other day for 6 days (n=6 mice per group). (D) On day 6, livers were formalin fixed and paraffin embedded, and stained with H&E. Livers were then evaluated by an experienced veterinary pathologist blinded to the composition of the groups, who found no evidence of liver toxicity observed microscopically (representative image shown, n=6 mice per group). (E) PCR of RNA extracted from livers on day 6 reveals that nanocomposite delivery of anti-miR122 PNA (D) significantly inhibits miR-122 and (F) modulates the expression of miR-122 direct target genes, Aldoa and Gys1, in addition to the indirect target gene MTTP. (Grey line indicates saline control). Cholesterol measurements on (G) high-density lipoprotein (HDL) and (H) low-density lipoprotein (LDL) fractions separated by FPLC from plasma collected on day 6 reveals decreased cholesterol in HDL following treatment with nanocomposites loaded with anti-miR-122 PNA. (*, **, and *** indicate p<0.05 when compared to free PNA, PSNPs, and empty vehicle control, respectively).
The organ biodistribution of PNA and the PSNP carrier were determined by excising the heart, lungs, liver, spleen, and kidneys 160 min after injection. Investigations into novel nucleic acid delivery systems typically track the fluorescently-labeled nucleic acid without tracking the carrier system. An advantage of using porous silicon nanocarriers is that their biodistribution can be tracked label-free using inductively coupled plasma-optical emission spectroscopy (ICP-OES). Figure 3B compares the biodistribution of fluorescently labeled PNA cargo with that of Si from the porous silicon nanocarriers. The biodistribution of Si is similar to that of the PNA for both uncoated and composite nanoparticles, suggesting that the PNA cargo may remain stably associated with the porous silicon carrier in the circulation and during initial tissue biodistribution. It is well established that nanoparticles larger than 200 nm in size are preferentially sequestered in the liver sinusoidal endothelium.[22] Due to their size, PSNPs and nanocomposites increased PNA delivery to the liver (where hepatocytes containing the target miR-122 are located) by ~16%, and reduced the amount of PNA in the kidneys by ~33% when compared with free PNA (Figure 3B). ICP-OES analysis reveals a ~20% increase in Si in the spleen of composite particles when compared to uncoated particles. The spleen, like the liver, is known to play a primary role in nanoparticle clearance. Additionally, PEGDB coated nanoparticles display 12% less Si accumulation in lungs compared with uncoated PSNPs 160 min after injection, and 29% less lung bioavailability over 24 h (Figure 3 and Supplementary Figure 4), corroborating in vitro data (Figure 1E–F) that PEGDB reduces flocculation which causes accumulation in the lungs.[23] A final observation is that while ~20% of all PNA is found in the kidneys, no more than 6% of all measured Si is detected in the kidneys by ICP-OES. Free PNA pharmacokinetics data suggests that the kidney is a preferential route of clearance, which is anticipated based on the small size of free PNA, below the renal cutoff. The difference in PNA and Si content suggests that a portion of the PNA cargo released from the PSNP carriers in circulation due to the simple, noncovalent PNA loading mechanism.
We next tested whether PSNP-polymer nanocomposites improve PNA bioactivity (miR-122 inhibition) in the liver. Female C57BL/6J mice (12 weeks of age, Jackson Laboratories) were treated with either saline, free anti-miR122 PNA (5 mg kg−1 PNA), PNA loaded into composite PSNPs (5 mg kg−1 PNA, 42 mg kg−1 PSNP composite), or an empty composite vehicle control (42 mg kg−1 PSNP composite). Importantly, the uncoated nanoparticles demonstrated poor colloidal stability in physiologic solutions (Figure 1E–F), and consequently we were unable to safely inject uncoated PSNPs intravenously at 5 mg kg−1 PNA dose for this study due to acute mortality. The acute toxicity for the 5 mg kg−1 uncoated treatment group could be due to particle aggregation resulting in blockage of pulmonary capillaries. This result highlights the importance of the colloidal stabilization of PSNPs by PEGDB, which significantly reduced particle accumulation in the lungs (Figure 3C and Supplementary Figure S4).
Mice were injected intravenously into the tail vein every other day for 6 days (3 injections), then sacrificed, and their livers were harvested for mRNA, and miRNA analysis (Figure 3D–F). There were no overt signs of toxicity for the composite nanoparticles, and levels of blood urea nitrogen (BUN) and alanine aminotransferase (ALT) measured on day 6 serum samples collected at the time of mouse sacrifice suggested that treatment did not cause kidney or liver toxicity relative to control treatment groups (Figure 3D and Supplemental Figure 5). Real-time PCR for miR-122 expression shows that composite PSNPs inhibited miR-122 by 46% relative to the empty vehicle control (Figure 3E). Additionally, treatment with nanocomposites caused an ~50% increase in expression of both Aldolase A (Aldoa) and Glycogen Synthase 1 (Gys1), validated miR-122 gene targets that encode for proteins which degrade cholesterol and synthesize glycogen, respectively (Figure 3F). Furthermore, expression of Microsomal Triglyceride Transfer Protein (MTTP), an indirect target of miR-122 known to be down-regulated upon miR-122 inhibition, was decreased by 36% relative to the vehicle control (Figure 3F).[24] Finally, the functional impact of miR-122 inhibition was assessed by analyzing the cholesterol content in the high-density lipoprotein (HDL) and low-density lipoprotein (LDL) serum fractions collected from mice on day 6 (Figure 3G–H). Consistent with the known function of miR-122,[19] inhibition of miR-122 using PNA-loaded nanocomposites caused an ~20% decrease in HDL cholesterol. This is, to our knowledge, the first demonstration of in vivo PNA-mediated miR-122 inhibition, and the nanocomposite PNA delivery technology exhibits miR-122 inhibition at 16× lower dose than RNA-based anti-miR122 antagomirs,[25] and 2.5× lower dose than a 2-O-methoxyethyl phosphorothioate antisense oligos.[19]
Systemic and intracellular pharmacokinetics limitations are the biggest challenges facing PNA-based therapeutics. Our in vitro and in vivo results confirm that free PNA suffers both from poor cellular internalization, poor systemic pharmacokinetics, and lack of intracellular bioavailability. Our in vitro data support the importance of overcoming intracellular delivery barriers. Despite a significantly higher level of cell uptake, the uncoated PSNP carriers produced significantly lower miRNA inhibition than composite particles with active endosomal escape capacity (Figure 2). Furthermore, the nanocomposite showed superior blood circulation time and systemic bioavailability (Figure 3) due to particle colloidal stabilization with PEG. Finally, PEGDB polymer coating enabled I.V. delivery of a 5× higher PNA dose than uncoated PSNPs. At this PNA dose (5 mg kg−1), composite particles successfully inhibited miR-122 in the liver, de-repressed the miR-122 target gene, Aldoa, and lowered plasma cholesterol levels. Thus, the nanocomposite design was strategically crafted to overcome both the key systemic and intracellular delivery barriers facing PNA.
Consequently, this is, to our knowledge, only the fourth report of in vivo miRNA silencing with a PNA therapeutic.[5, 6, 26] and the first report of an endosomolytic PNA delivery system. All other reports have focused on inhibition of miR-155 for treatment of lymphoma. Initial work by Fabani et al. modified PNA with positively-charged lysine residues and saw effective miR-155 inhibition at a 50 mg kg−1 PNA dose. In the study by Babar et al., PNA encapsulation into PLGA nanoparticles coated with a cell-penetrating peptide (0.35 wt% PNA loading) reduced the effective PNA dose to 1.5 mg kg−1. Cheng et. al. conjugated PNA to a pH-responsive peptide which enables targeted delivery of anti-miRNA PNA to the acidic tumor microenvironment. Additionally, the peptide utilizes an environmentally-activated non-endocytic cell uptake/membrane transduction mechanism. This enabled miR-155 inhibition at a 1 mg kg−1 dose in a mouse model of lymphoma. Our non-targeted PNA delivery system beat or approached the level of miRNA inhibitory potency of these different targeted delivery approaches, and we hypothesize that this new long-circulating nanocomposite can be made more potent by adding targeting ligands, such as GalNAc for hepatocyte targeting.[27]
In summary, this report showcases a new nanocomposite PNA delivery vehicle proven to overcome both systemic and intracellular delivery barriers. Porous silicon is leveraged as a highly porous scaffold amenable to high drug cargo loading; this substrate enables much higher and simpler drug loading relative to water in oil in water (W/O/W) emulsion methods commonly utilized to load hydrophilic cargo into hydrophobic polymer-based nano- and micro- carriers. Furthermore, simple electrostatic assembly was utilized for PSNP surface functionalization with a multifunctional polymer that enhances resultant particle colloidal stability, in vivo circulation, and intracellular bioavailability. This fabrication process is both facile and rapid, requiring only sequential centrifugal wash steps for composite purification. This approach would also be potentially amenable to adaptation to more controlled microfluidic synthesis techniques, such as those developed by Santos et. al.[6, 28] Furthermore, it is well-established that porous silicon can host a wide range of cargos.[29] Thus, we anticipate that this composite system can facilitate the intracellular activity of a broad range of therapeutic and diagnostic payloads. Finally, based on its high bioavailability and long blood circulation half-life, this system can also potentially be functionalized with active targeting ligands and further tuned to facilitate preferential delivery to defined cells and tissues. Thus, PSNP-polymer nanocomposites represent a promising material platform with potential high impact in miR inhibitory and other biologic nanomedicines.
Experimental
Detailed experimental methods can be found in the supporting information.
Nanocomposite Fabrication and Characterization
Porous silicon nanoparticles (PSNPs) were formed by ultrasonic fracture of porous silicon multilayers, followed by oxidation in H2O2 at 115°C for 4 hours. Oxidized particles were centrifugally washed 3× by pelleting at 14.1 × g for 15 min and replacing the supernatant with fresh ethanol. Following the final wash step, PSNPs were purified from large debris by centrifuging at 300 × g and collecting the smaller nanoparticles in the supernatant.
The surface of oxidized porous silicon is negatively charged (ζ= −20mV) at pH 5.5. At that pH, the PEG-(DMAEMA-co-BMA) is positively charged and largely unimeric in solution. Composite particles were formed electrostatically by mixing the two components together in de-ionized H2O (pH 5.5) while stirring at RT for 30 min, and the pH was subsequently raised to 7.4 using PBS. Coated PSNPs were purified from excess polymer by pelleting under centrifugation and removing the supernatant. The relative amounts of PEGDB and porous silicon within the composites were quantified by thermal gravimetric analysis (TGA). DLS and zeta potential measurements were performed using a Zetasizer NanoZS (Malvern Instruments, Worcestershire, UK) to assess changes in particle size and surface charge following polymer coating. Lastly, coated particle morphology and elemental composition were characterized by scanning transmission electron microscopy-energy-dispersive X-ray spectrometry (STEM-EDS), using a Tecnai Osiris microscope (FEI, Oregon, USA) at a 120 keV accelerating voltage.
PNA Synthesis and Loading
Cysteine-modified anti-miR122 PNA (NH2-ACA AAC ACC ATT GTC ACA CTC CA-cys-COOH) was synthesized from Rink Amide LL resin (EMD Millipore) using fluorenylmethyloxycarbonyl chloride (Fmoc) solid phase chemistry within a PS3 automated peptide synthesizer (Protein Technologies). Note that all PNA used in this study was modified by addition of a single cysteine at the C-terminus of the PNA. This was done to promote crosslinking of PNA following loading into PSNPs and reduce PNA diffusion from pores. PNA was purified by reverse-phase high-pressure liquid chromatography.
PNA was loaded into uncoated and composite PSNPs by non-covalent physical adsorption. Composite PSNPs were impregnated with PNA prior to polymer coating. PNA dissolved in deionized H2O was added to a solution of oxidized PSNPs in H2O at a 1:2 PNA:PSNP weight ratio. The PNA/PSNP solution was briefly ultrasonicated and then mixed on a shaker at room temperature for 3 hours. Finally, PNA-loaded PSNPs were frozen at −80°C and lyophilized overnight. Excess PNA was purified from drug loaded PSNPs by resuspending particles in H2O, centrifuging particles at 15k × g for 15 min, then removing the supernatant.
Cell Culture and In Vitro Bioactivity
Human hepatocellular carcinoma cells (Huh7) were cultured in Dubelcco’s modified eagle medium supplemented with 10% fetal bovine serum, 2% penicillin/streptomycin, and 2μg mL−1 ciprofloxacin. To characterize cellular uptake by confocal microscopy and flow cytometry, Huh7 cells were treated for 24 h with either free Oregon Green-labeled PNA, or labeled PNA loaded into uncoated, partially coated, and fully coated composite PSNPs at a 2μM PNA dose.
Huh7-psiCHECK-miR122 luciferase reporter cells were used to assay for anti-miR122 activity.[21] Cells were seeded at 3,000 cells per well in 96-well black-walled plates and allowed to adhere overnight. Cells were then treated in 10% serum for 24 hours at a 2 μM PNA dose. Luciferase expression was measured in triplicate using a Dual Luciferase Assay Kit (Promega Corporation, Madison, WI) according to the manufacturer’s protocol, and the luminescence was recorded an IVIS Lumina III imaging system (Xenogen Corporation, Alameda, CA, USA). Relative luciferase expression was calculated as the average ratio of Renilla to firefly luciferase expression for each of the three triplicates. Cytotoxicity was calculated from the constitutively expressed firefly luciferase luminescence.
In Vivo Pharmacokinetics and Biodistribution
CD-1 mice (10 weeks of age; Charles River) were injected intravenously into the tail vein with cy5-labeled anti-miR122. Retro-orbital blood collection was performed at 5 and 10 min after injection (n=8 per treatment group). In a second cohort of mice (n=5 per treatment group), retro-orbital blood collection was performed at 40 and 80 min after injection. Circulation half-life was determined from the amount of drug in the plasma collected retro-orbitally at 5, 10, 40 and 80 min.
Mice were euthanized 1 min, 20 min, 160 min, and 24 h after injection and organs were harvested for biodistribution analysis. An IVIS Lumina III imaging system was used to quantify Cy5 fluorescence in explanted lungs, heart, liver, kidney, and spleen using Living Image 4.4 quantification software. For Si content analysis, organs were weighed, homogenized in 3 mL of 20% ethanol in 1N NaOH and left for 48 h at room temperature for extraction of Si. Organ homogenate was then centrifuged at 42000 × g for 25 min. Following centrifugation, 1 mL of the supernatant was removed and diluted to 10 mL using de-ionized H2O. Finally, all samples were syringe filtered using a 0.2μm syringe filter and analyzed using inductively coupled plasma-optical emission spectroscopy (ICP-OES).
In Vivo miR-122 Inhibition
Female C57BL/6J mice (12 weeks age, Jackson Laboratories) were divided into the following 6 treatment groups (6 mice per group): saline, empty composite PSNPs, free anti-miR122 PNA (5 mg kg−1 PNA), PNA loaded into uncoated PSNPs (1 mg kg−1 PNA), or PNA loaded into composite PSNPs (1 and 5 mg kg−1 PNA). Mice were injected intravenously into the tail vein every other day for 6 days (3 injections). Mice injected with 5 mg kg−1 PNA in uncoated PSNPs experienced rapid mortality in pilot studies, and thus this group was omitted in the experimental design. Mice were sacrificed on day 6 and the livers were harvested for mRNA, miRNA, and toxicological analysis.
Blood taken from cardiac puncture on day 6 was submitted for toxicological analysis. Plasma collected from cardiac puncture on day 6 was used to analyze mouse plasma cholesterol. Mouse plasma was injected into an ®KTA Pure FPLC using a Superdex 200 increase 10/300 column (Amersham BioSciences). HDL and LDL fractions were pooled separately, and concentrated using Amicon Ultra 15 mL centrifugal filters with a MW cut-off of 3kDa. Total cholesterol in the concentrated HDL or LDL fractions was quantified using a Wako Kit, as per manufacturer’s instructions, and normalized to total protein content using Pierce BCA Protein Assay kit. Results are presented relative to cholesterol content in the HDL and LDL fractions of mice injected with saline only.
Mice were fed a standard chow diet ad libitum and had free access to water. All protocols were approved by the Institutional Animal Care and Use Committee of Vanderbilt University and done in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Ms. Rossane Delapp and Dr. David Kosson in Vanderbilt’s Department of Civil and Environmental Engineering for ICP-OES analysis of silicon content. The authors would also like to thank Dr. Alexander Deiters for kindly providing the luciferase reporter cell lines used in this study. This work was supported in part by the National Science Foundation (DMR-120701), the Department of Defense (DOD PRORP OR130302), the National Institutes of Health (R01 EB019409), and NSF Graduate Research Fellowships to KRB, TAW, TEK, KVK, and JSF. Confocal Imaging was performed in part through the use of the VUMC Cell Imaging Shared Resource, (supported by NIH grants CA68485, DK20593, DK58404, HD15052, DK59637 and Ey008126). DLS, TGA, and STEM-EDS were performed in the Vanderbilt Institute of Nanoscale Science and Engineering (VINSE). Financial support for STEM-EDS imaging was provided by NSF EPS 1004083. The authors also thank Daniel Balikov, John Martin, and Samantha Sarett for editing the manuscript.
Footnotes
Supporting Information is available online from Wiley InterScience or from the author.
Contributor Information
Kelsey R. Beavers, Interdisciplinary Graduate Program in Materials Science, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Thomas A. Werfel, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Tianwei Shen, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235 (USA).
Taylor E. Kavanaugh, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Kameron V. Kilchrist, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Dr. Jeremy W. Mares, Department of Electrical Engineering and Computer Science, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Joshua S. Fain, Department of Electrical Engineering and Computer Science, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Carrie B. Wiese, Department of Molecular Physiology & Biophysics, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Dr. Kasey C. Vickers, Department of Medicine/Division of Cardiovascular Medicine, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Dr. Sharon M. Weiss, Department of Electrical Engineering and Computer Science, Vanderbilt University, Nashville, Tennessee 37235 (USA)
Dr. Craig L. Duvall, Email: craig.duvall@vanderbilt.edu, Department of Biomedical Engineering, Vanderbilt University, Nashville, Tennessee 37235 (USA)
References
- 1.Nielsen PE. Pharmacol Toxicol. 2000;86:3. doi: 10.1034/j.1600-0773.2000.pto860102.x. [DOI] [PubMed] [Google Scholar]
- 2.Oh SY, Ju Y, Park H. Mol Cells. 2009;28:341. doi: 10.1007/s10059-009-0134-8. [DOI] [PubMed] [Google Scholar]
- 3.Beavers KR, Mares JW, Swartz CM, Zhao Y, Weiss SM, Duvall CL. Bioconjug Chem. 2014;25:1192. doi: 10.1021/bc5001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown PN, Yin H. Chem Commun (Camb) 2013;49:4415. doi: 10.1039/c2cc36540e. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fabani MM, Gait MJ. RNA. 2008;14:336. doi: 10.1261/rna.844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, Slack FJ. PNAS. 2012;109:E1695. doi: 10.1073/pnas.1201516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, Svoronos A, Braddock DT, Glazer PM, Engelman DM, Saltzman WM, Slack FJ. Nature. 2015;518:107. doi: 10.1038/nature13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mack GS. Nat Biotechnol. 2007;25:631. doi: 10.1038/nbt0607-631. [DOI] [PubMed] [Google Scholar]
- 8.Koppelhus U, Nielsen PE. ADDR. 2003;55:267. doi: 10.1016/s0169-409x(02)00182-5. [DOI] [PubMed] [Google Scholar]
- 9.McMahon BM, Mays D, Lipsky J, Stewart JA, Fauq A, Richelson E. Antisense Nucleic Acid Drug Dev. 2002;12:65. doi: 10.1089/108729002760070803. [DOI] [PubMed] [Google Scholar]
- 10.Leung AKL. Trends Cell Biol. 25:601. doi: 10.1016/j.tcb.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chin V, Collins BE, Sailor MJ, Bhatia SN. Adv Mater. 2001;13:1877. [Google Scholar]; Santos HA. Porous silicon for biomedical applications. Elsevier; 2014. [DOI] [PubMed] [Google Scholar]
- 12.Shahbazi MA, Almeida PV, Makila EM, Kaasalainen MH, Salonen JJ, Hirvonen JT, Santos HA. Biomaterials. 2014;35:7488. doi: 10.1016/j.biomaterials.2014.05.020. [DOI] [PubMed] [Google Scholar]
- 13.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. Mol Ther. 2013;21:149. doi: 10.1038/mt.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]; Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. PNAS. 1995;92:7297. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levchenko TS, Rammohan R, Lukyanov AN, Whiteman KR, Torchilin VP. Int J Pharm. 2002;240:95. doi: 10.1016/s0378-5173(02)00129-1. [DOI] [PubMed] [Google Scholar]
- 15.Camacho AI, Da Costa Martins R, Tamayo I, de Souza J, Lasarte JJ, Mansilla C, Esparza I, Irache JM, Gamazo C. Vaccine. 2011;29:7130. doi: 10.1016/j.vaccine.2011.05.072. [DOI] [PubMed] [Google Scholar]
- 16.Herranz-Blanco B, Liu D, Mäkilä E, Shahbazi MA, Ginestar E, Zhang H, Aseyev V, Balasubramanian V, Salonen J, Hirvonen J. Adv Funct Mater. 2015;25:1488. [Google Scholar]
- 17.Nelson CE, Kintzing JR, Hanna A, Shannon JM, Gupta MK, Duvall CL. ACS Nano. 2013;7:8870. doi: 10.1021/nn403325f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adolph EJ, Nelson CE, Werfel TA, Guo R, Davidson JM, Guelcher SA, Duvall CL. J Mater Chem B. 2014;2:8154. doi: 10.1039/C4TB00352G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP. Cell Metab. 2006;3:87. doi: 10.1016/j.cmet.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. Biomaterials. 2009;30:2440. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 21.Connelly CM, Thomas M, Deiters A. J Biomol Screen. 2012;17:822. doi: 10.1177/1087057112439606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanco E, Shen H, Ferrari M. Nat Biotech. 2015;33:941. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie G, Sun J, Zhong G, Shi L, Zhang D. Arch Toxicol. 2009;84:183. doi: 10.1007/s00204-009-0488-x. [DOI] [PubMed] [Google Scholar]
- 24.Tsai W-C, Hsu S-D, Hsu C-S, Lai T-C, Chen S-J, Shen R, Huang Y, Chen H-C, Lee C-H, Tsai T-F, Hsu M-T, Wu J-C, Huang H-D, Shiao M-S, Hsiao M, Tsou A-P. The Journal of Clinical Investigation. 122:2884. doi: 10.1172/JCI63455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Nature. 2005;438:685. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 26.Fabani MM, Abreu-Goodger C, Williams D, Lyons PA, Torres AG, Smith KGC, Enright AJ, Gait MJ, Vigorito E. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nair JK, Willoughby JLS, Chan A, Charisse K, Alam MR, Wang Q, Hoekstra M, Kandasamy P, Kel’in AV, Milstein S, Taneja N, O’Shea J, Shaikh S, Zhang L, van der Sluis RJ, Jung ME, Akinc A, Hutabarat R, Kuchimanchi S, Fitzgerald K, Zimmermann T, van Berkel TJC, Maier MA, Rajeev KG, Manoharan M. JACS. 2014;136:16958. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, Liu D, Shahbazi M-A, Mäkilä E, Herranz-Blanco B, Salonen J, Hirvonen J, Santos HA. Adv Mater. 2014;26:4497. doi: 10.1002/adma.201400953. [DOI] [PubMed] [Google Scholar]
- 29.Sailor MJ, Park J-H. Adv Mater. 2012;24:3779. doi: 10.1002/adma.201200653. [DOI] [PMC free article] [PubMed] [Google Scholar]; Savage DJ, Liu X, Curley SA, Ferrari M, Serda RE. Curr Opin Pharmacol. 2013;13:834. doi: 10.1016/j.coph.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.