

Abstract
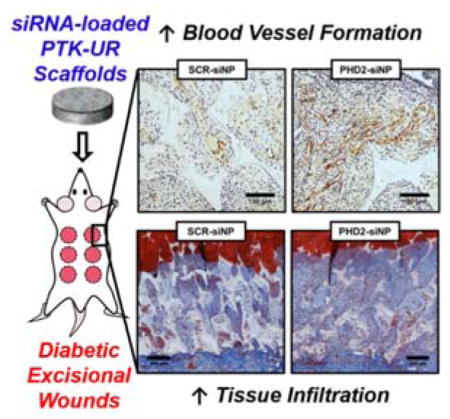
siRNA delivered from ROS-degradable tissue engineering scaffolds promotes diabetic wound healing in rats. Porous poly(thioketal-urethane) (PTK-UR) scaffolds implanted in diabetic wounds locally deliver siRNA that inhibits the expression of prolyl hydroxylase domain protein 2 (PHD2), thereby increasing the expression of pro-growth genes and increasing vasculature, proliferating cells, and tissue development in diabetic wounds.
Keywords: siRNA, wound healing, scaffold, diabetes, reactive oxygen species
Graphical abstract
Diabetes mellitus affects 9.3% of the US population and is increasing in prevalence,[1] with a doubling of incidence in the US from 1980–2012.[2] Patients with diabetes are more prone to cardiovascular and peripheral arterial disease and impaired wound healing, often exacerbating simple skin wounds towards chronic ulceration and in the worst cases, limb amputation. Approximately 25% of diabetics develop chronic ulcers,[3] and roughly 60% of non-traumatic lower-limb amputations for patients over 20 years of age occur in diabetics.[1] Impaired wound healing in diabetic patients is attributable to multiple factors,[4] but is especially affected by deterioration in the microvasculature and subsequent development of ischemia.[5] In normal wound healing, hypoxia due to injury-associated vascular disruption activates the transcription factor hypoxia-inducible factor-1α (HIF-1α), which is primarily regulated by prolyl hydroxylase domain protein 2 (PHD2). PHD2 actively triggers HIF-1α degradation under normoxia[6] but is inactive under hypoxic conditions, thereby stabilizing HIF-1α. Stabilized HIF-1α heterodimerizes with HIF-1β to promote expression of multiple reparative genes, including vascular endothelial growth factor (VEGF),[7] angiopoietin-1 (ANG-1),[8] stromal cell-derived factor-1 (SDF-1),[9] and others that induce cell proliferation/survival/recruitment and angiogenesis. Despite the presence of ischemia, HIF-1α is destabilized in diabetic wounds of both rodents[10] and human patients.[11] While inhibition of PHD2 through small molecule drugs has shown promise in re-establishing HIF-1α activity and improving healing outcomes in skin wounds,[10, 12] these compounds also inhibit other PHD isoforms (PHD1 and PHD3) and can result in off-target effects.[13] In particular, concurrent inhibition of PHD1 and PHD3 has been shown to increase accumulation of HIF-2α;[14] conversely to HIF-1α, increased levels of HIF-2α may impede wound closure and increase bacterial infection,[15] further motivating specific reduction of PHD2 activity rather than pan-inhibition of PHDs.
Small interfering RNA (siRNA) holds great potential as a therapeutic due to its precise mode of action (complementary base-pairing and degradation of specific messenger RNA sequences) and ability to effectively silence targeted gene expression.[16] However, the in vivo efficacy of siRNA has been limited by substantial delivery barriers, including degradation by endogenous nucleases, inability to access the cytoplasm, and insufficiently sustained bioactivity in the target tissue.[17] Much recent work has focused on the development of nanoparticle carriers that can shield siRNA from degradation[18] while facilitating intracellular payload delivery.[19] Furthermore, local delivery of siRNA avoids many of the challenges associated with systemic administration and helps to ensure that a sufficient siRNA dose reaches the target tissue while lessening the potential for side effects due to off-target gene silencing in healthy tissues.[20] To date, local siRNA delivery strategies for wound healing applications have targeted matrix metalloproteinase-9 (MMP-9),[21] connective tissue growth factor (CTGF),[22] p53,[23] and PHD2[24] using a host of biomaterial delivery depots, including layer-by-layer coatings on non-degradable bandages, alginate hydrogels, and acellular dermal matrix. Here we sought to expand our ongoing work to develop cell-degradable synthetic scaffolds that promote robust cellular infiltration and tissue regeneration and that can be also used for sustained, local drug or nanomedicine release.
The available chemistries used for fabrication of synthetic scaffolds offer the opportunity to produce templates for guiding new tissue growth in critically-sized defects with adjustable biodegradation mechanisms and rates; moreover, these materials can serve as a depot for delivering a diversity of therapeutic molecules including growth factors,[25] small molecule drugs,[12c, 26] or nanoparticles.[27] In order to leverage these scaffold properties, we have recently developed strategies for delivering siRNA-carrying nanoparticles (siNPs) from hydrolytically-biodegradable poly(ester urethane) (PEUR) tissue engineering scaffolds[27] and demonstrated in vivo knockdown of PHD2 with increased local angiogenesis in a subcutaneous mouse wound model.[28] Herein, we have applied this system in a more clinically-relevant diabetic rat skin excisional wound model. In addition, we have for the first time explored siNP delivery from a new poly(thioketal urethane) (PTK-UR) scaffold chemistry that features a cell-mediated (i.e. not hydrolytic) degradation mechanism driven by reactive oxygen species (ROS).[29] Relative to ester-based PEUR materials, the PTK-UR chemistry enables better matched rates of degradation and cell infiltration while more effectively inhibiting wound contraction.[30] Here, we pursued PTK-UR scaffolds to locally deliver siNPs for the in vivo knockdown of PHD2 to promote angiogenesis, cell proliferation, and an increased rate of new tissue formation within diabetic excisional skin wounds.
Porous, PTK-UR tissue engineering scaffolds were fabricated through reactive liquid molding of a PTK diol with lysine triisocyanate (LTI) (structures given in Figure 1A). Previous in vivo testing of PTK-UR materials used scaffolds fabricated with hexamethylene diisocyanate trimer (HDIt),[30] rather than LTI; HDIt is non-degradable compared to degradable LTI[31] and was previously used to experimentally isolate the degradation of the PTK polyol component. To generate a more fully degradable/resorbable biomaterial, LTI (which is degraded by both hydrolysis and oxidation)[31] was used as the isocyanate component for PTK-UR fabrication in these studies. For scaffold formation, the PTK polyol was combined with water (which reacts with the isocyanate to generate CO2 bubbles which create scaffold porosity), an amine catalyst with both gelling and foaming activity, and LTI (component amounts given in Table S1). PTK diol polymers were synthesized by a condensation polymerization with 2-mercaptoethyl ether (MEE) and 2,2 dimethoxypropane (DMP) with a p-toluene sulfonic acid catalyst (PTSA), followed by a post-polymerization hydroxyl end-group modification to yield a final MEE-PTK diol polymer with a molecular weight of 1100 Da (synthesis scheme in Figure S1). To assess the ROS-sensitivity of the fabricated PTK-UR materials, scaffolds were incubated in varying ROS-containing media (5 mM hydroxyl radical, superoxide, hydrogen peroxide, or peroxynitrite) for three days and then weighed to estimate degradation. Similar to previous results[30, 32], these PTK-based materials were particularly sensitive to hydroxyl radicals but were partially degraded over this timeframe by all the ROS tested (Figure S2). To serve as a material control for in vivo experiments, a well-characterized 900 Da polyester triol composed of 60% ε-caprolactone, 30% glycolide, and 10% D,L-lactide was also synthesized using previously described methods.[33]
Figure 1.

Chemical structures of (A) polythioketal (PTK) diol and lysine triisocyanate (LTI) which are the key precursors of ROS-degradable tissue scaffolds, and (B) the micelle-forming diblock copolymer DMAEMA-block-(DMAEMA-co-BMA-co-PAA).
We previously found that PTK-UR scaffolds fabricated from MEE-PTK diol polymers promoted robust tissue infiltration in vivo while also significantly improving wound stenting in subcutaneous rat wounds as compared to hydrolytically-degradable PEUR materials.[30] To test the initial in vivo performance of PTK-UR scaffolds against PEUR materials in a more clinically relevant wound model, hyperglycemia was induced in male Sprague-Dawley rats using streptozotocin (STZ, 45 mg/kg). After successful STZ-induced development of diabetes, six full-thickness excisional wounds were created in the rats’ dorsal skin with an 8 mm biopsy punch. Sterilized PTK-UR and PEUR scaffolds (1.5 mm thick, 8 mm diameter) were implanted into these wounds and allowed to heal for 4, 7, or 14 days before the animals were euthanized and processed for histological tissue evaluation. Representative gross views of the healing wounds with implanted scaffolds are shown in Figure S3. As determined from histological sections with hematoxylin and eosin (H&E) staining (Figure 2A), both implanted scaffold formulations had substantial tissue infiltration throughout the scaffold at day 4 post implantation. Tissue infiltration was defined as the cross-sectional area of the scaffold wound site that was occupied by granulation tissue (i.e. area not occupied by scaffold, intact dermis, or blank space). At day 7, both material formulations had significantly more tissue infiltration compared to day 4 levels, but the PTK-UR implants had both significantly greater infiltration (Figure 2B) and (similar to previous in vivo observations)[30] significantly greater stenting of the wound leading to a thicker layer of tissue growth within the PTK-UR scaffolds than within analogous PEURs (Figure 2C). The newly formed tissue was also evaluated in day 7 tissue sections by CD68 immunohistochemistry (IHC) to determine the presence of inflammatory cells. Quantification of IHC staining for this macrophage marker[34] revealed that there was no significant difference in macrophage presence between tissues surrounding PEUR and PTK-UR implants (Figure S4), suggesting that these materials elicited a similar inflammatory response. Both scaffold types were nearly fully resorbed by day 14, indicating that the use of LTI in the material formulation significantly increased the in vivo degradation rate of these scaffolds compared to similar implants made with minimally-degradable HDIt.[30–31] However, PTK-UR scaffolds promoted a highly favorable regenerative response with enhanced tissue infiltration, thicker tissue ingrowth, and similar presence of macrophages compared to conventional PEUR materials. These results supported the use of PTK-UR implants in subsequent studies.
Figure 2.

PEUR and PTK-UR scaffolds implanted into diabetic excisional wounds (A) were largely degraded by 14 days and covered by new epidermis. Implanted PTK-URs promoted (B) greater wound stenting/thicker granulation tissue formation and (C) supported more robust tissue infiltration over 7 days (mean ± SEM, n = 7 independent animals, *p<0.05).
To demonstrate the therapeutic potential of localized, scaffold-based siRNA delivery to clinically-relevant diabetic wounds, siRNA-carrying micellar nanoparticles (siNPs) were formulated with siRNA targeting PHD2 (PHD2-siNPs) or scrambled control siRNA (SCR-siNPs) and incorporated into PEUR and PTK-UR scaffolds. Both the scrambled and PHD2 siRNA sequences (Table S2) were pre-screened in vitro to ensure effective PHD2 gene silencing in rat cells as seen in Figure S5. The self-assembling, siRNA-condensing nanoparticles were composed of a diblock copolymer synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization, containing 2-(dimethylamino) ethyl methacrylate (DMAEMA), 2-propylacrylic acid (PAA), and butyl methacrylate (BMA) as previously described.[19, 27–28] As depicted in Figure 1B, the final polymer, DMAEMA67-b-(DMAEMA29-co-BMA75-co-PAA40), achieved a final molecular weight of 30,500 Da and was self-assembled into micellar nanoparticles. The addition of siRNA (5 nmol) to the micelle solution (1 mg polymer in 1 mg water) formed stable siNPs (Dh = 31 nm, ζ-potential = +20.2 mV), which were optimized to achieve pH-dependent endosomal escape and intracellular siRNA delivery.[19] After loading of the siRNA cargo, the excipient trehalose (60:1 weight ratio to siRNA) was added to the siNP solution to both stabilize the nanoparticles through lyophilization[35] and to increase in vivo siNP release from implanted scaffolds.[28] After lyophilization, the siNP-trehalose powder was mixed into the liquid PEUR or PTK-UR reaction (100 mg prepolymer), leading to homogenous distribution throughout the final 3D polymer structure following scaffold hardening. Corresponding to the rapid cellular infiltration seen in Figure 2, the specific dose of trehalose (5 wt% of the final scaffold weight) was chosen to facilitate relatively fast siNP release from implanted scaffolds with the aim of transfecting these early infiltrating cells. Initial testing of in vitro nanoparticle release from PTK-UR scaffolds using fluorescently-labeled double stranded DNA as a model for siRNA indicated that, similarly to in vitro siNP release data from PEUR scaffolds with the same dose of trehalose,[28] much of the siRNA payload was released over a two-day time course (Figure S6).
As an initial screen of the effectiveness of siNP delivery from PEUR and PTK-UR implants, no treatment scaffolds (NT) along with PHD2 and SCR siNP-loaded scaffolds were implanted into excisional wounds in STZ-diabetic rats and histologically evaluated after 7 days. Blood vessel area in H&E sections was quantified for PEUR and PTK-UR scaffolds with NT, SCR-siNP, and PHD2-siNP treatments. As seen in Figure S7, pilot in vivo data indicated that loading with PHD2-siNPs tended to cause increased vessel area in both PEUR and PTK-UR scaffolds. However, the NT PTK-UR scaffolds had a statistically higher vessel cross-sectional area than the NT PEUR scaffolds (Figure S7), indicating that PTK-UR scaffolds promoted more blood vessel growth than PEURs even without siNP treatment. Because PTK-URs encouraged both a more robust healing response (Figure 2) and enhanced baseline blood vessel formation (Figure S7) compared to the more conventional PEUR materials, a larger diabetic rat study with was carried out using only the better-performing PTK-UR scaffolds loaded with siNPs.
In the scaled up study, excisional wounds in STZ-diabetic rats were implanted with sterilized PTK-UR scaffolds (NT, SCR-siNP, or PHD2-siNP treatments, 0.5 nmol siRNA per SCR or PHD2-siNP scaffold). Tissue/scaffold samples from day 4 post implantation revealed 40% knockdown of PHD2 messenger RNA in PHD2-siNP scaffolds compared to NT and SCR-siNP implants (Figure 3A) as determined by quantitative real-time polymerase chain reaction (qRT-PCR, primer sequences listed in Table S2). The downstream transcriptional impact of PHD2 silencing was also confirmed, since both HIF-1α and VEGF protein levels were also significantly increased in the treatment group as quantified by western blot analysis compared to SCR-siNP scaffolds (Figure 3B–C). qRT-PCR of day 7 tissue samples did not show a significant decrease in PHD2 expression compared to NT or SCR-siNP implants (data not shown), potentially due to the relatively fast in vivo release of siNPs with added trehalose.[28] However, day 7 histological sections immunostained for collagen IV (Col IV), a marker of vascular basement membranes,[36] indicated a persistent increase in blood vessel density in PHD2-siNP scaffolds compared to SCR-siNP implants (Figure 4A). Enhanced vessel formation is a hallmark effect of PHD2 inhibition[24, 37] and is heavily implicated in improved wound healing outcomes. Furthermore, there was a higher density of vessels in PHD2-siNP scaffolds expressing α-smooth muscle actin (α-SMA), a marker of vessel maturation[38], as visualized by α-SMA IHC in Figure 4B. These data collectively demonstrate that a higher density of both immature and mature blood vessels form within the PHD2-siNP scaffolds compared to control materials.
Figure 3.

PTK-UR scaffolds loaded with PHD2-siNPs (A) significantly decrease PHD2 mRNA within the wound tissue scaffolds compared to NT and SCR-siNP implants, leading to (B) increased HIF-1α and (C) increased VEGF protein levels compared to SCR-siNP implants at day 4 post-implantation (mean ± SEM, n = 6 independent animals, *p<0.05).
Figure 4.

PHD2-siNP scaffolds featured (A) improved overall tissue vascularization as visualized by Col IV IHC, along with (B) more mature vessel formation as denoted by α-SMA IHC. PHD2-siNP scaffolds also (C) had increased cellular proliferation as visualized by Ki67 IHC, leading to (D) an overall increase in tissue infiltration into implanted scaffolds (mean ± SEM, n = 7 independent animals, *p<0.05).
Immunostaining for Ki67, a marker for cell proliferation,[36] also indicated that there was a significant increase in proliferating cells throughout the PHD2-siNP scaffolds (Figure 4C). Decreased PHD2 expression in cutaneous tissue is known to increase cell proliferation and migration while enhancing overall wound closure in mouse skin lesions.[39] Closer inspection suggested that the proliferating cells were largely granulation tissue-forming fibroblasts. To confirm cellular identity, tissue sections immunostained for S100A4, a fibroblast marker,[40] were compared to Ki67 IHC sections. As seen in representative images in Figure S8, comparing serial tissue sections immunostained for both markers indicated that regions of dense Ki67-positive proliferating cells also had high levels of expression of S100A4. These data suggest that PHD2-siNP mediated cellular proliferation is generating fibroblasts that can aid wound restoration.[41]
Alhough much past work has used surface wound closure percentage as a marker for overall healing in rodent models,[24a, 39] these biodegradable polyurethane scaffolds are designed to stent open contracting skin to decrease collagen alignment and scarring and improve the quality of new tissue formed[42], thus making topical measurements of wound closure a less relevant measure of healing in this system. This is highlighted in the macroscopic wound images in Figure S3, as wounds at day 7 are all roughly the same size between PEUR and PTK-UR implants due to the stenting effect of the scaffolds. Despite the similarity in apparent wound size, the amount of new tissue growth as gauged by histological evaluation was significantly different between the two formulations (Figure 2) thereby demonstrating the better sensitivity by measuring tissue growth within the wound histologically. To gauge overall wound healing of PHD2-siNP against SCR-siNP scaffolds at day 7, histological sections stained with Masson’s trichrome blue were evaluated for the level of tissue infiltration into the scaffold. PHD2-siNP scaffolds had a statistically significant increase in tissue infiltration compared to SCR-siNP scaffolds as depicted in Figure 4D, indicating that the local delivery of PHD2 siRNA from biodegradable scaffolds increased the amount of tissue within scaffolds and improved refilling of the diabetic wounds with vascularized, reparative granulation tissue.
In conclusion, porous PTK-UR scaffolds represent a promising biomaterial technology for the healing of chronic diabetic wounds both as a regenerative tissue engineering scaffold and as a drug depot for localized therapeutic delivery. Implanted PTK-UR scaffolds promote more robust tissue regeneration in diabetic wounds than commonly used polyester-based PEUR scaffolds. Furthermore, the local, scaffold-based delivery of siNPs to silence PHD2 expression is an effective and potent strategy for improving vascular development, cellular proliferation, and new tissue growth in diabetic wounds. These results represent a promising, clinically translatable approach to treat non-healing skin wounds, and ongoing work will further elucidate the impacts of PHD2 knockdown kinetics in this diabetic wound healing model and in more advanced pre-clinical settings.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Varun Prakesh for his contribution to this work. Funding was provided by the Vanderbilt University School of Engineering, the Department of Veterans Affairs, the NIH through grants R21EB012750, R01AR056138, R01EB019409, and T32DK101003, and the NSF through grant DMR-1006558. We acknowledge the Translational Pathology Shared Resource supported by NCI/NIH Cancer Center Support Grant 2P30 CA068485-14 and the Vanderbilt Mouse Metabolic Phenotyping Center Grant 5U24DK059637-13. The authors confirm that there are no known conflicts of interest associated with this publication and there has been no significant financial support for these efforts that could have influenced their outcome.
Footnotes
Supporting Information, including supplemental figures and an expanded description of the materials and methods used in this work, is available from the Wiley Online Library or from the author.
Contributor Information
John R. Martin, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Dr. Christopher E. Nelson, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Dr. Mukesh K. Gupta, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Dr. Fang Yu, Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Samantha M. Sarett, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Dr. Kyle M. Hocking, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
Alonda C. Pollins, Department of Plastic Surgery, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Dr. Lillian B. Nanney, Department of Plastic Surgery, Vanderbilt University Medical Center, Nashville, TN 37232, USA
Dr. Jeffrey M. Davidson, Department of Pathology, Microbiology, and Immunology, Vanderbilt University Medical Center, Nashville, TN 37232, USA. Medical Research Service, Veterans Affairs Tennessee Valley Healthcare System, Nashville, TN 37212, USA
Dr. Scott A. Guelcher, Department of Chemical and Biomolecular Engineering, Vanderbilt University, Nashville, TN 37235, USA
Dr. Craig L. Duvall, Email: craig.duvall@vanderbilt.edu, Department of Biomedical Engineering, Vanderbilt University, Nashville, TN 37235, USA
References
- 1.CDC. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. USDHHS; Atlanta, GA, USA: 2014. [Google Scholar]
- 2.Geiss LS, Wang J, Cheng YJ, Thompson TJ, Barker L, Li Y, Alright AL, Gregg EW. J Amer Med Assoc. 2014;312:1218–1226. doi: 10.1001/jama.2014.11494. [DOI] [PubMed] [Google Scholar]
- 3.Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Wound Repair Regen. 2009;17:763–771. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falanga V. Lancet. 2006;366:1736–1743. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 5.Catrina SB, Zheng X. Diabetes Metab Res Rev. 2016;32:179–185. doi: 10.1002/dmrr.2742. [DOI] [PubMed] [Google Scholar]
- 6.a) Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]; b) Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 9.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, Capla JM, Galiano RD, Levine JP, Gurtner GC. Nat Med. 2004;10:858–864. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 10.Botusan IR, Sunkari VG, Savu O, Catrina AI, Grünler J, Lindberg S, Pereira T, Ylä-Herttuala S, Poellinger L, Brismar K. P Natl Acad Sci USA. 2008;105:19426–19431. doi: 10.1073/pnas.0805230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catrina SB, Okamoto K, Pereira T, Brismar K, Poellinger L. Diabetes. 2004;53:3226–3232. doi: 10.2337/diabetes.53.12.3226. [DOI] [PubMed] [Google Scholar]
- 12.a) Zhang Y, Strehin I, Bedelbaeva K, Gourevitch D, Clark L, Leferovich J, Messersmith PB, Heber-Katz E. Sci Transl Med. 2015;7:290ra292. doi: 10.1126/scitranslmed.3010228. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chang EI, Loh SA, Ceradini DJ, Chang EI, Lin S-e, Bastidas N, Aarabi S, Chan DA, Freedman ML, Giaccia AJ, Gurtner GC. Circulation. 2007;116:2818–2829. doi: 10.1161/CIRCULATIONAHA.107.715847. [DOI] [PubMed] [Google Scholar]; c) Chen H, Jia P, Kang H, Zhang H, Liu Y, Yang P, Yan Y, Zuo G, Guo L, Jiang M, Qi J, Liu Y, Cui W, Santos HA, Deng L. Adv Healthc Mater. 2016;5:907–918. doi: 10.1002/adhm.201501018. [DOI] [PubMed] [Google Scholar]
- 13.a) Fraisl P, Aragones J, Carmeliet P. Nat Rev Drug Discov. 2009;8:139–152. doi: 10.1038/nrd2761. [DOI] [PubMed] [Google Scholar]; b) Kim SY, Yang EG. Molecules. 2015;20:20551–20568. doi: 10.3390/molecules201119717. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chan MC, Holt-Martyn JP, Schofield CJ, Ratcliffe PJ. Mol Aspects Med. 2016;47–48:54–75. doi: 10.1016/j.mam.2016.01.001. [DOI] [PubMed] [Google Scholar]; d) Schneider M, Harnoss JM, Strowitzki MJ, Radhakrishnan P, Platzer L, Harnoss JC, Hank T, Cai J, Ulrich A. Hypoxia. 2015;3:1–14. doi: 10.2147/HP.S60872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cowburn AS, Alexander LEC, Southwood M, Nizet V, Chilvers ER, Johnson RS. J Invest Dermatol. 2014;134:801–808. doi: 10.1038/jid.2013.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dykxhoorn D, Palliser D, Lieberman J. Gene Ther. 2006;13:541–552. doi: 10.1038/sj.gt.3302703. [DOI] [PubMed] [Google Scholar]
- 17.a) White PJ. Clin Exp Pharmacol P. 2008;35:1371–1376. doi: 10.1111/j.1440-1681.2008.04992.x. [DOI] [PubMed] [Google Scholar]; b) Dominska M, Dykxhoorn DM. J Cell Sci. 2010;123:1183–1189. doi: 10.1242/jcs.066399. [DOI] [PubMed] [Google Scholar]
- 18.a) Miteva M, Kirkbride KC, Kilchrist KV, Werfel TA, Li H, Nelson CE, Gupta MK, Giorgio TD, Duvall CL. Biomaterials. 2015;38:97–107. doi: 10.1016/j.biomaterials.2014.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sarett SM, Kilchrist KV, Miteva M, Duvall CL. J Biomed Mater Res A. 2015;103:3107–3116. doi: 10.1002/jbm.a.35413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Convertine AJ, Benoit DSW, Duvall CL, Hoffman AS, Stayton PS. J Control Release. 2009;133:221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, Stayton PS. Biomacromolecules. 2010;11:2904–2911. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Sarett SM, Nelson CE, Duvall CL. J Control Release. 2015;218:94–113. doi: 10.1016/j.jconrel.2015.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Krebs MD, Alsberg E. Chem Eur J. 2011;17:3054–3062. doi: 10.1002/chem.201003144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castleberry SA, Almquist BD, Li W, Reis T, Chow J, Mayner S, Hammond PT. Adv Mater. 2015 doi: 10.1002/adma.201503565. [DOI] [PubMed] [Google Scholar]
- 22.Castleberry SA, Golberg A, Sharkh MA, Khan S, Almquist BD, Austen WG, Jr, Yarmush ML, Hammond PT. Biomaterials. 2016;95:22–34. doi: 10.1016/j.biomaterials.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen PD, Tutela JP, Thanik VD, Knobel D, Allen JRJ, Chang CC, Levine JP, Warren SM, Saadeh PB. Wound Repair Regen. 2010;18:553–559. doi: 10.1111/j.1524-475X.2010.00638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Wetterau M, George F, Weinstein A, Nguyen PD, Tutela JP, Knobel D, Cohen BAO, Warren SM, Saadeh PB. Wound Repair Regen. 2011;19:481–486. doi: 10.1111/j.1524-475X.2011.00697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vandegrift MT, Szpalski C, Knobel D, Weinstein A, Ham M, Ezeamuzie O, Warren SM, Saadeh PB. J Surg Res. 2015;195:360–367. doi: 10.1016/j.jss.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 25.a) Li B, Davidson JM, Guelcher SA. Biomaterials. 2009;30:3486–3494. doi: 10.1016/j.biomaterials.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Almquist BD, Castleberry SA, Sun JB, Lu AY, Hammond PT. Adv Healthc Mater. 2015;4:2090–2099. doi: 10.1002/adhm.201500403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hafeman AE, Zienkiewicz KJ, Carney E, Litzner B, Stratton C, Wenke JC, Guelcher SA. J Biomat Sci-Polymer E. 2010;21:95–112. doi: 10.1163/156856209X410256. [DOI] [PubMed] [Google Scholar]
- 27.Nelson CE, Gupta MK, Adolph EJ, Shannon JM, Guelcher SA, Duvall CL. Biomaterials. 2012;33:1154–1161. doi: 10.1016/j.biomaterials.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson CE, Kim AJ, Adolph EJ, Gupta MK, Yu F, Hocking KM, Davidson JM, Guelcher SA, Duvall CL. Adv Mater. 2014;26:607–614. doi: 10.1002/adma.201303520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Nat Mater. 2010;9:923–928. doi: 10.1038/nmat2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin JR, Gupta MK, Page JM, Yu F, Davidson JM, Guelcher SA, Duvall CL. Biomaterials. 2014;35:3766–3776. doi: 10.1016/j.biomaterials.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafeman AE, Zienkiewicz KJ, Zachman AL, Sung HJ, Nanney LB, Davidson JM, Guelcher SA. Biomaterials. 2011;32:419–429. doi: 10.1016/j.biomaterials.2010.08.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shim MS, Xia Y. Angew Chem Int Ed. 2013;52:6926–6929. doi: 10.1002/anie.201209633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hafeman AE, Li B, Yoshii T, Zienkiewicz K, Davidson JM, Guelcher SA. Pharm Res. 2008;25:2387–2399. doi: 10.1007/s11095-008-9618-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray PJ, Wynn TA. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.a) Kaushik JK, Bhat R. J Biol Chem. 2003;278:26458–26465. doi: 10.1074/jbc.M300815200. [DOI] [PubMed] [Google Scholar]; b) Adolph EJ, Nelson CE, Werfel TA, Guo R, Davidson JM, Guelcher SA, Duvall CL. J Mater Chem B. 2014;2:8154–8164. doi: 10.1039/C4TB00352G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo R, Ward CL, Davidson JM, Duvall CL, Wenke JC, Guelcher SA. Biomaterials. 2015;54:21–33. doi: 10.1016/j.biomaterials.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a) Zhang X, Yan X, Cheng L, Dai J, Wang C, Han P, Chai Y. PLoS ONE. 2013;8:e84548. doi: 10.1371/journal.pone.0084548. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zimmermann AS, Morrison SD, Hu MS, Li S, Nauta A, Sorkin M, Meyer NP, Walmsley GG, Maan ZN, Chan DA. PLOS ONE. 2014;9:e93373. doi: 10.1371/journal.pone.0093373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brudno Y, Ennett-Shepard AB, Chen RR, Aizenberg M, Mooney DJ. Biomaterials. 2013;34:9201–9209. doi: 10.1016/j.biomaterials.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kalucka J, Ettinger A, Franke K, Mamlouk S, Singh RP, Farhat K, Muschter A, Olbrich S, Breier G, Katschinski DM, Huttner W, Weidemann A, Wielockx B. Mol Cell Biol. 2013;33:3426–3438. doi: 10.1128/MCB.00609-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Opalenik SR, Davidson JM. FASEB J. 2005;19:1561–1563. doi: 10.1096/fj.04-2978fje. [DOI] [PubMed] [Google Scholar]
- 42.a) Adolph EJ, Hafeman AE, Davidson JM, Nanney LB, Guelcher SA. J Biomed Matet Res A. 2012;100A:450–461. doi: 10.1002/jbm.a.33266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lee SY, Oh JH, Kim JC, Kim YH, Kim SH, Choi JW. Biomaterials. 2003;24:5049–5059. doi: 10.1016/s0142-9612(03)00411-3. [DOI] [PubMed] [Google Scholar]; c) Adolph EJ, Guo R, Pollins AC, Zienkiewicz K, Cardwell N, Davidson JM, Guelcher SA, Nanney LB. J Biomed Matet Res B. 2015 doi: 10.1002/jbm.b.33515. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lorden ER, Miller KJ, Bashirov L, Ibrahim MM, Hammett E, Jung Y, Medina MA, Rastegarpour A, Selim MA, Leong KW, Levinson H. Biomaterials. 2015;43:61–70. doi: 10.1016/j.biomaterials.2014.12.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.