

Abstract
Intellectual disability is a common and highly heterogeneous disorder etiologically. In a multiplex consanguineous family, we applied autozygosity mapping and exome sequencing and identified a novel homozygous truncating mutation in PUS3 that fully segregates with the intellectual disability phenotype. Consistent with the known role of Pus3 in isomerizing uracil to pseudouridine at positions 38 and 39 in tRNA, we found a significant reduction in this post-transcriptional modification of tRNA in patient cells. Our finding adds to a growing list of intellectual disability disorders that are caused by perturbation of various tRNA modifications, which highlights the sensitivity of the brain to these highly conserved processes.
Keywords: pseudouridine, post-transcriptional modification, intellectual disability, exome
INTRODUCTION
Intellectual disability (ID) is defined as significant limitations both in intellectual functioning and in adaptive behavior as expressed in conceptual, social, and practical adaptive skills, originating before the age 18 (Association 2013). ID is a highly prevalent condition with estimates ranging 1–2% across populations (Maulik et al. 2011). While chromosomal abnormalities are seen in at least 10% of ID patients (Miller et al. 2010), large genomic sequencing studies suggest that single gene mutations are far more frequent and account for another 50%, almost always de novo dominant in severe cases (de Ligt et al. 2012; Gilissen et al. 2014; Rauch et al. 2012; Vissers et al. 2010). The contribution of autosomal recessive mutations to ID is unknown but likely to be significant as inferred by the large number of candidate genes identified by recent large scale studies (Alazami et al. 2015; Karaca et al. 2015).
The genes that are mutated in ID patients encode highly diverse groups of proteins, which suggests critical and non-redundant roles of numerous biological processes in normal cognition. One such process that emerged from the discovery of Mendelian forms of ID is tRNA modification. For example, the most common form of autosomal recessive ID in Arabia was found to be due to mutation of ADAT3, which edits adenosine to inosine at the wobble position 34 of mature tRNA (Alazami et al. 2013; El-Hattab et al. 2016; Gerber and Keller 1999). More recently, we have identified a form of severe microcephalic primordial dwarfism with global developmental delay that is caused by mutation of WDR4, a subunit of the methyltransferase that catalyzes 7-methylguanosine modification of residue G46 of tRNA (Shaheen et al. 2015). These findings suggest that normal brain development and function is highly sensitive to perturbation of tRNA modification, presumably by virtue of impaired protein synthesis.
In this study, we show that a homozygous truncation mutation in PUS3 segregating with ID results in impaired isomerization of uridine to pseudouridine (Ψ) in patient tRNA. PUS3 is a member of the highly conserved TruA/Pus3 family of pseudouridylases, which are important for healthy growth in bacteria and yeast (Bekaert and Rousset 2005; Carbone et al. 1991; Chang et al. 1971; Lecointe et al. 2002; Tsui et al. 1991), and catalyze pseudouridine formation at specific uridine residues in the anticodon-stem loop of tRNAs in all kingdoms of life (Blaby et al. 2011; Chen and Patton 2000; Han et al. 2015; Hur and Stroud 2007; Lecointe et al. 1998; Singer et al. 1972). These findings expand the list of tRNA modification defects linked to ID.
MATERIALS AND METHODS
Human Subjects
Evaluation of affected members by a board-certified clinical geneticist included obtaining medical and family histories, clinical examination, neuroimaging and clinical laboratory investigations. After obtaining a written informed consent for enrollment in an IRB-approved project (KFSHRC RAC#2121053), venous blood was collected in EDTA and sodium heparin tubes for DNA extraction and establishment of lymphoblastoid cell lines (IV:3 15DG1746, IV:4 15DG1747, IV:5 15DG1750, control 15DG0421), respectively. A separate consent to publish photographs was also obtained.
Autozygosity Mapping and Exome Sequencing
Determination of the autozygome of each available family member and the exclusively shared autozygome among the affected members was essentially as described before (Alkuraya 2012). Briefly, runs of homozygosity ≥2Mb were used as surrogates of autozygosity and determined using AutoSNPa based on a genomewide SNP genotyping file generated using Axiom SNP Array following the manufacturer’s instructions (Affymetrix). Exome sequencing was done with an Illumina HiSeq2000TM following exome enrichment using the Agilent SureSelectXT Human All Exon 50Mb Kit. Read alignment was carried out with BWA and variant calling with GATK. Annotation was according to an in-house pipeline that was previously described in combination with autozygome analysis (Alkuraya 2013; Group 2015). Briefly, we only considered homozygous coding/splicing variants, novel/very rare (MAF<0.001) within the shared autozygome of the three affected individuals and such variants were subsequently confirmed by Sanger sequencing and segregation was tested in all available family members.
Extraction of total RNA and purification of tRNAPhe from human cells
LCLs were grown at 37°C in 5% CO2 in RPMI 1640 medium containing FBS (15%), penicillin (1 U/mL), streptomycin (1 μg/mL), and amphotericin b (0.5 μg/mL) to a density of ~ 1.0 × 106 cells/mL, and bulk RNA from ~1.2 × 108 cells was extracted with TRIzol (Life Technologies) according to manufacturer’s instructions. Then tRNAPhe was purified from (1~1.25 mg) bulk RNA using a 5′ biotinylated oligonucleotide (Integrated DNA Technologies) complementary to nucleotides 53–76 of the tRNA, as previously described (Jackman et al. 2003).
HPLC analysis of tRNA
Purified tRNA (1.25 μg) was digested with P1 nuclease and phosphatase as previously described (Jackman et al. 2003), and nucleosides were subjected to HPLC analysis at pH 7.0 as previously described (Guy et al. 2012).
RESULTS
Identification of A Recessive Form of Syndromic Intellectual Disability
Index (IV:3) is a 15 ½ year-old girl with a history of global developmental delay since infancy and failure to thrive (birth growth parameters were normal). She currently ambulates with assistance and has severe ID. There is a history of seizures at 10 months of age but she has been seizure-free on multiple antiepileptic medications. Family history is notable for first cousin parents and two similarly affected sisters (figure 1A) (see below). Examination revealed severe growth deficiency and microcephaly: weight 21 kg (−7.5SD), OFC 50.5 cm (−3.3SD), and height 124 cm (−5.9SD). She has coarse facial features, bilateral strabismus, grey sclera and extensive Mongolian spots (figure 1B). Negative results include urine organic acids and GAGs, Tandem MS, plasma lactate, uric acid, lipid profile, CDG screening, and molecular karyotyping. Brain MRI at 7 years of age showed mild ventriculomegaly with multiple arachnoid cysts in the middle cranial fossae bilaterally and the right cerebellopontine angle with the largest in the right middle cranial fossa, which measures 5.0 × 3.0 cm and causes mild mass effect on the right temporal lobe. In addition, there was volume loss in the frontal lobes anteriorly, and multiple T2/FLAIR white matter signal abnormalities in the subcortical white matter. Current evaluation shows that she is in the severe range of ID (IQ 30).
Figure 1.
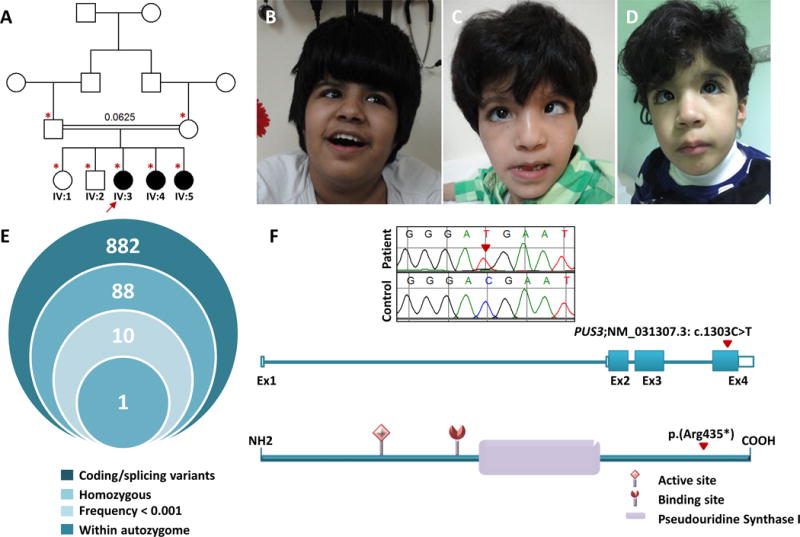
Identification of an autosomal recessive Syndromic Intellectual Disability and the causative variant. A) Pedigree of the family showing the consanguineous nature of the parents. The index is indicated by an arrow, and asterisks denote individuals whose DNA was available for analysis. B, C and D) facial images of the three sister in the family (IV:3, IV:4 and IV:5 respectively) showing coarse facial features, bilateral strabismus, grey sclera and extensive Mongolian spots. E) Stacked Venn illustrating the exome filtration scheme and the number of surviving variants in each filtration steps. F) Upper panel: Sequence chromatograms of PUS3 nonsense variant identified in the family (control tracing is shown for comparison and the location of the mutation is denoted by red triangle). Middle panel Schematic representation of PUS3 transcript, (red triangle indicate the site of the mutations (NM_031307.3: c.1303C>T)). Lower panel: Schematic of PUS3 showing the position of domain” Pseudouridine Synthase I” and the active and binding sites as well as the location of the mutation (p.Arg435*).
IV:4 is the 12-year-old sister of IV:3 who similarly presented with a history of global developmental delay, failure to thrive (birth growth parameters were normal) and severe ID. She had no seizures. Examination revealed coarse facies but less striking compared to the sister, severe growth deficiency with borderline microcephaly (weight 16.2 kg (−4.1SD), height 119 cm (−4.3SD), OFC 50 cm (−2.3SD), strabismus, grey sclera and widespread Mongolian spots (figure 1C). Brain MRI showed signal abnormalities within the globus pallidus and dentate nucleus bilaterally with nonspecific symmetrical hyperintensities in the frontal lobes in subcortical location. In addition, there was evidence of dysgenesis of corpus callosum, mild diffuse brain atrophy and a prominent cisterna magna. Similar to her older sister currently she is in the severe range of ID.
IV:5 is the 3.3-year-old sister of the index who also presented with global developmental delay and failure to thrive (birth growth parameters were normal) but no seizures. Assessment at 2 years showed severe axial and appendicular hypotonia with poor interactions. Body weight was 12 kg (48th centile), height was 88 cm (74th centile) and head circumference was 47 cm (37th centile). Brain MRI at 2 years was unremarkable apart from small arachnoid cyst seen along the anterolateral surface of the left cerebellar hemisphere in the posterior fossa. Recent evaluation at the age of 3.3 years showed that her weight is 13.02 kg (16th centile), length 89.5cm (4th centile) and OFC 45.5cm (−2.1SD). She had strabismus, grey sclerae and Mongolian spots (figure 1D). She had global developmental delay and her IQ was in the profound range (<20).
A Homozygous Truncating Mutation in PUS3 Segregates with a Syndromic Form of Intellectual Disability
Autozygosity mapping revealed that two autozygous intervals on chromosome 11 are exclusively shared by the three affected sisters in the family (Figure S1). Exome sequencing revealed a single coding/splicing novel variant within these critical intervals (figure 1E). Specifically, we identified a homozygous nonsense variant in PUS3; Chr11(GRCh37):g.125763823G>A; NM_031307.3: c.1303C>T: p.(Arg435*) that fully segregated with the phenotype in the family as confirmed by Sanger sequencing (figure 1F). This variant is absent in 1,500 Saudi exomes as well as in the ExAC Browser.
PUS3 protein is a member of the phylogenetically conserved TruA/Pus3 family of proteins that catalyzes isomerization of specific uridines to Ψ in the anticodon-stem loop of tRNAs, at U38, U39, and U40 in bacteria (Hur and Stroud 2007; Singer et al. 1972); at U38 and U39 in the yeast Saccharomyces cerevisiae (Lecointe et al. 1998); at U39 (and likely U38) in the archaeon Haloferax volcanii (Blaby et al. 2011); and based on in vitro studies, at U39 and to some extent U38 in murine PUS3 (Chen and Patton 2000).
The Arg435 residue at which the stop codon occurs in this family is located in the C-terminal portion of the 481 amino acid PUS3 protein, at the end of a conserved region found widely in teleost vertebrates, but not in the elephant shark, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, Dictyostelium discoideum, S. cerevisiae and E. coli (figure. 2A), while the subsequent 46 residues of PUS3 comprise a region that is widely conserved in mammals.
Figure 2.
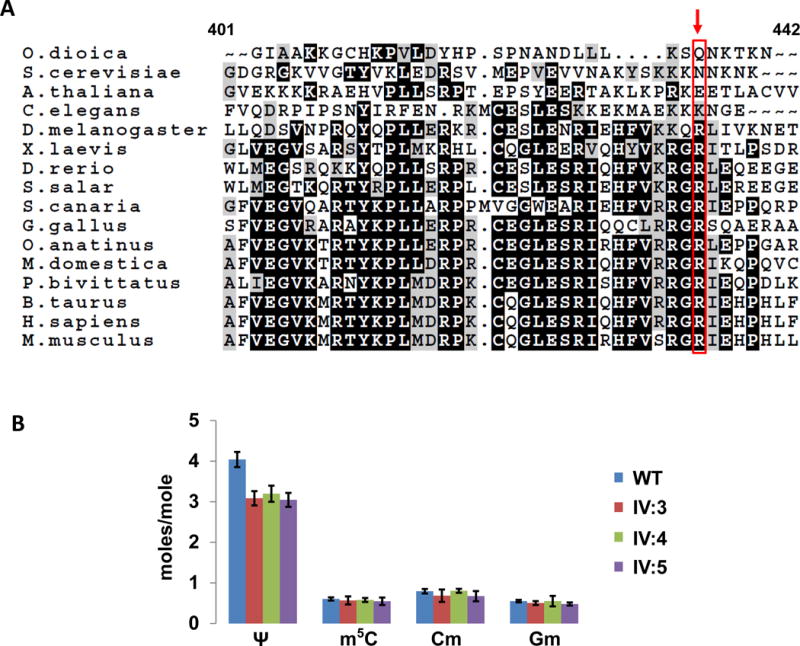
A) Alignment of the C-terminal region of human PUS3. The figure shows the alignment of human PUS3 (residues 401–442) with the corresponding region from other eukaryotes as indicated. The arrow indicates site of the truncation mutation at Arg435*. B) tRNAPhe purified from LCLs of patients with the Arg435* mutation have reduced Ψ relative to that from control LCLs. Representative data are from Table 1 in the text, to show that Ψ levels are reduced in patient LCLs (by 1 mole/mole), whereas levels of each of three other modifications are not.
Cells Derived from Patients with a Homozygous Nonsense Mutation in PUS3 Have Decreased Levels of Pseudouridine in tRNA
To further define the defect of the Arg435* allele of PUS3 and its relationship to ID in this family, we generated lymphoblastoid cell lines (LCLs) derived from the three affected individuals (IV:3, IV:4, IV:5) as well as control LCLs (WT) from a healthy unrelated individual (cell lines from healthy related individuals were not obtained), and analyzed the pseudouridine (Ψ) content of a representative tRNA in which U39 is normally modified to Ψ39. This modification is found in all 6 of the 18 characterized human tRNAs with an encoded U39, including tRNAPhe (Machnicka et al. 2013), which we chose for analysis because of the six gene copies that specify identical tRNA products. As anticipated if the PUS3 mutation was responsible for Ψ39 formation, we found that tRNAPhe purified from the WT LCL had almost exactly one additional mole of Ψ/mole tRNA, compared to tRNAPhe from LCLs of each of the three affected individuals (4.04 moles/mole, compared to 3.09, 3.20, and 3.05 moles/mole). By contrast, the levels of the other modifications of tRNAPhe (Cm, Gm, m2G, m1A, m7G, and m5C) were all very similar in all of the LCLs including WT (figure. 2B, Table 1). Thus, our results strongly suggest that PUS3 with the Arg435* mutation is inactive in the isomerization of U39 to Ψ, linking this defect to the ID, consistent with the known biological effects of Pus3 (see below).
Table 1.
Analysis of modified nucleotides in tRNAPhe from control (WT) and patient LCLs
| Modificationa | Moles Expected | WT | IV:3 | IV:4 | IV:5 |
|---|---|---|---|---|---|
| Ψ | 4 | 4.04 ± 0.19 | 3.09 ± 0.18 | 3.20 ± 0.20 | 3.05 ± 0.17 |
| m5C | 1 | 0.60 ± 0.04 | 0.57 ± 0.10 | 0.58 ± 0.05 | 0.55 ± 0.09 |
| m1A | 2 | 0.92 ± 0.07 | 0.96 ± 0.17 | 1.02 ± 0.08 | 0.93 ± 0.17 |
| Cm | 1 | 0.80 ± 0.05 | 0.69 ± 0.15 | 0.80 ± 0.05 | 0.67 ± 0.13 |
| m7G | 1 | 0.70 ± 0.23 | 0.49 ± 0.11 | 0.53 ± 0.07 | 0.47 ± 0.08 |
| Gm | 1 | 0.55 ± 0.03 | 0.50 ± 0.05 | 0.55 ± 0.13 | 0.48 ± 0.04 |
| M2G | 1 | 0.68 ± 0.01 | 0.59 ± 0.10 | 0.65 ± 0.07 | 0.54 ± 0.08 |
Mean and standard deviation based on three individual growths and RNA preparations
DISCUSSION
Pseudouridine is the most abundant post-transcriptional modification of non-coding RNA observed to date across all domains of life (Cantara et al. 2011; Charette and Gray 2000). Pseudouridine possesses an additional free NH group at position 3 allowing for additional hydrogen bonding with other molecules, and it is thought that the presence of this class of modified nucleotides plays a role in increasing the stability of RNA (Auffinger and Westhof 1998). In characterized human tRNAs, pseudouridine is found at 13 different locations (residues 13, 20a, 20b, 31, 32, 27, 28, 35, 38, 39, 54, 55, and e2), and different pseudouridylases are required for modification at each of the different sites or sets of sites. PUS3 was originally identified as the S. cerevisiae ortholog of E.coli TruA, which synthesizes Ψ at U38, U39 and U40 (Kammen et al. 1988; Singer et al. 1972). However, the yeast ortholog was found to possess activity only at positions 38 and 39 (Lecointe et al. 1998), and similar results have been obtained in the mammalian ortholog protein, which prefers U39 (Chen and Patton 2000).
Lack of PUS3 activity is clearly physiologically consequential. Bacterial truA mutants have significant growth defects (Chang et al. 1971; Tsui et al. 1991), while S. cerevisiae pus3Δ mutants are slow growing and temperature-sensitive (Carbone et al. 1991; Lecointe et al. 2002), primarily due to reduced function of tRNAGln(UUG) (Han et al. 2015), and have reduced −1 frameshifting (Bekaert and Rousset 2005). Since tRNAPhe from LCLs with the Arg435* allele of PUS3 had almost exactly 1 mole/mole less Ψ than control LCLs, we infer that the Arg435* allele nearly completely knocks out PUS3 function due to truncation of the conserved domain and the last 45 amino acids, although the mechanism is not clear.
Our understanding of the physiological context of pseudouridine synthesis, like many other biological processes, was informed by Mendelian disorders that are caused by mutations in genes involved in this process. In 1998, a severe multisystem disorder known as dyskeratosis congenita (DKC) was found to be caused by mutations in DKC1, which encodes a protein that both pseudouridylates rRNA and maintains telomere length (Heiss et al. 1998; Mochizuki et al. 2004). More relevant to the family we present is the finding that a stop mutation in PUS1, which is another tRNA pseudouridylase, was associated with mitochondrial myopathy and sideroblastic anemia because PUS1 pseudouridylates tRNA, although the activity of PUS1 on mitochondrial tRNA may have influenced the mitochondrial-specific phenotype (Bykhovskaya et al. 2004; Fernandez-Vizarra et al. 2007).
The phenotype we observe in the context of PUS3 deficiency is largely brain-specific. The expression profile of PUS3 in mammals is unknown but available datasets suggest a wide tissue distribution (Expression Atlas, EBI). Therefore, it is possible that brain-specific phenotype reflects a higher sensitivity to reduced translational efficiency by the brain compared to other organs (Torres et al. 2014). This would be consistent with the finding by us and others that mutations in other genes involved in tRNA modification result in phenotypes with predilection to the brain e.g. ADAT3, WDR4, TRM10, NSUN2, and FTSJ1 (Abbasi-Moheb et al. 2012; Alazami et al. 2013; El-Hattab et al. 2016; Fahiminiya et al. 2014; Freude et al. 2004; Gillis et al. 2014; Guy et al. 2015; Igoillo-Esteve et al. 2013; Khan et al. 2012; Martinez et al. 2012; Ramser et al. 2004; Shaheen et al. 2015). Similarly, many disease-causing mutations in tRNA synthetases and in the ribosome manifest their effects in functional disturbance of the nervous system (Antonellis et al. 2003; Antonellis and Green 2008; Brooks et al. 2014; Yao and Fox 2013).
In conclusion, we show that a nonsense mutation in PUS3 segregates with an apparently novel autosomal recessive form of ID. Our finding that this mutation results in functional deficiency of PUS3 and a reduction in the availability of tRNA species that are pseudouridylated suggests a disease mechanism that is consistent with the recent identification of ID phenotypes caused by defects in other forms of tRNA modification.
Acknowledgments
We thank the family for their enthusiastic participation. We also thank the Sequencing and Genotyping Facilities at KFSHRC for their technical help. This work was supported in part by KACST grant 13-BIO1113-20 (FSA) and National Institutes of Health grant GM052347 (EMP).
Footnotes
Conflict of Interest
Authors declare no conflict of interest
References
- Abbasi-Moheb L, Mertel S, Gonsior M, Nouri-Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli-Nieh S, Cremer K. Mutations in NSUN2 cause autosomal-recessive intellectual disability. The American Journal of Human Genetics. 2012;90:847–855. doi: 10.1016/j.ajhg.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alazami AM, Hijazi H, Al-Dosari MS, Shaheen R, Hashem A, Aldahmesh MA, Mohamed JY, Kentab A, Salih MA, Awaji A. Mutation in ADAT3, encoding adenosine deaminase acting on transfer RNA, causes intellectual disability and strabismus. Journal of medical genetics. 2013;50:425–430. doi: 10.1136/jmedgenet-2012-101378. [DOI] [PubMed] [Google Scholar]
- Alazami AM, Patel N, Shamseldin HE, Anazi S, Al-Dosari MS, Alzahrani F, Hijazi H, Alshammari M, Aldahmesh MA, Salih MA. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell reports. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Current protocols in human genetics. 2012:6.12.1–6.12.13. doi: 10.1002/0471142905.hg0612s75. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Human genetics. 2013;132:1197–1211. doi: 10.1007/s00439-013-1344-x. [DOI] [PubMed] [Google Scholar]
- Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin S-Q, Jordanova A, Kremensky I, Christodoulou K, Middleton LT. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. The American Journal of Human Genetics. 2003;72:1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonellis A, Green ED. The role of aminoacyl-tRNA synthetases in genetic diseases*. Annu Rev Genomics Hum Genet. 2008;9:87–107. doi: 10.1146/annurev.genom.9.081307.164204. [DOI] [PubMed] [Google Scholar]
- Association AP. Diagnostic and statistical manual of mental disorders (DSM-5®) American Psychiatric Pub; 2013. [DOI] [PubMed] [Google Scholar]
- Auffinger P, Westhof E. Effects of pseudouridylation on tRNA hydration and dynamics: a theoretical approach. Modification and Editing of RNA. 1998:103–112. [Google Scholar]
- Bekaert M, Rousset J-P. An extended signal involved in eukaryotic− 1 frameshifting operates through modification of the E site tRNA. Molecular cell. 2005;17:61–68. doi: 10.1016/j.molcel.2004.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaby IK, Majumder M, Chatterjee K, Jana S, Grosjean H, de Crécy-Lagard V, Gupta R. Pseudouridine formation in archaeal RNAs: The case of Haloferax volcanii. Rna. 2011;17:1367–1380. doi: 10.1261/rna.2712811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SS, Wall AL, Golzio C, Reid DW, Kondyles A, Willer JR, Botti C, Nicchitta CV, Katsanis N, Davis EE. A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X-linked microcephaly in humans. Genetics. 2014;198:723–733. doi: 10.1534/genetics.114.168211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) The American Journal of Human Genetics. 2004;74:1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–201. doi: 10.1093/nar/gkq1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone MLA, Solinas M, Sora S, Panzeri L. A gene tightly linked to CEN6 is important for growth of Saccharomyces cerevisiae. Current genetics. 1991;19:1–8. doi: 10.1007/BF00362080. [DOI] [PubMed] [Google Scholar]
- Chang GW, Roth JR, Ames BN. Histidine regulation in Salmonella typhimurium VIII. Mutations of the hisT gene. Journal of bacteriology. 1971;108:410–414. doi: 10.1128/jb.108.1.410-414.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charette M, Gray MW. Pseudouridine in RNA: what, where, how, and why. IUBMB life. 2000;49:341–351. doi: 10.1080/152165400410182. [DOI] [PubMed] [Google Scholar]
- Chen J, Patton JR. Pseudouridine synthase 3 from mouse modifies the anticodon loop of tRNA. Biochemistry. 2000;39:12723–12730. doi: 10.1021/bi001109m. [DOI] [PubMed] [Google Scholar]
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, Vulto-van Silfhout AT, Koolen DA, de Vries P, Gilissen C. Diagnostic exome sequencing in persons with severe intellectual disability. New England Journal of Medicine. 2012;367:1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Saleh MA, Hashem A, Al-Owain M, Asmari AA, Rabei H, Abdelraouf H, Hashem M, Alazami AM, Patel N. ADAT3-related intellectual disability: Further delineation of the phenotype. American Journal of Medical Genetics Part A. 2016 doi: 10.1002/ajmg.a.37578. [DOI] [PubMed] [Google Scholar]
- Fahiminiya S, Almuriekhi M, Nawaz Z, Staffa A, Lepage P, Ali R, Hashim L, Schwartzentruber J, Abu Khadija K, Zaineddin S. Whole exome sequencing unravels disease-causing genes in consanguineous families in Qatar. Clinical genetics. 2014;86:134–141. doi: 10.1111/cge.12280. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) Journal of medical genetics. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freude K, Hoffmann K, Jensen L-R, Delatycki MB, des Portes V, Moser B, Hamel B, van Bokhoven H, Moraine C, Fryns J-P. Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine–binding protein cause nonsyndromic X-linked mental retardation. The American Journal of Human Genetics. 2004;75:305–309. doi: 10.1086/422507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science. 1999;286:1146–9. doi: 10.1126/science.286.5442.1146. [DOI] [PubMed] [Google Scholar]
- Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, Leach R, Klein R, Tearle R, Bo T, Pfundt R, Yntema HG, de Vries BB, Kleefstra T, Brunner HG, Vissers LE, Veltman JA. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- Gillis D, Krishnamohan A, Yaacov B, Shaag A, Jackman JE, Elpeleg O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J Med Genet. 2014;51:581–6. doi: 10.1136/jmedgenet-2014-102282. [DOI] [PubMed] [Google Scholar]
- Group SM. Comprehensive gene panels provide advantages over clinical exome sequencing for Mendelian diseases. Genome biology. 2015;16 doi: 10.1186/s13059-015-0693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Podyma BM, Preston MA, Shaheen HH, Krivos KL, Limbach PA, Hopper AK, Phizicky EM. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA. 2012;18:1921–33. doi: 10.1261/rna.035287.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy MP, Shaw M, Weiner CL, Hobson L, Stark Z, Rose K, Kalscheuer VM, Gecz J, Phizicky EM. Defects in tRNA Anticodon Loop 2′-O-Methylation Are Implicated in Nonsyndromic X-Linked Intellectual Disability due to Mutations in FTSJ1. Human mutation. 2015;36:1176–1187. doi: 10.1002/humu.22897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Kon Y, Phizicky EM. Functional importance of Psi38 and Psi39 in distinct tRNAs, amplified for tRNAGln(UUG) by unexpected temperature sensitivity of the s2U modification in yeast. RNA. 2015;21:188–201. doi: 10.1261/rna.048173.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss NS, Knight SW, Vulliamy TJ, Klauck SM, Wiemann S, Mason PJ, Poustka A, Dokal I. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nature genetics. 1998;19:32–38. doi: 10.1038/ng0598-32. [DOI] [PubMed] [Google Scholar]
- Hur S, Stroud RM. How U38, 39, and 40 of many tRNAs become the targets for pseudouridylation by TruA. Molecular cell. 2007;26:189–203. doi: 10.1016/j.molcel.2007.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Genin A, Lambert N, Desir J, Pirson I, Abdulkarim B, Simonis N, Drielsma A, Marselli L, Marchetti P, Vanderhaeghen P, Eizirik DL, Wuyts W, Julier C, Chakera AJ, Ellard S, Hattersley AT, Abramowicz M, Cnop M. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet. 2013;9:e1003888. doi: 10.1371/journal.pgen.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Montange RK, Malik HS, Phizicky EM. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA. 2003;9:574–85. doi: 10.1261/rna.5070303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammen HO, Marvel CC, Hardy L, Penhoet EE. Purification, structure, and properties of Escherichia coli tRNA pseudouridine synthase I. Journal of Biological Chemistry. 1988;263:2255–2263. [PubMed] [Google Scholar]
- Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Akdemir ZC, Gonzaga-Jauregui C, Erdin S, Bayram Y, Campbell IM. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron. 2015;88:499–513. doi: 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Rafiq MA, Noor A, Hussain S, Flores JV, Rupp V, Vincent AK, Malli R, Ali G, Khan FS. Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal-recessive intellectual disability. The American Journal of Human Genetics. 2012;90:856–863. doi: 10.1016/j.ajhg.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecointe F, Namy O, Hatin I, Simos G, Rousset J-P, Grosjean H. Lack of pseudouridine 38/39 in the anticodon arm of yeast cytoplasmic tRNA decreases in vivo recoding efficiency. Journal of Biological Chemistry. 2002;277:30445–30453. doi: 10.1074/jbc.M203456200. [DOI] [PubMed] [Google Scholar]
- Lecointe F, Simos G, Sauer A, Hurt EC, Motorin Y, Grosjean H. Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of Ψ38 and Ψ39 in tRNA anticodon loop. Journal of Biological Chemistry. 1998;273:1316–1323. doi: 10.1074/jbc.273.3.1316. [DOI] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Oglou OO, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic acids research. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabriel S, Frye M, Al-Gazali L, Gleeson JG. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J Med Genet. 2012;49:380–5. doi: 10.1136/jmedgenet-2011-100686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Research in developmental disabilities. 2011;32:419–436. doi: 10.1016/j.ridd.2010.12.018. [DOI] [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. The American Journal of Human Genetics. 2010;86:749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki Y, He J, Kulkarni S, Bessler M, Mason PJ. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10756–10761. doi: 10.1073/pnas.0402560101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramser J, Winnepenninckx B, Lenski C, Errijgers V, Platzer M, Schwartz C, Meindl A, Kooy R. A splice site mutation in the methyltransferase gene FTSJ1 in Xp11. 23 is associated with non-syndromic mental retardation in a large Belgian family (MRX9) Journal of medical genetics. 2004;41:679–683. doi: 10.1136/jmg.2004.019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. The Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, Ismail SI, Emam BA, Phizicky EM, Alkuraya FS. Mutation in WDR4 impairs tRNA m7G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome biology. 2015;16:1–11. doi: 10.1186/s13059-015-0779-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CE, Smith GR, Cortese R, Ames BN. Mutant tRNA His ineffective in repression and lacking two pseudouridine modifications. Nat New Biol. 1972;238:72–4. doi: 10.1038/newbio238072a0. [DOI] [PubMed] [Google Scholar]
- Torres AG, Batlle E, de Pouplana LR. Role of tRNA modifications in human diseases. Trends in molecular medicine. 2014;20:306–314. doi: 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Tsui H, Arps P, Connolly D, Winkler M. Absence of hisT-mediated tRNA pseudouridylation results in a uracil requirement that interferes with Escherichia coli K-12 cell division. Journal of bacteriology. 1991;173:7395–7400. doi: 10.1128/jb.173.22.7395-7400.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P. Wieskamp N, del Rosario M. A de novo paradigm for mental retardation. Nature genetics. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO molecular medicine. 2013;5:332–343. doi: 10.1002/emmm.201100626. [DOI] [PMC free article] [PubMed] [Google Scholar]