

Abstract
The transcriptional intermediary factor 1β (TIF1β) is a corepressor for KRAB-domain-containing zinc finger proteins and is believed to play essential roles in cell physiology by regulating chromatin organization at specific loci through association with chromatin remodeling and histone-modifying activities and recruitment of heterochromatin protein 1 (HP1) proteins. In this study, we have engineered a modified embryonal carcinoma F9 cell line (TIF1βHP1box/-) expressing a mutated TIF1β protein (TIF1βHP1box) unable to interact with HP1 proteins. Phenotypic analysis of TIF1βHP1box/- and TIF1β+/- cells shows that TIF1β–HP1 interaction is not required for differentiation of F9 cells into primitive endoderm-like (PrE) cells on retinoic acid (RA) treatment but is essential for further differentiation into parietal endoderm-like (PE) cells on addition of cAMP and for differentiation into visceral endoderm-like cells on treatment of vesicles with RA. Complementation experiments reveal that TIF1β–HP1 interaction is essential only during a short window of time within early differentiating PrE cells to establish a selective transmittable competence to terminally differentiate on further cAMP inducing signal. Moreover, the expression of three endoderm-specific genes, GATA6, HNF4, and Dab2, is down-regulated in TIF1βHP1box/- cells compared with wild-type cells during PrE differentiation. Collectively, these data demonstrate that the interaction between TIF1β and HP1 proteins is essential for progression through differentiation by regulating the expression of endoderm differentiation master players.
Keywords: Transcriptional silencing, KRAB zinc finger proteins, F9 EC cells, retinoic acid, cAMP, nuclear compartmentalization
It has become clear within the last two decades that regulation of chromatin higher-order structures by histone modification and chromatin remodeling is essential for genome programming during early embryogenesis, tissue-specific gene expression, and global gene silencing (for review, see Li 2002). The isolation and biochemical characterization of several complexes involved in chromatin remodeling, including SWI/SNF, NuRD, and ISWI, led to major breakthroughs into the mechanisms of chromatin structure regulation (for review, see Narlikar et al. 2002). The identification of chromatin-modifying enzymes (e.g., histone acetylases, deacetylases, and methyltransferases) and determination of their substrate specificities suggested the existence of a histone code (Jenuwein and Allis 2001). However, how these activities are coordinated to respond properly to any environmental stimulus in vivo remains largely unknown.
TIF1β (Le Douarin et al. 1996), also known as KAP-1 (Friedman et al. 1996) or KRIP-1 (Kim et al. 1996), is a member of an emerging family of transcriptional regulators designated as TIF1 (transcriptional intermediary factor 1). The TIF1 family includes TIF1α (Le Douarin et al. 1995), TIF1γ (Venturini et al. 1999; Yan et al. 2004) in mammals, and Bonus in Drosophila (Beckstead et al. 2001). TIF1 proteins are defined by the presence of two conserved amino acid regions: an N-terminal RBCC (RING finger, B boxes, coiled-coil) motif that is involved in homo- and heterodimerization (Peng et al. 2002), and a C-terminal bromodomain preceded by a PHD (plant homeodomain) finger. These latter two motifs are often associated and present in a number of transcriptional cofactors acting at the chromatin level (Aasland et al. 1995; Jeanmougin et al. 1997; Zeng and Zhou 2002).
Recent genetic studies in mice have provided evidence that TIF1β is a developmental regulatory protein that exerts cellular function(s) essential for early embryonic development (Cammas et al. 2000) and for spermatogenesis (Weber et al. 2002). Furthermore, TIF1β displays several biochemical properties suggesting that it could be a coordinator in epigenetic regulation of transcription: (1) TIF1β is a universal corepressor for a large family of transcription factors, the Krüppel associated box (KRAB)-domain containing zinc finger proteins (Friedman et al. 1996; Kim et al. 1996; Moosmann et al. 1996; Abrink et al. 2001); (2) TIF1β is an intrinsic component of two chromatin remodeling and histone deacetylase complexes, N-CoR1 and NuRD (Underhill et al. 2000; Schultz et al. 2001); (3) TIF1β directly interacts with the histone methyltransferase SETDB1, which specifically methylates Lys 9 of histone H3 preferentially within euchromatin (Schultz et al. 2002); and (4) TIF1β interacts with members of the heterochromatin protein 1 (HP1) family (Le Douarin et al. 1996; Nielsen et al. 1999; Ryan et al. 1999).
HP1 is a structurally and functionally highly conserved protein with family members found in a variety of eukaryotic organisms ranging from Schizosaccharomyces pombe to humans (Eissenberg et al. 1990; Wang et al. 2000). These proteins participate in chromatin packaging and have a well-established function in heterochromatin-mediated silencing (for review, see Eissenberg and Elgin 2000). Mice and humans each have three different HP1 proteins (HP1α, β, and γ) that are associated, although not exclusively, with pericentromeric heterochromatin (Nielsen et al. 1999). The structure of the HP1-like proteins comprises a conserved N-terminal region called chromo-domain (CD) and a conserved C-terminal chromo shadow domain (CSD) separated by a less conserved hinge region (Eissenberg 2001). The HP1 CD binds methylated Lys 9 of histone H3 (Lachner et al. 2001; Nakayama et al. 2001), as well as the histone fold motif of histone H3 (Nielsen et al. 2001a). HP1s interact with a large number of proteins and in particular with several proteins known to function at the transcriptional level through a specific pentapeptide PxVxL called HP1 box (Le Douarin et al. 1996; Thiru et al. 2004). These proteins include the chromatin remodeling factor BRG1 (Nielsen et al. 2002), the TBP-associated factor TAF130 (Vassallo and Tanese 2002), the retinoblastoma protein Rb (Nielsen et al. 2001b), and the transcriptional intermediary factors TIF1α and TIF1β (Le Douarin et al. 1996; Nielsen et al. 1999). It is currently speculated that HP1 serves as a bridging protein, connecting histones through interaction with the CD to nonhistone chromosomal proteins through interaction with the CSD (Li et al. 2002). We and others have previously demonstrated that the interaction between TIF1β and HP1 is required for the TIF1β transcriptional repression activity, which also requires histone deacetylase activity (Nielsen et al. 1999; Ryan et al. 1999). In mouse embryonal carcinoma (EC) F9 cells that resemble the pluripotent inner cell mass cells of the early embryo and can be induced to differentiate into primitive endoderm-like (PrE) cells or into parietal endoderm-like (PE) cells on treatment with retinoic acid (RA) alone or RA plus cAMP, respectively (Strickland and Mahdavi 1978; Strickland et al. 1980), we have shown that the interaction between TIF1β and HP1 proteins was essential for the relocation of TIF1β from eu- to heterochromatin that accompanies PrE differentiation (Cammas et al. 2002). Thus TIF1β–HP1 interaction may play an essential role during cell differentiation.
In this paper, we report the first demonstration of the functional relevance of the interaction between TIF1β and HP1 proteins. We have introduced by homologous recombination in F9 EC cells a mutation in the TIF1β HP1 box motif that had previously been shown to disrupt the interaction between TIF1β and HP1 proteins (Cammas et al. 2002). We demonstrate that TIF1β–HP1 interaction is essential for F9 cell differentiation into PE cells and into visceral endoderm-like (VE) cells obtained by treatment of vesicles with RA.
Results
Targeted mutation of the TIF1β HP1 box
To disrupt the interaction between TIF1β and HP1 proteins in F9 EC cells, we specifically mutated the HP1 box coding sequence of one TIF1β allele and inactivated the other TIF1β allele. Two targeting vectors, pTIF1βLHL:HP1box and pTIF1βLNL:L (Fig. 1A), were constructed. pTIF1βLNL:L was obtained by introduction of a PGK-Neo selection cassette flanked by two LoxP sites in intron 3 and a LoxP site in intron 14 (Fig. 1A; see Materials and Methods; Cammas et al. 2000). pTIF1βLHL:HP1box was obtained by introduction of a PGK-Hygro selection cassette flanked by two LoxP sites in intron 3 and mutation of the TIF1β HP1 box-coding sequence within exon 12 (substitution of two amino acids, V488L490/AA), which was previously shown to disrupt the interaction between TIF1β and HP1α, β, and γ (Fig. 1A; Materials and Methods; Cammas et al. 2002). This mutation generated an Eco47III restriction site in the targeted TIF1β allele (E47, Fig. 1A).
Figure 1.

Targeted mutation of the TIF1β HP1box motif. (A) Diagram showing the genomic map of TIF1β; the targeting constructs for the disruption of the first TIF1β allele and for the mutation in the HP1box motif of the second TIF1β allele; and the targeted alleles before (L3 and L2HP1box, respectively) and after (L- and LHP1box, respectively) Cre-mediated excision of the LoxP-sites-flanked sequences. Exons are represented as numbered boxes and introns as connected lines. The LoxP sites are represented by open triangles and the PGK-Neo, PGK-hygro, and diphterin toxin A (DTA) cassettes are indicated. The 5′ and 3′ probes have previously been described (Cammas et al. 2000). The size of the DNA fragments expected with the 5′ probe on digestion with EcoRV and with the 3′ probe on digestion with BamHI are indicated. Relevant restriction sites are EcoRV (V), EcoRI (E), BamHI (B), HindIII (H), XhoI (X), and Eco47III (E47). (B) Southern blot analysis of DNAs derived from wild-type (WT), targeted TIF1β+/L3, and TIF1βL3/L2HP1box F9 cells and from the corresponding TIF1β+/L- and TIF1βL-/LHP1box F9 cells that had the Lox-P-flanked DNA sequences deleted by Cre-mediated excision. Genomic DNA was digested with EcoRV or BamHI as indicated, blotted, and hybridized with the 5′, 3′ probes as indicated. (C) PCR strategy for amplification of wild-type (WT), deleted (L-), and mutated (LHLHP1box and LHP1box) TIF1β alleles. DNA samples were subjected to PCR amplification using a mixture of three primers (YD208, VR211, and VR211; see Materials and Methods) or two primers surrounding the HP1box motif (VR216 and TV211; see Materials and Methods). (D) PCR amplification with YD208 and VR211 produced a 152-bp DNA fragment (wild-type [WT]) and a 171-bp DNA fragment (LHP1box) whereas PCR amplification with YD208 and TV210 produced a 390-bp DNA fragment for the deleted TIF1β allele (L-). (Upper band) PCR amplification produced a 726-bp fragment with the wild-type (WT), L3, LHP1box, and L2HP1box TIF1β alleles. (Lower bands) This 726-bp DNA fragment was digested by Eco47III and produced two smaller bands of 305 and 421 bp specifically for LHP1box and L2HP1box alleles (see Materials and Methods). (E) TIF1β protein levels in wild-type (WT), TIF1β+/L-, and TIF1βL-/LHP1box F9 cells. Increasing amounts of whole-cell extracts (3–30 μg) were analyzed by Western blotting using the anti-TIF1β monoclonal antibody 1TB3 directed against the C terminus (amino acids 123–834) or the anti-RXRα polyclonal antibody (pAb) RPRXα. (F) TIF1βHP1box does not interact with HP1. WCE from wild-type (WT), TIF1β+/-, and TIF1βHP1box/- F9 cells were analyzed by Western blotting either directly (input) or following immunoprecipitation with a TIF1β mAb (TIF1β IP). The control IP was done with an anti-Flag antibody. Western blots probed with HP1α, HP1β, HP1γ, and TIF1β mAbs are shown. Inputs correspond to 1/10 the amount of cell extracts used for IP.
F9 cells were electroporated with pTIF1βLNL:L. One G-418-resistant clone, TIF1βL3/+, with homologous recombination of a single copy of the targeting vector as assessed by Southern blot with three different probes (3′, 5′, and Neo probes [Fig. 1A,B]; data not shown) was isolated and was transfected with the pTIF1βLHL:HP1box targeting construct. A clone resistant to both G-418 and hygromycin and having integrated a single copy of pTIF1βLHL:HP1box into the second TIF1β allele (5′ and Hygro probes [Fig. 1B]; PCR analysis with primers flanking the HP1box, followed by digestion with Eco47III [Fig. 1C,D]; data not shown) was isolated and called TIF1βL3/L2HP1box. To excise the LoxP-flanked sequences, we transiently transfected TIF1βL3/L2HP1box cells with the Cre-recombinase encoding expression vector pPGK-Cre, yielding a clone, TIF1βLHP1box/L-, with complete excision in one allele, whereas the other allele had the HP1box motif point mutations (Southern blot analysis with three different probes: 5′, Neo, and Hygro [Fig. 1B]; data not shown; PCR analysis followed by Eco47III digestion [Fig. 1C,D]). The heterozygous clone TIF1β+/L- was obtained by transfection of TIF1β+/L3 cells with pPGK-Cre (Fig. 1A,B).
The level of TIF1β gene expression in both TIF1β+/L- and TIF1βLHP1box/L- F9 cell lines was compared with the level of TIF1β expression in wild-type F9 cells. A TIF1β monoclonal antibody (mAb) revealed a decrease by ∼50% in TIF1β+/L- and TIF1βLHP1box/L- F9 cells in comparison with wild-type F9 cells. Thus, the HP1 box mutation had no effect on the level of expression of the mutated TIF1β allele (Fig. 1E). TIF1β+/L- and TIF1βLHP1box/L- F9 cell lines, thereafter designated as TIF1β+/- and TIF1βHP1box/- cells, respectively, expressed the wild-type (TIF1β+) and mutant (TIF1βHP1box) proteins, respectively.
The HP1 box mutation has previously been shown to disrupt the interaction between TIF1β and HP1α, HP1β, and HP1γ (Weber et al. 2002). To investigate whether the TIF1βHP1box mutant protein was associated with HP1 proteins in TIF1βHP1box/- cells, we performed immunoprecipitation (IP). Whole-cell extracts (WCE) from wild-type and TIF1βHP1box/- F9 cells were immunoprecipitated with TIF1β mAb. As indicated by Western blot analysis, HP1α, HP1β, and HP1γ were found in wild-type immunoprecipitates, but not in TIF1βHP1box/- immunoprecipitates (Fig. 1F). Thus, the TIF1βHP1box mutant protein is not associated with HP1 proteins within TIF1βHP1box/- cells.
TIF1β interaction with HP1 proteins is not required for PrE cell differentiation
Treatment of wild-type, TIF1β+/-, and TIF1βHP1box/- cells with 1 μM RA led to an enlargement and flattening of the cells, which are morphological features characteristic of F9 cell differentiation into PrE cells (Fig. 2, panels a–f). To confirm the PrE identity of these cells, we analyzed expression of Troma-1, a marker for F9 cell differentiation (Kemler et al. 1981; Verheijen et al. 1999). Wild-type, TIF1β+/-, and TIF1βHP1box/- cells were grown for 4 d in the absence or presence of 1μM RA and analyzed by immunocytochemistry. Troma-1 was weakly expressed and localized exclusively in nuclei of nontreated wild-type, TIF1β+/-, and TIF1βHP1box/- cells (Fig. 3A, panels a,b,e,f,i,j), whereas it was highly expressed and distributed in the cytoplasm of RA-treated cells (Fig. 3A, panels c,d,g,h,k,l), indicating that these cells have features of PrE cells.
Figure 2.

TIF1βHP1box/- F9 cells differentiate into PrE cells but not into PE or VE cells. At day 0, 104 wild-type (WT), TIF1β+/-, and TIF1βHP1box/- cells were plated, and they were treated as described at day 1. (A) Wild-type (WT, panels a,d,g), TIF1β+/- (panels b,e,h), and TIF1βHP1box/- (panels c,f,i) cells were cultured either with vehicle (no treatment; panels a,b,c), in the presence of 1 μM tRA (panels d,e,f), or in the presence of 1 μM tRA + 250 μM dbcAMP (panels g,h,i). (B) Wild-type (WT, panels a,d), TIF1β+/- (panels b,e), and TIF1βHP1box/- (panels c,f) cells were cultured in bacterial Petri dishes with vehicle (no treatment; panels a–c) or in the presence of 50 nM RA (panels d–f) Cells were photographed at day 6 under a phase contrast microscope. Bar, 100 μm.
Figure 3.

The relocation of TIF1β from eu- to heterochromatin is not required for PrE differentiation. (A) Wild-type (WT), TIF1β+/-, and TIF1βHP1box/- F9 cells were grown on glass coverslips, treated with 1 μM tRA for 4 d, fixed, and hybridized with an anti-Troma-1 mAb as indicated. Nuclei were visualized by Hoechst staining. Projection of three confocal sections through nondifferentiated and differentiated wild-type, TIF1β+/-, and TIF1βHP1box/- F9 cells is shown. Bar, 50 μm. (B) TIF1βHP1box does not relocate from eu- to heterochromatin during PrE differentiation. TIF1β+/- and TIF1βHP1box/- were grown on glass coverslips for 4 d in the presence or absence of 1 μM tRA, fixed, and hybridized with an anti-Troma-1 mAb and the anti-TIF1β pAb PF64. The DNA content was visualized by Hoechst staining. Single confocal sections through differentiated (panels g–l) and nondifferentiated cells (panels a–f) are shown. Bars, 5 μm.
We previously reported that PrE differentiation is accompanied by relocation of TIF1β from eu- to heterochromatin (Cammas et al. 2002). The localization of TIF1βHP1box was analyzed during PrE differentiation. wild-type, TIF1β+/-, and TIF1βHP1box/- cells were grown for 4 d in the absence or presence of 1 μM RA and analyzed by immunocytochemistry. In nontreated cells, TIF1β labeling was rather uniformly distributed within the nucleoplasm and largely excluded from the nucleoli of wild-type, TIF1β+/-, and TIF1βHP1box/- cells (Fig. 3B, panels a,b,d,e; data not shown). After 4 d of RA treatment, wild-type and TIF1β+/- cells displayed an enrichment of TIF1β within heterochromatin characterized by bright spots of Hoechst staining, whereas TIF1βHP1box remained diffusely distributed throughout TIF1βHP1box/- nuclei (Fig. 3B, panels g,h,j,k). The PrE identity of these cells exhibiting diffused distribution of TIF1βHP1box was confirmed by expression of Troma-1 (Fig. 3B, panels i,l). Taken together, these results indicate that the interaction between TIF1β and HP1 proteins is not required for PrE differentiation.
TIF1β interaction with HP1 proteins is required for terminal differentiation into PE cells and into VE cells
Wild-type, TIF1β+/-, and TIF1βHP1box/- cells were treated for 4 d with RA, and then with 250 μM dbcAMP and 1 μM RA for 2 d. wild-type and TIF1β+/- cells differentiated into cells exhibiting a very stringent core body and neuronal-like outgrowths that are morphological changes characteristic of PE differentiation (Fig. 2, panels g,h). In contrast, TIF1βHP1box/- cells differentiated into PrE cells but did not further differentiate into PE cells (Fig. 2, panels d–f,i). Expression of Troma-1 in these cells confirmed their differentiated status (Fig. 4A, panel f). To investigate the RA + cAMP responsiveness of TIF1βHP1box/- cells at the molecular level, we analyzed the expression of thrombomodulin (TM), a molecular marker unique to PE cells (Weiler-Guettler et al. 1992). RNA from wild-type, TIF1β+/-, and TIF1βHP1box/- cells grown for 6 d with either vehicle or 1 μM RA or for 4 d with 1 μM RA followed by addition of 250 μM dbcAMP for 2 d were analyzed by semiquantitative RT–PCR. No TM expression was detected in either nontreated or RA-treated cell lines (Fig. 4B; lanes 1–6). In contrast, TM expression was induced after treatment with RA-db-cAMP in wild-type and TIF1β+/- cells, whereas it could not be detected in TIF1βHP1box/- cells (Fig. 4B; lanes 7–9). Thus, in contrast to wild-type and TIF1β+/- cells, TIF1βHP1box/- cells do not undergo PE differentiation after treatment with RA-dbcAMP. Note that TM expression was lower in TIF1β+/- cells when compared with wild-type cells, in agreement with the observation that ∼20%–30% of the TIF1β+/- cell population failed to morphologically differentiate into PE cells, indicating that the correct gene dosage of TIF1β is required for full differentiation into PE cells (data not shown). It is noteworthy that TIF1βHP1box/- cells treated for 6 d with RA and dbcAMP added simultaneously were also unable to undergo PE differentiation (data not shown). Altogether, these results demonstrate that differentiation of TIF1βHP1box/- cells on RA-dbcAMP treatment is blocked at the PrE stage, and therefore that the interaction between TIF1β and HP1 proteins is essential for differentiation of PrE cells into PE cells.
Figure 4.

TIF1βHP1box/- cells do not differentiate into PE cells. (A) TIF1β+/- and TIF1βHP1box/- cells were grown on glass coverslides, treated with vehicle (no treatment) or with 1 μM RA + 250 μM dbcAMP for 6 d, fixed, and hybridized with an anti-Troma-1 mAb antibody. DNA content was visualized by Hoechst staining. Bar, 5 μm. (B) RNA from wild-type (WT), TIF1β+/-, and TIF1βHP1box/- F9 cells treated for 96 h with either the vehicle or 1 μM tRA or for 6 d with 1 μM tRA + 250 μM dbcAMP were subjected to semiquantitative RT–PCR analysis with the PE differentiation marker thrombomodulin (TM)-specific primers. The HPRT RT–PCR was used as an internal control. (C) TIF1β transiently concentrates within pericentromeric heterochromatin during PE differentiation. Wild-type (WT) cells were grown on glass coverslides in the presence of either 1 μM RA (open bars) or 1 μM RA plus 250 μM dbcAMP (black bars) for 1, 2, 3, 4, and 6 d; fixed; and hybridized with an anti-TIF1β mAb. The percentage of cells displaying TIF1β heterochromatic foci is plotted. (D) TIF1β expression is decreased during PE differentiation. Wild-type (WT) and TIF1βHP1box/- cells were grown for 4 d in the presence of 1 μM RA followed by 2 d in the presence of 1 μM RA plus 250 μM dbcAMP. Cells were collected each day and WCE of wild-type (TIF1β) of TIF1βHP1box/- (TIF1βHP1box) cells were analyzed by Western blot with a TIF1β-specific mAb. Actin was used as an equal loading control.
To evaluate the requirement of the TIF1β–HP1 interaction for VE differentiation, we grew wild-type, TIF1β+/-, and TIF1βHP1box/- cells in bacterial Petri dishes to allow formation of vesicles and treated them for 6 d with 50 nM RA as described previously (Casanova and Grabel 1988). Wild-type and TIF1β+/- cells treated with RA formed vesicles with an outer layer of large stringent cells typical of VE differentiation (Fig. 2B, panels d,e). In contrast, TIF1βHP1box/- cells rapidly degenerated from 2 d (data not shown) to 6 d of RA treatment at a stage at which there were only few residual vesicles left, as illustrated in Figure 2B, panel f. This effect is specific of VE differentiation and not of the growth conditions because TIF1βHP1box/- cells grown on bacterial Petri dishes in the absence of RA treatment formed vesicles resembling wild-type and TIF1β+/- vesicles (Fig. 2B, panels a–c). These results indicate that the interaction between TIF1β and HP1 is essential for VE differentiation.
We have previously shown and confirmed in the present study that association of TIF1β to heterochromatin was not observed within differentiated PE nuclei (Fig. 4A, panel b; Cammas et al. 2002). However, PE differentiation is a two-step differentiation process including an RA-dependent phase followed by a cAMP-dependent phase. It is therefore conceivable that TIF1β could concentrate within heterochromatin at specific stages during the process of PE differentiation. To address this question, we treated F9 cells for 6 d with either 1 μM RA alone or with 1 μM RA plus 250 μM dbcAMP. Cells were analyzed each day of the differentiation processes by immunofluorescence with a TIF1β mAb. As previously observed (Cammas et al. 2002), RA treatment led to relocation of TIF1β from eu- to heterochromatin in a progressively increasing number of nuclei to reach a maximum of ∼50% after 4 d of treatment (Fig. 4C). This percentage remained unchanged at all subsequent points (Fig. 4C; data not shown). In the presence of RA plus dbcAMP, TIF1β also relocates from eu- to heterochromatin until 3 d of treatment to reach a maximum of ∼20%–25%. Thereafter, the percentage of cells with heterochromatic TIF1β decreased to reach ∼0%–5% after 6 d of PE differentiation (Fig. 4C). The kinetics of replacement of the cell population with heterochromatic TIF1β by a cell population with diffused nuclear TIF1β correlated with the morphological changes associated with the differentiation of PrE cells into PE cells (data not shown). It is noteworthy that the homogenous localization of TIF1β within PE nuclei is also observed when F9 cells are sequentially treated 4 d with 1 μM RA followed by 2 d with 1 μM RA plus 250 μM dbcAMP (data not shown). Altogether, these results demonstrated that TIF1β concentration within heterochromatin is a transient status during PE differentiation, whereas it is permanently established during PrE differentiation. Interestingly, TIF1β relocation from eu- to heterochromatin also occurs during VE differentiation of F9 cells (data not shown). These results suggest that the relocation of TIF1β from eu- to heterochromatin during the RA-treated phase of endodermal differentiation might play a role in the regulation of TIF1β concentration within differentiated cell nuclei. The level of TIF1β and TIF1βHP1box was assessed in wild-type and TIF1βHP1box/- cells, respectively, during PE differentiation. Western blot analysis with a TIF1β mAb revealed that TIF1β concentration is constant for up to 2 d of RA treatment and then decreases of 10-fold after 4 d of treatment (Fig. 4D, cf. lanes 5 and 1). The level of TIF1β further decreased by 1.7-fold after treatment with dbcAMP (Fig. 4D, lanes 6,7). In TIF1βHP1box/- cells, the level of TIF1βHP1box was also decreased after 4 d of RA treatment (fourfold) but did not further decrease after RA-dbcAMP treatment (Fig. 4D). This expression of TIF1β within F9 PE cells is in contrast with our previous observation that TIF1β transcript was not detected, by in situ hybridization, within PE and VE layers of embryonic day 6.5 (E6.5) and E7.5 embryos (Cammas et al. 2000). We therefore analyzed TIF1β expression within embryos by immunohistochemistry using the TIF1β pAb, PF64. This study revealed that, as in F9 cells, TIF1β is expressed in both PE and VE of E6.5 and PE of E7.5 embryos (data not shown).
Ectopic expression of TIF1β into TIF1βHP1box/- cells restores their ability to differentiate into PE cells
To unequivocally demonstrate that the failure of TIF1βHP1box/- cells to differentiate into PE cells reflects the requirement of an interaction between TIF1β and HP1 proteins, we investigated whether ectopically expressed TIF1β or TIF1βHP1box mutant proteins into TIF1βHP1box/- cells could restore PE differentiation. TIF1βHP1box/- cells were transfected with three constructs to perform doxycycline (Dox)-inducible expression of TIF1β: (1) an expression cassette encoding the reverse tetracycline-controlled transactivator (rtTA; Gossen et al. 1995); (2) a construct containing the mouse Flag-tagged TIF1β or TIF1βHP1box cDNAs (f.TIF1β and f.TIF1βHP1box, respectively) under the control of tetracycline operators; and (3) a PGK-Hygro selection cassette-expressing vector (Fig. 5A). Clones resistant to hygromycin were amplified, grown in the presence or absence of 1 μg/mL Dox for 3 d, and analyzed by Western blotting with an anti-Flag mAb. Two cell lines expressing either f.TIF1β or f.TIF1βHP1box proteins selectively in the presence of Dox were isolated and designated TIF1βHP1box/-/rtTA-f.TIF1β and TIF1βHP1box/-/rtTA-f.TIF1βHP1box, respectively (Fig. 5B). Western blotting using a TIF1β-specific mAb showed that in the presence of Dox, TIF1βHP1box/-/rtTA-f.TIF1β cells express a total amount of TIF1β protein similar to wild-type cells, whereas TIF1βHP1box/-/rtTA-f.TIF1βHP1box cells express a slightly lower level (Fig. 5B, lane TIF1β). The ability of these two cell lines to undergo PE differentiation on RA-dbcAMP treatment was analyzed in the absence or presence of 1 μg/mL Dox. TIF1βHP1box/-/rTA-f.TIF1β cells treated with RA-dbcAMP in the presence of Dox differentiated into PE cells as assessed by morphological analysis, whereas the same cells differentiated only into PrE cells in the absence of Dox (Fig. 5C, panels f,b). In contrast, TIF1βHP1box/-/rTA-f.TIF1βHP1box cells treated with RA-dbcAMP in the presence or absence of Dox only differentiated into PrE-like cells (Fig. 5C, panels d,h). These results were confirmed by the expression of the PE marker TM. As expected, TM was expressed in RA-dbcAMP-treated wild-type, but not TIF1βHP1box/- cells, independently of the presence of Dox (Fig. 5D, lanes 1–8). In TIF1βHP1box/-/rTA-f.TIF1β cells, but not in TIF1βHP1box/-/rTA-f.TIF1βHP1box cells, TM was induced specifically in the presence of RA-dbcAMP and Dox, thus confirming that ectopic expression of TIF1β enabled TIF1βHP1box/- cells to differentiate into PE cells, whereas TIF1βHP1box did not (Fig. 5D, lanes 9–16). Note that the level of TM induction in TIF1βHP1box/-/rTA-F.TIF1β was reduced in comparison with wild-type cells, confirming that PE differentiation is sensitive to TIF1β dosage.
Figure 5.

Expression of ectopic TIF1β cDNA in TIF1βHP1box/- F9 cells rescues the ability of these cells to differentiate into PE cells. (A) TIF1βHP1box/- F9 cells were cotransfected with a vector allowing the expression of the reverse tetracycline activator (rtTA) cDNA under the control of the human cytomegalovirus (hCMV) promoter/enhancer and with a vector driving the expression of either the Flag-tagged TIF1β or the Flag-tagged TIF1βHP1box cDNAs under the control of the hCMV/tetracycline operator (hCMV/Tet-Op) in parallel with a plasmid carrying the PGK-hygro cassette as a selection marker. (B) One stable hygromycin-resistant cell line expressing either Flag-TIF1β or Flag-TIF1βHP1box proteins only after induction with 1 μg/mL doxycyclin (Dox) as assessed by Western blot analysis using an anti-Flag mAb were chosen (TIF1βHP1box/-/rTA-f.TIF1β and TIF1βHP1box/-/rTA-f.TIF1βHP?box). Total amount of TIF1β (TIF1β wild-type [WT] and TIF1βHP1box) was assessed using a TIF1β mAb. (C) TIF1βHP1box/-/rTA-f.TIF1β and TIF1βHP1box/-/rTA-f.TIF1βHP?box F9 cells were grown for 6 d without (panels a–d) or with (panels e–h) 1 μg/mL Dox, in the absence (panels a,c,e,g) or presence (panels b,d,g,h) of 1 μM RA + 250 μM dbcAMP. Cells were photographed under a phase contrast microscope. Bar, 100 μm. (D) RNA from wild-type (WT), TIF1βHP1box/-/rTA-f.TIF1β, and TIF1βHP1box/-/rTA-f.TIF1βHP1box cells induced by 1 μM RA + 250 μM dbcAMP in the absence or presence of 1 μg/mL Dox were subjected to RT–PCR analysis with TM-specific primers. The HPRT RT–PCR was used as an internal control.
Taken together, these results firmly establish that TIF1β interaction with HP1 proteins is indispensable to enable F9 cells to differentiate into PE cells, whereas it is not required for PrE differentiation.
Interaction between TIF1β and HP1 is essential during RA-induced PrE differentiation to enable further PE differentiation
To establish whether the TIF1β–HP1 interaction was required throughout PE differentiation, we took advantage of the temporally controlled inducibility of expression of f.TIF1β cDNA in TIF1βHP1box/-/rtTA-f.TIF1β cells. These cells were grown in PE differentiation-inducing conditions: (1) 3 d in the absence of treatment (Fig. 6A, d-3 to d0), followed by (2) 4 d in the presence of 1 μM RA (Fig. 6A, d0 to d4), and finally (3) 2 d in the presence of 1 μM RA plus 250 μM dbcAMP (Fig. 6A, d4 to d6). f.TIF1β expression was induced in these cells by addition of 1 μg/mL Dox at various times of the differentiation process (Fig. 6A). Western blot analysis using an anti-Flag antibody revealed that f.TIF1β was expressed during different phases of the differentiation process (Fig. 6B). Actin was used as an equal loading control (data not shown).
Figure 6.

TIF1β interaction with HP1 is essential during RA-induced PrE differentiation to enable further differentiation into PE cells. (A) Scheme representing the treatment of TIF1βHP1box/-/rTA-f.TIF1β F9 cells. TIF1βHP1box/-/rTA-f.TIF1β F9 cells were first grown for 3 d without treatment (d-3 to d0), followed by 4 d of treatment with 1 μM tRA (d0 to d4) and 2 d with 1 μM tRA + 250 μM dbcAMP (d4 to d6). (Lanes 1–11) Dox was added to the medium during the indicated periods of time. (B) Flag-TIF1β protein level in TIF1βHP1box/-/rTA-f.TIF1β F9 cells. WCE were prepared each day from d-2 to d4 and at day d6. (C) The percentage of clones containing cells with morphological features of PE cells at day d6 is plotted for each growing condition. Each bar represents the mean + standard deviation of data from three independent differentiation experiments. For each point, >200 clones were counted. (D) RNA from TIF1βHP1box/-/rTA-f.TIF1β cells treated in the previously described conditions were subjected to semiquantitative RT–PCR analysis with TM-specific primers. The HPRT RT–PCR was used as an internal control.
To quantify the extent of rescue of the PE phenotype by f.TIF1β expression, we performed morphological analysis in the previously described growing conditions (Fig. 6A), with cell dilutions allowing us to count individually each clone arising from single cells. The percentage of clones containing PE cells in each growing condition is plotted in Figure 6C. Note that in the same experimental conditions, 100% of wild-type cells underwent PE differentiation (data not shown). This morphological analysis indicated that no PE cells were detected in TIF1βHP1box/- cells expressing TIF1β only from d-2 to d1 (Fig. 6B,C, lane 3), whereas PE differentiation was induced in cells expressing TIF1β from d0 (Fig. 6B,C, lane 8). Therefore, expression of f.TIF1β before RA treatment is neither necessary (Fig. 6B,C, lanes 7,8) nor sufficient (Fig. 6B,C, lane 3) to enable TIF1βHP1box/-/rtTA-f.TIF1β cells to undergo PE differentiation. In contrast, TIF1β must be expressed between at least the first and second day of RA treatment in order for PE differentiation to occur (Fig. 6B,C, lanes 5–9). Strikingly, cells that expressed TIF1β only after 2–3 d of RA treatment were mostly blocked at the PrE stage of differentiation (Fig. 6B,C, lanes 10,11), demonstrating that the TIF1β–HP1 interaction was essential for PE differentiation well before the addition of dbcAMP. This morphological analysis was confirmed at the molecular level by the expression of TM, which was only induced in cells expressing f.TIF1β between at least the first and second day of RA treatment (Fig. 6D, lanes 5–9).
Altogether, these results indicated that the essential role of the interaction between TIF1β and HP1 proteins for PE differentiation is restricted to an early time window during the initial RA-dependent phase of the differentiation process.
Interaction between TIF1β and HP1 is essential to regulate expression of endoderm-specific genes
To further investigate the RA responsiveness of TIF1βHP1box/- cells, we analyzed the expression of several genes, including (1) genes known to play essential roles in mouse endoderm differentiation: the transcription factor genes GATA6 (Morrisey et al. 1998), HNF4 (Chen et al. 1994), and Oct3/4 (Niwa et al. 2000) and the signal transduction adapter protein Disabled-2 (Dab2; Yang et al. 2002); and (2) genes whose expression is known to be induced by RA treatment of F9 cells: the transcription factor genes RARβ2 and Hoxb1, and genes encoding the components of the extracellular matrix procolIagen type IV (ColIV) and laminin B1 (LamB1; Harris and Childs 2002). Wild-type, TIF1β+/-, and TIF1βHP1box/- cells were either not treated or treated for 24 h or 48 h with 1 μM RA. These time points corresponded to the window of time during which TIF1β–HP1 interaction is indispensable for PE differentiation. Expression was analyzed by semiquantitative RT–PCR analysis. We found that, in the absence of RA treatment, only two genes, HNF4 and Oct3/4, had a detectable level of expression and were both down-regulated in TIF1βHP1box/- cells compared with wild-type cells (seven- and threefold, respectively; Fig. 7, lanes 1–3). For Oct3/4, the difference between wild-type and TIF1βHP1box/- cells was maintained constant in the presence of RA treatment, even though, as expected, Oct3/4 expression was repressed by RA treatment in wild-type and TIF1β+/- cells (2.5-fold after 24 h of RA treatment; Fig. 7, cf. lanes 4,5 and 1,2). For all of the other genes tested, RA induced their expression in wild-type, TIF1β+/-, and TIF1βHP1box/- cells (Fig. 7, cf. lanes 4–9 and 1–3). However, the extent of induction was highly dependent on cell genotypes. After 24 h of RA treatment, GATA6 and HNF4 expression was, respectively, four and three times lower in TIF1βHP1box/- cells in comparison with wild-type cells, whereas the expression of all of the other genes was not significantly different between the two cell lines. After 48 h of RA treatment, GATA6, Dab2, HNF4, ColIV, and lamB1 displayed reduced expression in TIF1βHP1box/- cells in comparison with wild-type cells (Fig. 7; eight-, seven-, 40-, 1.5-, and twofold, respectively). In sharp contrast, expression of RARβ2 was equivalent in wild-type and TIF1βHP1box/- cells, and Hoxb1 expression was higher in TIF1βHP1box/- cells in comparison with wild-type cells (Fig. 7, threefold).
Figure 7.
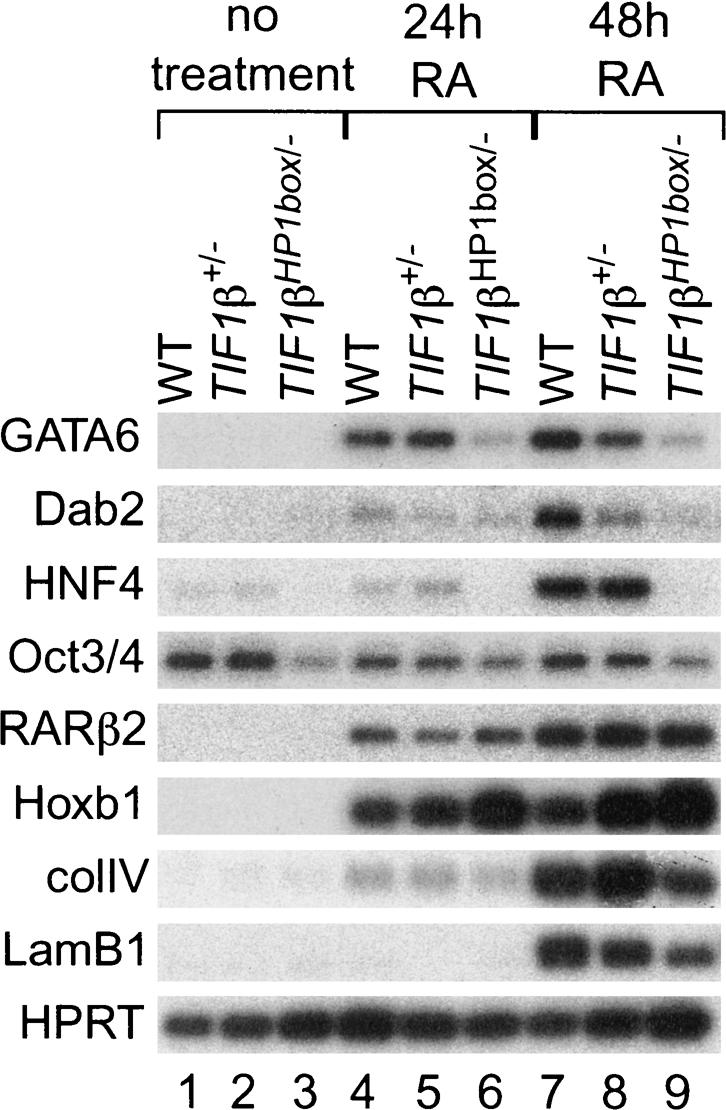
Interaction between TIF1β and the HP1s is essential for accurate expression of endoderm-specific genes during PrE differentiation. Wild-type (WT), TIF1β+/-, and TIF1βHP1box/- cells were treated 48 h with vehicle (-) or 24 or 48 h with 1 μM RA. Total RNA extracted from these cells was submitted to semiquantitative RT–PCR analysis for the expression of GATA6, Dab-2, HNF4, oct3/4, RARβ2, Hoxb1, ColIV, and lamB1. HPRT RT–PCR was used as an internal control.
Taken together, these results indicate that the interaction between TIF1β and HP1 proteins is essential to regulate the expression of specific genes in both nondifferentiated and differentiated F9 cells, and in particular is critical for the induction of endoderm master players in response to RA treatment.
Discussion
We previously reported that TIF1β relocates from eu- to heterochromatin during RA-induced PrE differentiation of F9 cells via its interaction with HP1 proteins (Cammas et al. 2002). In this study, we demonstrate, by specifically mutating in EC F9 cells the HP1-interacting PxVxL motif of TIF1β, that the association between TIF1β and HP1 proteins is essential for F9 cells to differentiate into PE and VE cells.
The TIF1β–HP1 association is required for progression through differentiation
Differentiation is a highly regulated process that comprises different phases including self-renewal, commitment, initiation, and progression through terminal differentiation. These different phases correlate with specific combinations of gene induction and repression orchestrated by a limited number of master players (for review, see Fisher 2002). When grown as a monolayer, EC F9 cells treated either sequentially or in combination with RA plus cAMP differentiate first into PrE cells, and then into terminally differentiated PE cells (Strickland et al. 1980). This progression through differentiation is marked by characteristic morphological changes as well as by a bimodal expression pattern of hundreds of genes (Harris and Childs 2002). We have shown here that the disruption of the interaction between TIF1β and HP1 does not affect the ability of EC F9 cells (TIF1βHP1box/- cells) to morphologically differentiate into PrE cells, but completely abrogates their capacity to differentiate into PE cells on further treatment with cAMP, indicating that the TIF1β–HP1 interaction is not required to initiate differentiation, but is essential at later stages to allow terminal differentiation. Furthermore, using a temporally controlled system of TIF1β expression to re-establish the interaction between TIF1β and HP1 proteins within TIF1βHP1box/- cells, we have established that (1) TIF1β expression during the noninduced phase up to the first day of RA treatment, or during a period extending from the second day after RA treatment through the end of the differentiation process, is neither necessary nor sufficient to allow TIF1βHP1box/- cells to differentiate into PE cells; and (2) TIF1β expression during the noninduced phase up to the second day of RA treatment or during a period of time extending from the first day of RA treatment through the end of the differentiation process enables TIF1βHP1box/- cells to differentiate into PE cells. Altogether, these results indicate that (1) the interaction between TIF1β and HP1 proteins is essential only during a short window of time early within differentiating PrE cells, whereas it is not required during the cAMP-dependent phase of PE differentiation; and (2) the deleterious effect of the TIF1β HP1box mutation on PE cell differentiation, once established in PrE early differentiating cells, cannot be corrected by re-establishing the interaction between TIF1β and HP1 proteins. This strongly suggests that the TIF1β–HP1 association is required within early differentiating PrE cells to establish a selective, transmittable competence to respond to the further cAMP differentiation signal.
TIF1β–HP1 interaction is involved in control of expression of endoderm-specific genes
Gene expression analysis in differentiating PrE cells has revealed that expression of GATA6, HNF4, Dab2, and Oct3/4 is severely down-regulated in the absence of interaction between TIF1β and HP1 proteins, whereas the expression of RARβ2 and Hoxb1 was either not modified or moderately increased, respectively. In keeping with the competence of TIF1βHP1box/- cells to morphologically differentiate into PrE cells, these data demonstrate that the TIF1β–HP1 association does not play a general role in F9 cell RA responsiveness, but rather is critical for the regulation of a specific set of genes. Of particular interest, GATA6, HNF4, Oct3/4, and Dab2, but not RARβ2 or Hoxb1, have been shown to play essential roles in endoderm differentiation during early mouse embryogenesis (Chen et al. 1994; Gavalas et al. 1998; Morrisey et al. 1998, Dupe et al. 1999; Koutsoukaris et al. 1999; Niwa et al. 2000; Yang et al. 2002). Mice having either GATA6, HNF4, or Dab2 genes inactivated can develop normally until the blastocyst stage displaying normal PrE differentiation, but fail to gastrulate with specific defects into extraembryonic visceral endoderm cells, demonstrating that none of these factors is essential for primitive endoderm differentiation, but rather they are master players for the progression of extraembryonic endoderm differentiation (Chen et al. 1994; Morrisey et al. 1998; Koutsoukaris et al. 1999; Yang et al. 2002). We therefore propose that the inability of TIF1βHP1box/- cells to differentiate into PE cells is a consequence of the down-regulation of one of these genes. Furthermore, differential expression of HNF4 between wild-type and TIF1βHP1box/- cells is detectable prior differential expression of GATA6, although it has previously been shown that GATA6 is epistatic to HNF4 during endoderm differentiation (Morrisey et al. 2000), strongly suggesting that GATA6 and HNF4 genes are independently down-regulated in TIF1βHP1box/- cells. From the available literature and our present findings, we propose that the interaction between TIF1β and HP1 proteins plays an essential role during PrE differentiation for proper induction of GATA6, HNF4, and/or Dab2 that are essential for PE and VE differentiation. We have previously shown that TIF1β is not critical for the formation of either PE- or VE-like layers within early embryos (Cammas et al. 2000). Whether TIF1β is required for appropriate differentiation of embryonic PE cells remains to be determined, but we have clearly shown that TIF1β is critical for the complete differentiation of the VE layer into squamous cells (Cammas et al. 2000), strongly suggesting that the inappropriate endoderm differentiation ability of TIF1βHP1box/- F9 cells has some functional relevance.
The TIF1β–HP1 association represses a factor that negatively controls the expression of endoderm-specific genes
How are TIF1β–HP1 interactions involved in the control of expression of endoderm-specific genes? From studies showing that TIF1β interacts with the transcriptional repressors KRAB-ZFPs and recruits histone deacetylase and methyltransferase complexes together with HP1 proteins to specific chromatin sites, it is currently assumed that TIF1β acts as a corepressor of KRAB-ZFPs (Ayyanathan et al. 2003; and see introduction). It is therefore unlikely that the TIF1β–HP1 association directly regulates the expression of the genes that are down-regulated in TIF1βHP1box/- cells. Rather, we propose that the TIF1β–HP1 association indirectly controls the expression of these endoderm-specific genes by repressing the expression of a factor(s) that normally down-regulates their expression. This factor(s) could belong to different classes of regulators: (1) It could be a transcriptional repressor, the expression of which would maintain the expression of endoderm-specific genes either low (HNF4) or fully repressed (GATA6) in nondifferentiated cells, whereas its repression by TIF1β–HP1 after RA treatment would allow the induction of endoderm-specific gene expression. Similar mechanisms of regulation of differentiation progression by transcriptional repressors have been described for different differentiation pathways. For example, the repressor REST (Ballas et al. 2001) and the nuclear receptor corepressor N-CoR (Hermanson et al. 2002) play essential roles in neural stem cell self-renewal and their decreased expression led to neuronal and astroglial cell differentiation, respectively. The transcription factor Pax-5 has been shown to play essential roles in B-lineage commitment by repressing lineage-promiscuous transcription (Nutt et al. 1999; Mikkola et al. 2002); (2) It could correspond to factor(s) that inactivate, either by posttranslational modification or by degradation, an activator(s) of endoderm-specific gene expression. In this respect, we note that RXRα phosphorylation has been shown to be crucial for the expression of several RA-responsive genes in F9 cells (Bastien et al. 2002) and also that the ligand-dependent degradation of RARs and RXRs by the ubiquitin proteasome pathway could be important for the magnitude and duration of the effect of retinoid signals in F9 cells (Kopf et al. 2000). (3) It could be a chromatin silencing factor(s) such as histone methyltransferase, deacetylase, and/or DNA methyltransferase, which have all been shown to be involved in control of embryonic development and cellular differentiation (for reviews, see Ansel et al. 2003; Ehrlich 2003). The identification of the genes that are misexpressed in TIF1βHP1box/- cells during the early phase of PrE differentiation will allow discrimination between these various possibilities.
It is noteworthy that the window of time during which the TIF1β–HP1 interaction is essential for progression through differentiation strikingly correlates with the normal onset of TIF1β relocation from eu- to heterochromatin domains (Cammas et al. 2002). We therefore propose that, in the presence of RA, TIF1β–HP1 triggers the translocation of the direct target genes from eu- to heterochromatin for their silencing. In this respect, we note that Matsuda et al. (2001) have shown the colocalization of two KRAB-ZFP proteins, KRAZ1 and KRAZ2, with TIF1β and HP1 proteins within pericentromeric heterochromatin of a fibroblast population. We note also that the translocation of specific euchromatic genes near or within heterochromatin has been proposed for the transcription factor Ikaros that regulates the expression of genes involved in T-cell activation (Brown et al. 1997, 1999).
In conclusion, this report is the first demonstration that the interaction between TIF1β and HP1 proteins plays a critical role in vivo, that is, for the progression of F9 cells through terminal differentiation. Although EC F9 cells represent a relatively simple model of differentiation, it is likely that the regulatory mechanism uncovered in this study will apply to other differentiation pathways. It is indeed noteworthy that we have also observed TIF1β relocation from eu- to heterochromatin during differentiation of ES cells into cardiomyocytes and neuronal cell types (Cammas et al. 2002), which suggests that the TIF1β–HP1 association is also playing a pivotal role in these differentiation pathways.
Materials and methods
Details on individual plasmid constructs, which were all verified by sequencing, are available on request.
Construction of the targeting vector
The targeting vector pTIF1βLNL:L for inactivation of the first TIF1β allele was described previously (Cammas et al. 2000), with the addition of a positive selection cassette containing the coding sequence for the diphterin toxin A (DTA) under the control of the TK promoter (DTA cassette; McCarrick et al. 1993; gift from J.W. McCarrick, Wistar Institute, Philadelphia, PA) at the 3′ end of the construct. The pTIF1β(LNL:L) ClaI fragment was ligated in the ClaI site of pSL1190-DTA to give pTIFβLNL:L. To mutate the HP1 box motif on the second allele of TIF1β, we exchanged the PGK-Neo cassette of pTIF1β(LNL:L) with a PGK-Hygro (gift from D. Metzger, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch, France) and introduced the previously described HP1 box mutation (Cammas et al. 2002) in the TIF1β gene. The HindIII PGK-HygroMod fragment was ligated in purified HindIII TIF1B11 fragment to give TIF1B40 (see Cammas et al. 2000). The double mutation in the HP1 box of TIF1β (leading to the V488A/L490A substitutions) was generated by site-directed PCR mutagenesis with the following internal primers (the codon change is shown in boldface): ABP88 (5′-GAAGGTGCCACGCGCGAGCGCTGAAC GCCTGG-3′) and ABP89 (5′-CCAGGCGTTCAGCGCTCGC GCGTGGCACCTTC-3′). The mutation in the HP1 box was introduced by PCR in TIF1B4 (see Cammas et al. 2000). Two PCR reactions were first performed with oligonucleotides ACC242 (5′-CACCAGGAACACATTTTGGCG-3′) and ABP89 and with oligonucleotides ACC243 (5′-GGGACGGCATG GTTCATGGC-3′) and ABP88 using TIF1B4 as template. These two PCR fragments were then mixed and used as the template for a third PCR with oligonucleotides ACC242 and ACC243. This PCR fragment was then digested with EcoRI and NdeI and ligated in NdeI/EcoRI-digested TIF1B4 plasmid, to give TIF1B41. The 4.5-kb XhoI/XhoI fragment from TIF1B41 was then cloned in the XhoI site of TIF1B40 to give TIF1B23. The DTA cassette was added to this construct as described earlier to give pTIF1βLHL:L.
For the temporally controlled expression of the Flag-tagged TIF1β and TIF1βHP1box cDNAs, the reverse tetracycline system was used. The expression vector pDG1-rtTA, in which the expression of the rtTA (Gossen et al. 1995) is under the control of the PGK promoter, was a gift from Dr. Metzger. The Flag-tagged TIF1β and TIF1βHP1box cDNA EcoRI fragments from pCX-Flag-TIF1β and pCX-Flag-TIF1βHP1box (Cammas et al. 2002) were ligated in EcoRI-digested PUHD10.3 (Gossen and Bujard 1992) vector containing the hCMV/Tet-Operator sequence (gift from Pr. Bujard, Max-Planck Institute, Heidelberg, Germany) resulting in TIF1B16.
Antibodies
mAbs used include mouse anti-TIF1β mAb, 1Tb3, raised against recombinant Escherichia coli expressed mouse TIF1β (123–834; Nielsen et al. 1999); the mouse anti-flag mAb 2FLB11; mouse anti-RPB1 mAb, 1PB-7C2, raised against the heptad repeat CTD-containing peptide of the RPB1 largest subunit of the human RNA polymerase II (Nguyen et al. 1996); the anti-HP1α mAb 2HP-2G9; the HP1β mAb 1MOD-1A9; the anti-HP1γ mAb, 2 Mod-1G6 (Nielsen et al. 1999), rat anti-Endo A, Troma-1 (kindly provided by Dr. R. Kemler, Department of Molecular Embryology, Max-Planck Institute, Freiburg, Germany). The rabbit polyclonal antibodies (pAbs) used include anti-TIF1β pAb, PF64, raised against TIF1β (amino acids 141–155; Cammas et al. 2002) and the anti-RXRα pAb RPRXα (gift from Dr. Rochette-Egly, Institut de Génétique et de Biologie Moléculaire et Cellulaire, Illkirch, France).
Cell culture and establishment of stable cell lines
Wild-type and mutant F9 cells were grown as monolayers on gelatinized surfaces in Dulbecco modified Eagle medium (DMEM, Gibco) supplemented with 10% fetal calf serum as previously described (Boylan and Gudas 1991). To induce PrE and PE differentiations, we plated cells at a density of 102–103 cells/cm2 on cell culture dishes and treated them with 1.0 μM all-trans RA (Sigma) alone or in combination with 250 μM dibutyryl cAMP (dbcAMP, Sigma) or with vehicle (stem, no treatment) for the indicated times, with a change of media every 2 d. To induce VE differentiation, we plated cells at a density of 102 cells/cm2 on bacterial Petri dishes and treated them with 50 nM RA or with vehicle (stem, no treatment) for the indicated times, with a change of media every 2 d. Cells were counted with a particle counter (Coulter Z2).
To establish an F9 cell line with one TIF1β null allele and one TIF1β mutated in the HP1 box motif, we transfected 5 × 106 exponentially growing F9 EC cells (106 per 10-cm plate) by electroporation (200 V, 975 μF) with 5 μg of the targeting vector pTIFβL:LNL linearized with SfiI. After selection with G-418 (500 μg/mL, Sigma), G-418-resistant clones were expanded, and their genomic DNA was prepared, digested with BamH1, and analyzed by Southern blotting with the 3′ probe (a 1.2-kb DNA fragment downstream of the TIF1β sequence included in the targeting vector; Cammas et al. 2000). Positive clones for homologous recombination were further analyzed by Southern blot after EcoRV digestion and hybridization with the 5′ probe (a 1.5-kb EcoRI genomic fragment upstream of the TIF1β sequence included in the targeting vector; Cammas et al. 2000) and a Neo probe and called TIF1βL3/+. This clone was transfected by electroporation with 5 μg of the targeting vector pTIFβL:LHL linearized with SfiI. After selection with G-418 (500 μg/mL) and hygromycin B (250 μg/mL; Roche), clones resistant to G-418 and hygromycin were expanded, and their genomic DNA was prepared, digested with EcoRV, and analyzed by Southern blotting with the 5′ probe and the Hygro probe. Positive clones for homologous recombination were further analyzed by PCR with oligonucleotides VR216 (5′-ATGGAAGTACAAGAGGGATAT GGC-3′) and TV211 (5′-TGAGCTGGTACTGCCACTAGG-3′), followed by digestion with Eco47III to detect the mutation in the HP1 box motif. The TIF1βL3/L2HP1box clone was transfected by electroporation with 10 μg of the PGK-Cre expression vector (gift from Dr. Metzger). Cells were redistributed 72 h following the transfection at a concentration of 250 cells/10-cm plate and grown in the absence of any selection for 10 d. Clones were expanded, and their genomic DNA was prepared and analyzed by PCR with YD208/TV210/VR211 as described previously to detect the excision of the LoxP-flanked sequences (Cammas et al. 2000).
To establish TIF1βHP1box/- cell lines expressing f.TIF1β or f.TIF1βHP1box, we coelectroporated TIF1βHP1box/- F9 cells with 2 μg of pDG1-rtTA linearized by Aat2, 2 μg TIF1B16 or TIF1B18 (Cammas et al. 2002) linearized by AatII, and 0.4 μg of PGK-NeoALS1 (gift from Dr. Metzger) linearized by BsaI. After G-418 selection, cells were expanded and treated with 1 μg/mL Dox for 3 d. Clones expressing f.TIF1β or f.TIF1βHP1box only in the presence of Dox were selected by Western blotting using the anti-Flag mAb.
Immunofluorescence and confocal microscopy
Cells were grown on gelatinized glass coverslips, washed once with phosphate-buffered saline (PBS), and fixed with 2% paraformaldehyde in PBS (pH 7.5) for 10 min at room temperature. Samples were then permeabilized twice with 0.1% Triton X-100 (PBS-Tx) for 5 min at room temperature, washed in PBS, and incubated for 16 h at room temperature with primary antibodies appropriately diluted in PBS. Cells were washed twice with PBS-Tx for 5 min at room temperature and incubated for 1 h with fluorochrome-conjugated secondary antibodies in PBS-Tx. Slides were washed twice (5 min/wash) in PBS-Tx, stained for DNA with Hoechst 33258 at 5 μg/mL, and mounted in PBS 5% propyl gallate 80% glycerol. Image acquisition was performed using a Leica TCS-4D confocal scanning microscope.
Immunoprecipitation and Western blot analysis
Isolation of whole-cell extracts from F9 cells and Western blot detection were performed as previously described (Chiba et al. 1997a; Nielsen et al. 1999).
RT–PCR
RNA extraction was performed with a water-saturated phenol/guanidinium thiocyanate solution as described previously (Rothblum and Xie 1991). Semiquantitative RT–PCRs were performed as described previously (Chiba et al. 1997b). The PCR primers used for amplification are available on request. End-labeled oligonucleotides were generated for probing the PCR products on Southern blots.
Acknowledgments
We are grateful to R. Kemler for providing the rat monoclonal antibody Troma-1 against Endo A. We thank M. Cerviño and O. Wendling for technical assistance; M. Beglin, J.-L. Vonesh, and D. Hentsch for confocal laser scanning microscopy; S. Vicaire and D. Stephan for DNA sequencing; A. Staub and F. Ruffenach for oligonucleotide synthesis; and M. Oulad-Abdelghani for antibody supply. We also acknowledge K. Khechoumian for helpful discussions. This work was supported by the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale, l'Hôpital Universitaire de Strasbourg (H.U.S.), the association pour la Recherche sur le Cancer, the Collège de France, Bristol-Myers-Squibb, and grants from the European Commission (Biomed 2 program [PL962433]). F.C. was supported by a fellowship from the Fondation pour la Recherche Médicale (FRM).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.302904.
References
- Aasland R., Gibson, T.J., and Stewart, A.F. 1995. The PHD finger: Implications for chromatin-mediated transcriptional regulation. Trends Biochem. Sci. 20: 56-59. [DOI] [PubMed] [Google Scholar]
- Abrink M., Ortiz, J.A., Mark, C., Sanchez, C., Looman, C., Hellman, L., Chambon, P., and Losson, R. 2001. Conserved interaction between distinct Kruppel-associated box domains and the transcriptional intermediary factor 1 β. Proc. Natl. Acad. Sci. 98: 1422-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansel K.M., Lee, D.U., and Rao, A. 2003. An epigenetic view of helper T cell differentiation. Nat. Immunol. 4: 616-623. [DOI] [PubMed] [Google Scholar]
- Ayyanathan K., Lechner, M.S., Bell, P., Maul, G.G., Schultz, D.C., Yamada, Y., Tanaka, K., Torigoe, K., and Rauscher III, F.J. 2003. Regulated recruitment of HP1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: A mammalian cell culture model of gene variegation. Genes & Dev. 17: 1855-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N., Battaglioli, E., Atouf, F., Andres, M.E., Chenoweth, J., Anderson, M.E., Burger, C., Moniwa, M., Davie, J.R., Bowers, W.J., et al. 2001. Regulation of neuronal traits by a novel transcriptional complex. Neuron 31: 353-365. [DOI] [PubMed] [Google Scholar]
- Bastien J., Adam-Stitah, S., Plassat, J-L., Chambon, P., and Rochette-Egly, C. 2002. The phosphorylation site located in the A region of retinoic X receptor is required for the antiproliferative effect of retinoic acid (RA) and the activation of RA target genes in F9 cells. J. Biol. Chem. 277: 28683-28689. [DOI] [PubMed] [Google Scholar]
- Beckstead R., Ortiz, J.A., Sanchez, C., Prokopenko, S.N., Chambon, P., Losson, R., and Bellen, H.J. 2001. Bonus, a Drosophila homolog of TIF1 proteins, interacts with nuclear receptors and can inhibit βFTZ-F1-dependent transcription. Mol. Cell 7: 753-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan J.F. and Gudas, L.J. 1991. Overexpression of the cellular retinoic acid binding protein-I (CRABP-I) results in a reduction in differentiation-specific gene expression in F9 teratocarcinoma cells. J. Cell Biol. 112: 965-979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown K.E., Guest, S.S., Smale, S.T., Hahm, K., Merkenschlager, M., and Fisher, A.G. 1997. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell 91: 845-854. [DOI] [PubMed] [Google Scholar]
- Brown K.E., Baxter, J., Graf, D., Merkenschlager, M., and Fisher, A.G. 1999. Dynamic repositioning of genes in the nucleus of lymphocytes preparing for cell division. Mol. Cell 3: 207-217. [DOI] [PubMed] [Google Scholar]
- Cammas F., Mark, M., Dollé, P., Dierich, A., Chambon, P., and Losson, R. 2000. Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 127: 2955-2963. [DOI] [PubMed] [Google Scholar]
- Cammas F., Oulad-Abdelghani, M., Vonesch J.-L., Chambon, P., and Losson, R. 2002. Cell differentiation induces TIF1β association with centromeric heterochromatin through HP1 interaction. J. Cell Sci. 115: 3439-3448. [DOI] [PubMed] [Google Scholar]
- Casanova J.E. and Grabel, L.B. 1988. The role of cell interactions in the differentiation of teratocarcinoma-derived parietal and visceral endoderm. Dev. Biol. 129: 124-139. [DOI] [PubMed] [Google Scholar]
- Chen W.S., Manova, K., Weinstein, D.C., Duncan, S.A., Plump, A.S., Prezioso, V.R., Bachvarova, R.F., and Darnell Jr., J.E. 1994. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes & Dev. 8: 2466-2477. [DOI] [PubMed] [Google Scholar]
- Chiba H., Clifford, J., Metzger, D., and Chambon, P. 1997a. Specific and redundant functions of retinoid X Receptor/Retinoic acid receptor heterodimers in differentiation, proliferation, and apoptosis of F9 embryonal carcinoma cells. J. Cell Biol. 139: 735-747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ____. 1997b. Distinct retinoid X receptor/retinoic acid receptor heterodimers are differentially involved in the control of expression of retinoid target genes in F9 embryonal carcinoma cells. Mol. Cell. Biol. 17: 3013-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupe V., Ghyselinck, N.B., Wendling, O., Chambon, P., and Mark, M. 1999. Key roles of retinoic acid receptors alpha and beta in the patterning of the caudal hindbrain, pharyngeal arches and otocyst in the mouse. Development 126: 5051-5059. [DOI] [PubMed] [Google Scholar]
- Ehrlich M. 2003. Expression of various genes is controlled by DNA methylation during mammalian development. J. Cell. Biochem. 88: 899-910. [DOI] [PubMed] [Google Scholar]
- Eissenberg J.C. 2001. Decisive factors: A transcription activator can overcome heterochromatin silencing. Bioessays 23: 767-771. [DOI] [PubMed] [Google Scholar]
- Eissenberg J.C. and Elgin, S.C. 2000. The HP1 protein family: Getting a grip on chromatin. Curr. Opin. Genet. Dev. 10: 204-210. [DOI] [PubMed] [Google Scholar]
- Eissenberg J.C., James, T.C., Foster-Hartnett, D.M., Hartnett, T., Ngan, V., and Elgin, S.C. 1990. Mutation in a heterochromatin-specific chromosomal protein is associated with suppression of position-effect variegation in Drosophila melanogaster. Proc. Natl. Acad. Sci. 87: 9923-9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A.G. 2002. Cellular identity and lineage choice. Nat. Rev. Immunol. 2: 977-982. [DOI] [PubMed] [Google Scholar]
- Friedman J.R., Fredericks, W.J., Jensen, D.E., Speicher, D.W., Huang, X.P., Neilson, E.G., and Rauscher III, F.J. 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes & Dev. 10: 2067-2078. [DOI] [PubMed] [Google Scholar]
- Gavalas A., Studer, M., Lumsden, A., Rijli, F.M., Krumlauf, R., and Chambon, P. 1998. Hoxa1 and Hoxb1 synergize in patterning the hindbrain, cranial nerves and second pharyngeal arch. Development 125: 1123-1136. [DOI] [PubMed] [Google Scholar]
- Gossen M. and Bujard, H. 1992. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. 89: 5547-5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M., Freundlieb, S., Bender, G., Muller, G., Hillen, W., and Bujard, H. 1995. Transcriptional activation by tetracyclines in mammalian cells. Science 268: 1766-1769. [DOI] [PubMed] [Google Scholar]
- Harris T.M. and Childs, G. 2002. Global gene expression patterns during differentiation of F9 embryonal carcinoma cells into parietal endoderm. Funct. Integr. Genomics 2: 105-119. [DOI] [PubMed] [Google Scholar]
- Hermanson O., Jepsen, K., and Rosenfeld, M.G. 2002. N-CoR controls differentiation of neural stem cells into astrocytes. Nature 419: 934-939. [DOI] [PubMed] [Google Scholar]
- Jeanmougin F., Wurtz, J.-M., Le Douarin, B, Chambon, P., and Losson, R. 1997. The bromodomain revisited. Trends Biochem. Sci. 22: 151-153. [DOI] [PubMed] [Google Scholar]
- Jenuwein T. and Allis, C.D. 2001. Translating the histone code. Science 294: 1074-1080. [DOI] [PubMed] [Google Scholar]
- Kemler R., Brulet, P., Schnebelen, M.T., Gaillard, J., and Jacob, F. 1981. Reactivity of monoclonal antibodies against intermediate filament proteins during embryonic development. J. Embryol. Exp. Morphol. 64: 45-60. [PubMed] [Google Scholar]
- Kim S.S., Chen, Y.M., O'Leary, E., Witzgall, R., Vidal, M., and Bonventre, J.V. 1996. A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins, Proc. Natl. Acad. Sci. 93: 15299-15304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopf E., Plassat, J.L., Vivat, V., de The, H., Chambon, P., and Rochette-Egly, C. 2000. Dimerization with retinoid X receptors and phosphorylation modulate the retinoic acid-induced degradation of retinoic acid receptors alpha and gamma through the ubiquitin–proteasome pathway. J. Biol. Chem. 275: 33280-33288. [DOI] [PubMed] [Google Scholar]
- Koutsourakis M., Langeveld, A., Patient, R., Beddington, R., and Grosveld, F. 1999. The transcription factor GATA6 is essential for early extraembryonic development. Development 126: 723-732. [PubMed] [Google Scholar]
- Lachner M., O'Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116-120. [DOI] [PubMed] [Google Scholar]
- Le Douarin B., Zechel, C., Garnier, J.-M., Lutz, Y., Tora, L., Pierrat, B., Heery, D., Gronemeyer, H., Chambon, P., and Losson, R. 1995. The N-terminal part of TIF1, a putative mediator of the ligand-dependent activation function (AF-2) of nuclear receptors, is fused to B-raf in the oncogenic protein T18. EMBO J. 14: 2020-2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin B., Nielsen, A.L., Garnier, J.-M., Ichinose, H., Jeanmougin, F., Losson, R., and Chambon, P. 1996. A possible involvement of TIF1α and TIF1β in the epigenetic control of transcription by nuclear receptors. EMBO J. 15: 6701-6715. [PMC free article] [PubMed] [Google Scholar]
- Li E. 2002. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 3: 662-673. [DOI] [PubMed] [Google Scholar]
- Li Y., Kirschmann, D.A., and Wallrath, L.L. 2002. Does heterochromatin protein 1 always follow code? Proc. Natl. Acad. Sci. 99: 16462-16469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda E., Agata, Y., Sugai, M., Katakai, T., Gonda, H., and Shimizu, A. 2001. Targeting of Kruppel-associated box-containing zinc finger proteins to centromeric heterochromatin. Implication for the gene silencing mechanisms. J. Biol. Chem. 276: 14222-14229. [DOI] [PubMed] [Google Scholar]
- McCarrick III J.W., Parnes, J.R., Seong, R.H., Solter, D., and Knowles, B.B. 1993. Positive-negative selection gene targeting with the diphtheria toxin A-chain gene in mouse embryonic stem cells. Transgenic Res. 2: 183-190. [DOI] [PubMed] [Google Scholar]
- Mikkola I., Heavey, B., Horcher, M., and Busslinger, M. 2002. Reversion of B cell commitment upon loss of Pax5 expression. Science 297: 110-113. [DOI] [PubMed] [Google Scholar]
- Moosmann P., Georgiev, O., Le Douarin, B., Bourquin, J.P., and Schaffner, W. 1996. Transcriptional repression by RING finger protein TIF1β that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 24: 4859-4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey E.E., Tang, Z., Sigrist, K., Lu, M.M., Jiang, F., Ip, H.S., and Parmacek, M.S. 1998. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes & Dev. 12: 3579-3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrisey E.E., Musco, S., Chen, M.Y., Lu, M.M., Leiden, J.M., and Parmacek, M.S. 2000. The gene encoding the mitogen-responsive phosphoprotein Dab2 is differentially regulated by GATA-6 and GATA-4 in the visceral endoderm. J. Biol. Chem. 275: 19949-19954. [DOI] [PubMed] [Google Scholar]
- Nakayama J., Rice, J.C., Strahl, B.D., Allis, C.D., and Grewal, S.I. 2001. Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110-113. [DOI] [PubMed] [Google Scholar]
- Narlikar G.J., Fan, H.Y., and Kingston, R.E. 2002. Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475-487. [DOI] [PubMed] [Google Scholar]
- Nguyen V.T., Giannoni, F., Dubois, M.F., Seo, S.J., Vigneron, M., Kedinger, C., and Bensaude, O. 1996. In vivo degradation of RNA polymerase II largest subunit triggered by alpha-amanitin. Proc. Natl. Acad. Sci. 24: 2924-2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen A.L., Ortiz, J.A., You, J., Oula-Abdelghani, M., Khechumian, R., Gansmuller, A., Chambon, P., and Losson, R. 1999. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 18: 6385-6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen A.L., Oulad-Abdelghani, M., Ortiz, J.A., Remboutsika, E., Chambon, P., and Losson R. 2001a. Heterochromatin formation in mammalian cells: Interaction between histones and HP1 proteins. Mol. Cell 7: 729-739. [DOI] [PubMed] [Google Scholar]
- Nielsen S.J., Schneider, R., Bauer, U.M., Bannister, A.J., Morrison, A., O'Carroll, D., Firestein, R., Cleary, M., Jenuwein, T., Herrera, R.E., et al. 2001b. Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561-565. [DOI] [PubMed] [Google Scholar]
- Nielsen A.L., Sanchez, C., Ichinose, H., Cervino, M., Lerouge, T., Chambon, P., and Losson, R. 2002. Selective interaction between the chromatin-remodeling factor BRG1 and the heterochromatin-associated protein HP1α. EMBO J. 21: 5797-5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H., Miyazaki, J., and Smith, A.G. 2000. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24: 372-376. [DOI] [PubMed] [Google Scholar]
- Nutt S.L., Heavey, B., Rolink, A.G., and Busslinger, M. 1999. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556-562. [DOI] [PubMed] [Google Scholar]
- Peng H., Feldman, I., and Rauscher III, F.J. 2002. Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors: A potential mechanism for regulating the switch between coactivation and corepression. J. Mol. Biol. 320: 629-644. [DOI] [PubMed] [Google Scholar]
- Rothblum L.I. and Xie, W. 1991. Rapid, small-scale RNA isolation from tissue culture cells. Biotechniques 11: 323-327. [PubMed] [Google Scholar]
- Ryan R.F., Schultz, D.C., Ayyanathan, K., Singh, P.B., Friedman, J.R., Fredericks, W.J., and Rauscher III, F.J. 1999. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 19: 4366-4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Friedman, J.R., and Rauscher III, F.J. 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes & Dev. 15: 428-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz D.C., Ayyanathan, K., Negorev, D., Maul, G.G., and Rauscher III, F.J. 2002. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes & Dev. 16: 919-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland S. and Mahdavi, V. 1978. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell 15: 393-403. [DOI] [PubMed] [Google Scholar]
- Strickland S., Smith, K.K., and Marotti, K.R. 1980. Hormonal induction of differentiation in teratocarcinoma stem cells: Generation of parietal endoderm by retinoic acid and dibutyryl cAMP. Cell 21: 347-355. [DOI] [PubMed] [Google Scholar]
- Thiru A., Nietlispach, D., Mott, H.R., Okuwaki, M., Lyon, D., Nielsen, P.R., Hirshberg, M., Verreault, A., Murzina, N.V., and Laue, E.D. 2004. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 23: 489-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill C., Qutob, M.S., Yee, S.-P., and Torchia, J. 2000. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 275: 40463-40470. [DOI] [PubMed] [Google Scholar]
- Vassallo M.F. and Tanese, N. 2002. Isoform-specific interaction of HP1 with human TAFII130. Proc. Natl. Acad. Sci. 99: 5919-5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini L., You, J., Stadler, M., Galien, R., Lallemand, V., Koken, M.H.M., Mattei, M.G., Ganser, A., Chambon, P., Losson, R., et al. 1999. TIF1γ, a novel member of the transcriptional intermediary factor 1 family. Oncogene 18: 1209-1217. [DOI] [PubMed] [Google Scholar]
- Verheijen M.H., Wolthuis, R.M., Bos, J.L., and Defize, L.H. 1999. The Ras/Erk pathway induces primitive endoderm but prevents parietal endoderm differentiation of F9 embryonal carcinoma cells. J. Biol. Chem. 274: 1487-1494. [DOI] [PubMed] [Google Scholar]
- Wang G., Ma, A., Chow, C.M., Horsley, D., Brown, N.R., Cowell, I.G., and Singh, P.B. 2000. Conservation of heterochromatin protein 1 function. Mol. Cell. Biol. 20: 6970-6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber P., Cammas, F., Gerard, C., Metzger, D., Chambon, P., Losson, R., and Mark, M. 2002. Germ cell expression of the transcriptional co-repressor TIF1β is required for the maintenance of spermatogenesis in the mouse. Development 129: 2329-2337. [DOI] [PubMed] [Google Scholar]
- Weiler-Guettler H., Yu, K., Soff, G., Gudas, L.J., and Rosenberg, R.D. 1992. Thrombomodulin gene regulation by cAMP and retinoic acid in F9 embryonal carcinoma cells. Proc. Natl. Acad. Sci. 89: 2155-2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K., Dollé, P., Mark, M., Lerouge, T., Wendling, O., Chambon, P., and Losson, R. 2004. Molecular cloning, genomic structure, and expression analysis of mouse Transcriptional Intermediary Factor 1 gamma (Tif1γ) gene. Gene 334: 3-13. [DOI] [PubMed] [Google Scholar]
- Yang D.H., Smith, E.R., Roland, I.H., Sheng, Z., He, J., Martin, W.D., Hamilton, T.C., Lambeth, J.D., and Xu, X.X. 2002. Disabled-2 is essential for endodermal cell positioning and structure formation during mouse embryogenesis. Dev. Biol. 251: 27-44. [DOI] [PubMed] [Google Scholar]
- Zeng L. and Zhou, M.M. 2002. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 513: 124-128. [DOI] [PubMed] [Google Scholar]