

Abstract
An amide-functionalized metal organic framework (MOF) material, MFM-136, shows a high CO2 uptake of 12.6 mmol g–1 at 20 bar and 298 K. MFM-136 is the first example of an acylamide pyrimidyl isophthalate MOF without open metal sites and, thus, provides a unique platform to study guest binding, particularly the role of free amides. Neutron diffraction reveals that, surprisingly, there is no direct binding between the adsorbed CO2/CH4 molecules and the pendant amide group in the pore. This observation has been confirmed unambiguously by inelastic neutron spectroscopy. This suggests that introduction of functional groups solely may not necessarily induce specific guest–host binding in porous materials, but it is a combination of pore size, geometry, and functional group that leads to enhanced gas adsorption properties.
Recent developments in materials chemistry and crystal engineering have shown that metal–organic frameworks (MOFs) have promising properties that complement or compete favorably with zeolites and activated carbons in various applications.1 MOFs are crystalline porous coordination polymers consisting of polyatomic organic ligands linked to metal ions/clusters by covalent bonds.2 MOFs have shown great promise for gas adsorption and storage owing to their high porosity and internal surface area, and tunable functionality on the pore surface for selective gas binding. Generation of open metal sites3 and incorporation of pendant functional groups4 at the pore surface are two dominant methods of functionalizing MOF cavities. For example, MOFs with open Cu(II) sites can show strong adsorption affinity to molecular H2.5 Recently, the detailed binding mechanisms to saturated and unsaturated light hydrocarbons have been rationalized in a hydroxyl-functionalized MOF.6 Within the field of carbon capture, materials functionalized with amines (−NH2), imines (−NH), and amides (−CONH) dominate, largely because of their potential to form specific interactions with CO2, leading to highly selective CO2 uptakes. Although high CO2 adsorption has been observed in a number of amine-, imine- and amide-functionalized MOFs,7 molecular insight into the direct binding between adsorbed CO2 molecules and porous host (especially toward these functional groups) is largely lacking. Recently, direct H2N(δ−)···(δ+)CO2 binding has been observed in a Zn(II) MOF incorporating amine groups that protrude into the pore, providing structural insight into the observed high CO2 adsorption in this material.4
The incorporation of pendent amide (−CONH−) and/or amine groups into MOFs is thus regarded generally as a promising approach to enhance CO2 uptake due to the formation of hydrogen bonds with amides serving as both hydrogen bond acceptors (via C=O) and donors (via N–H). A series of amide-functionalized MOFs have been synthesized and shown to exhibit high CO2 uptakes and selectivities.7b,8 Likewise, computational studies attribute this to the specific binding and formation of hydrogen bonds between adsorbed CO2 molecules and free amide or amine groups thus enhancing adsorption affinity and selectivity for CO2.7c,8c,9 However, to date there are few physical investigations on the precise role of amides in CO2 binding in MOFs. The challenge of such investigations is further increased in MOF systems containing open metal sites owing to the inevitable competition for guest binding between the open metal sites and the organic functional group(s) in the pore. Here, we report the synthesis, structure, and gas adsorption properties of an amide-functionalized pyrimidyl Cu(II)-isophthalate MOF, MFM-136, which shows a high CO2 adsorption capacity (12.6 mmol g–1 at 20 bar and 298 K). In MFM-136, all Cu(II) sites are fully coordinated to carboxylate and pyrimidyl groups, affording a pore environment without open metal sites. This gives an ideal environment for studying the binding interaction between amides and adsorbed CO2 molecules since it eliminates the competitive binding of CO2 on the open Cu(II) sites. In this situation, we can clearly evaluate the precise role of free amides in guest binding in the pore. Combined neutron diffraction and inelastic neutron spectroscopy have revealed the preferred binding sites for CO2 in the pore and the corresponding host–guest binding dynamics. Surprisingly, there is no direct binding between adsorbed CO2/CD4 and free amides in this case. This is supported by grand canonical Monte Carlo (GCMC) simulations.
Solvothermal reaction of 5-[4-(pyrimidin-5-yl)benzamido]isophthalic acid (H2L, Figure 1a) with Cu(NO3)2·3H2O in DMF at 80 °C for 16 h yields MFM-136 as green single crystals. MFM-136 crystallizes in space group R32 and shows a 3D binodal (3,6)-connected network with a rare eea-topology.8f,10 In MFM-136, the binuclear [Cu2(O2CR)4] paddlewheels coordinate to two pyrimidyl nitrogen atoms from two different ligands at both axial positions, resulting in the absence of open Cu(II) sites in the entire structure (Figure 1). The metal–ligand connectivity in MFM-136 affords two types of cages (A and B). Cage A is surrounded by 12 [Cu2(O2CR)4] paddlewheel units and six linkers and has a prolate-ellipsoid shape (length 24.9 Å, width 10.6 Å). Cage B is enclosed by six [Cu2(O2CR)4] paddlewheel units and six linkers and has a more spherical shape (length 16.2 Å, width 12.5 Å). The overall structure is an alternate packing of these two types of cages to give a highly porous and robust framework material with a void fraction of 54% and BET surface area of 1634 m2 g–1 (Figure S6).
Figure 1.

(a) Chemical structure of ligand H2L. (b) Coordination environment of L2– in MFM-136. (c) View along the c-axis of the Kagome lattice in MFM-136. (d) View of the alternate packing of large cages A (blue) and small cages B (orange) in MFM-136. Views of (e) the large cage A and (f) small cage B both along the b-axis. Colors: C, black; H, white; O, red; N, blue; Cu, teal.
At 273 K, the CO2 sorption isotherms of desolvated MFM-136 show an uptake of 7.3 mmol g–1 at 1 bar and 14.3 mmol g–1 at 20 bar, representing the highest CO2 uptake in monoamide-functionalized MOFs reported to date (Table S2). Methane adsorption in MFM-136 gives a lower uptake of 2.9 mmol g–1 at 1 bar and 8.3 mmol g–1 at 20 bar at 273 K. The experimental CO2 adsorption isotherms show good agreement with grand canonical Monte Carlo (GCMC) simulations (Figure 2). In contrast, MFM-136 shows negligible N2 uptake under the same conditions, leading to selectivities for CO2/N2 and CO2/CH4 of 27:1 and 6.3:1, respectively, at 273 K. The isosteric heats of adsorption for CO2 and CH4 in MFM-136 are calculated using the Virial method as 25.6 and 16.0 kJ mol–1, respectively, at low surface coverage. The selective CO2 uptake in MFM-136 is lower than the leading ultramicroporous MOFs;11 however, the high capacity indicates MFM-136 remains a promising candidate in the separation of CO2 over CH4 and N2.12 The lack of open Cu(II) sites in the pores of MFM-136 prevents strong binding to water molecules, which often triggers framework collapse or hydrolysis in MOFs containing open metal sites.13 Previously reported MOFs containing amides in the absence of open metals sites have exhibited high CO2 capacities;14 however, the role of the amides in CO2 binding was not defined structurally.
Figure 2.

Experimental and simulated adsorption isotherms for CO2 in desolvated MFM-136 at 273 and 298 K.
It is reported that the excellent uptake of CO2 in amide-functionalized MOFs is a consequence of specific CO2-amide interactions based upon hydrogen bond formation between the amide −NH(δ+) and the O(δ−) of CO2.7c,8a To gain experimental insight, preferred binding sites in MFM-136 have been determined by in situ neutron powder diffraction (NPD) as a function of gas loading (CO2 and CD4). NPD patterns were recorded at 7 K for the desolvated material and at loadings of 1.8 and 2.3 CO2/Cu and 1.1 CD4/Cu. Fourier difference map analysis of the NPD patterns revealed positions of the adsorbed CO2 and CD4 molecules, which were further developed by Rietveld refinement. All binding sites were checked carefully for their unambiguous presence in the final structural model; i.e., a parallel refinement without each of the binding sites was carried out to confirm the presence of each site by comparing the R factors and the residual peaks.
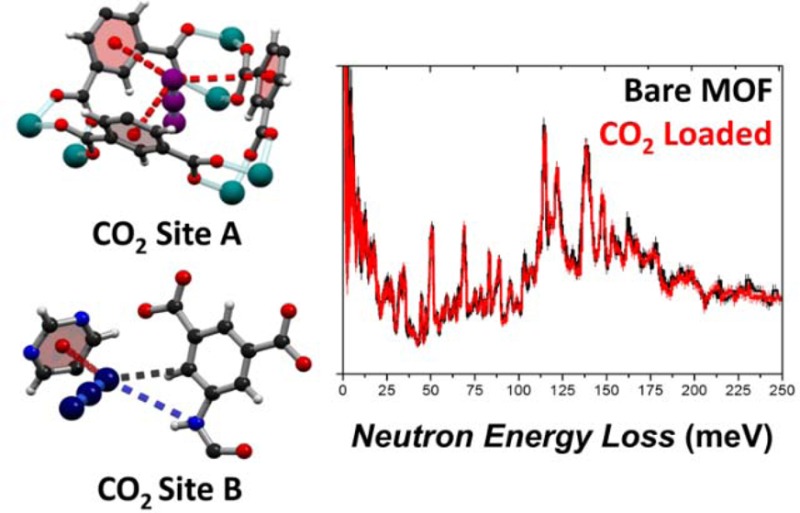
The NPD data at a loading of 1.8 CO2/Cu reveals eight binding sites A–H distributed between cages A and B (Figure 3). The CO2 molecules are constrained to be linear with equal C–O bond lengths, while their crystallographic occupancies and positions (including orientations) have been refined. At low CO2 loading, adsorbate–adsorbate interactions will be negligible meaning that the site occupancies directly reflect the binding strength between CO2 and the framework. Three CO2 sites A–C have significantly higher occupancies (0.65, 0.44, and 0.40, respectively) than the remaining five sites D–H (ranging within 0.26–0.11); surprisingly none of the sites makes an apparent hydrogen bonding interaction with the amide moiety. Site A resides on a 3-fold symmetry axis in the center of a triangular pocket formed by a [(Cu2)3(isophalate)3] unit, where the CO2 makes three identical long contacts to the phenyl rings [O1ACO2···ring centroid = 4.21(2) Å]. Site B lies close to a pyrimidine ring (O1BCO2···ring centroid = 3.22(9) Å) and makes a shorter contact with the aromatic C–H group on a phenyl ring (O1BCO2···C32 = 3.27(7) Å) along with a longer distance to an amide N–H (O1BCO2···N29 = 3.96(8) Å). Site C lies close to an isophalate phenyl ring (O3CCO2···ring centroid = 2.99(6) Å). The shortest contact between a guest CO2 and amide nitrogen atom is observed for the low occupancy site F (occupancy = 0.15) where the guest accepts a weak hydrogen bond (N29···O3FCO2 = 3.22(20) Å; < N–H···O = 122(5)°). The absence of CO2 molecules adjacent to the uncoordinated pyrimidine nitrogen atom is not surprising, as the site is sterically hindered by a neighboring phenyl ring; however, no such impediment exists around the amide N–H site which is fully accessible to the guest molecules. The preference of CO2 to make multiple long-range contacts with phenyl and pyrimidyl rings rather than accepting hydrogen bonds from the amide is striking and contrary to the assumptions which have previously informed the design philosophy of MOF materials for CO2 capture. Upon increasing the guest loading to 2.3 CO2/Cu(II), the occupancy of site A nears saturation (0.96) and remains distinctly higher than that of remaining sites B–H (range 0.50–0.18). In the structure of MFM-136 loaded with 1.1 CD4/Cu(II), an equivalent site to site A in the center of the triangular pocket (C1ACD4···ring centroid = 4.33(2) Å) is observed to have the highest occupancy of 0.36. Additional CD4 binding sites with lower occupancies were observed without notable interaction to the MOF host (Figure S19). To date, crystallographic characterizations of adsorbed gas molecules in MOFs have been mostly limited to one or two binding sites for materials with narrow pores.3−6 Simultaneous refinement of a large number of sites as reported in this work is made possible by the neutron diffraction data which give equal prominence to the light guests (particularly for CD4) and heavy framework.
Figure 3.

Binding sites of guests in MFM-136 at loadings of 1.8 CO2/Cu(II) and 1.1 CD4/Cu(II) elucidated from Rietveld refinement of NPD data. Colors: carbon, black; hydrogen, white; oxygen, red; nitrogen, blue; copper, teal; CO2/CD4 guests, purple/dark blue/green for sites A/B/C, respectively. Refined chemical occupancies of guest molecules inset.
The absence of adsorbed CO2 molecules at the pendant amide group could be due to the transition between “dynamic” and “kinetic” products in which the adsorbed CO2 has great mobility to translate/diffuse along the pore and form interactions with amide groups in a “come and go” fashion. The static crystallographic experiment can only paint a picture averaged over an extended time scale. Hence, only more stable environments of CO2 can be seen from the diffraction study. Thus, to gain direct insight into the binding dynamics of adsorbed CO2 molecules and the free amide groups, inelastic neutron spectroscopy (INS) was measured for MFM-136 as a function of CO2 loading (Figure 4). INS spectra for the bare MOF show multiple features which have been identified via DFT calculations (Figure S20). Specifically, the peak at 69 meV corresponds to the out-of-plane wagging modes of the N–H group, and peaks around 110–160 meV originate from the motion of aromatic C–H groups and deformational modes of the phenyl rings. Comparison of the INS spectra for bare and CO2-loaded MFM-136 shows very small changes to the overall vibrational peaks except for a guest–host stiffening effect as evidenced by a global shift of peaks to slightly higher energy. Indeed, the N–H motion (69 meV) has no detectable changes upon CO2 loading, while the aromatic C–H groups show small changes as confirmed by the difference spectra (Figure 4b), including a small increase in intensity at 116 meV (assigned as out-of-plane C–H bending on the isophthalate ring) and a decrease at 136 meV (assigned as in-plane C–H bending on all phenyl rings). This result is in excellent agreement with the NPD study and reaffirms the conclusion made from the diffraction experiment that direct CO2 binding to the amide groups in the pore is absent.
Figure 4.
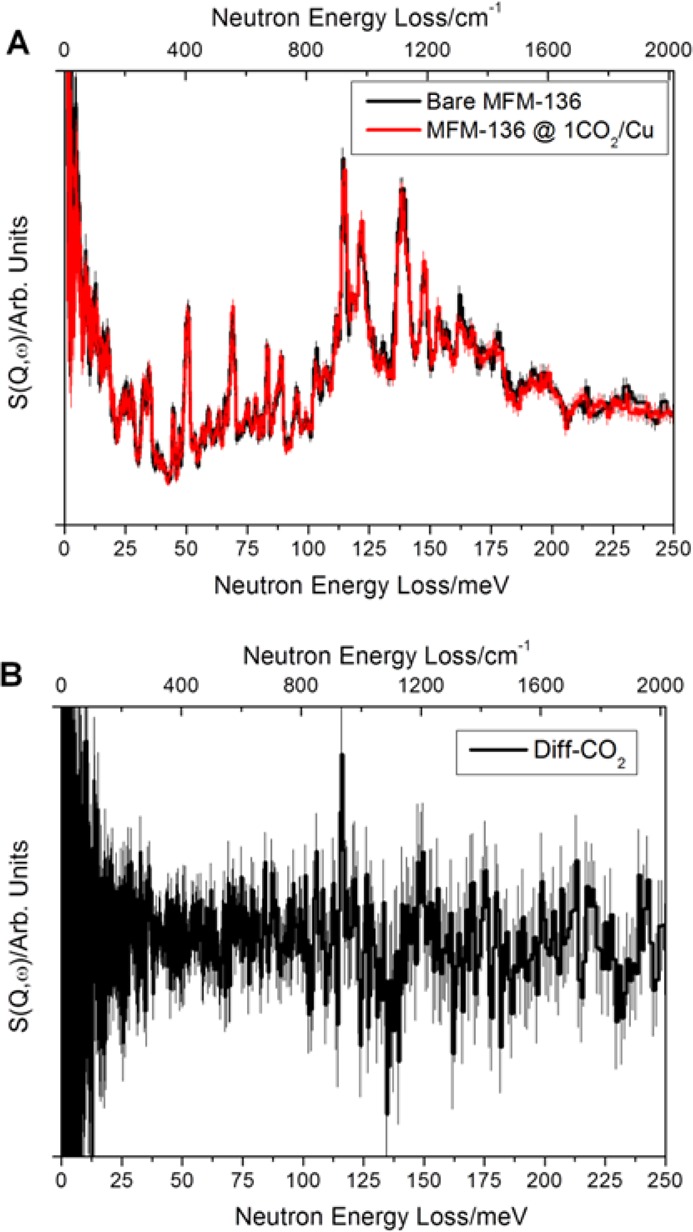
(a) Overlay of the INS spectra for bare and CO2-loaded MFM-136; (b) difference INS spectrum for the bare and CO2-loaded MFM-136.
Analysis of the CO2-MOF interaction energy landscape determined during the GCMC simulation of the isotherm shows that the strongest predicted guest adsorption locations are in agreement with site A, followed by sites around the periphery of cage A corresponding to sites B–E (Figure S21). As in the NPD and INS studies, no strong adsorption was observed in the regions surrounding the N–H group.
In summary, a (3,6)-connected pyrimidyl isophthalate acylamide decorated MOF with a rare eea-topology has been synthesized. The amide-functionalized MOF exhibits high CO2 uptake capacities and selectivity over CH4 and N2. Although it was anticipated that the amide moieties would actively participate in gas adsorption, the NPD and INS data reveal otherwise. The strongest binding site for both adsorbed CO2 and CD4 molecules are at the phenyl-isophthalate rings, and there is an absence of direct binding between adsorbed gas molecules and the pendent amide group in the pore. This has been confirmed by INS which shows retention of vibrational motion of the amide group upon CO2 binding. This study indicates that introduction of functional groups in MOF structures may not necessarily result in the formation of strong binding sites for gas molecules. Future investigation of the impact of a combination of functional groups and pore geometry is currently underway.
Acknowledgments
We thank EPSRC, ERC, Universities of Manchester and Nottingham for funding. O.B. thanks Niger Delta University and TETFund, Nigeria for a PhD scholarship. We thank STFC/ISIS for access to Beamlines TOSCA and WISH and providing computing resources on SCARF computer cluster. We thank Drs S. Rudić and D. Khalyavin for help at ISIS. E.B. acknowledges access to the High Performance Computing Facility at the University of Nottingham.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b08059.
The authors declare no competing financial interest.
Supplementary Material
References
- Allendorf M. D.; Stavila V. CrystEngComm 2015, 17, 229. 10.1039/C4CE01693A. [DOI] [Google Scholar]
- Guillerm V.; Kim D.; Eubank J. F.; Luebke R.; Liu X.; Adil K.; Lah M. S.; Eddaoudi M. Chem. Soc. Rev. 2014, 43, 6141. 10.1039/C4CS00135D. [DOI] [PubMed] [Google Scholar]
- a Bloch E. D.; Queen W. L.; Krishna R.; Zadrozny J. M.; Brown C. M.; Long J. R. Science 2012, 335, 1606. 10.1126/science.1217544. [DOI] [PubMed] [Google Scholar]; b Xiang S.; Zhou W.; Zhang Z.; Green M. A.; Liu Y.; Chen B. Angew. Chem., Int. Ed. 2010, 49, 4615. 10.1002/anie.201000094. [DOI] [PubMed] [Google Scholar]; c Queen W. L.; Hudson M. R.; Bloch E. D.; Mason J. A.; Gonzalez M. I.; Lee J. S.; Gygi D.; Howe J. D.; Lee K.; Darwish T. A.; James M.; Peterson V. K.; Teat S. J.; Smit B.; Neaton J. B.; Long J. R.; Brown C. M. Chem. Sci. 2014, 5, 4569. 10.1039/C4SC02064B. [DOI] [Google Scholar]
- Vaidhyanathan R.; Iremonger S. S.; Shimizu G. K. H.; Boyd P. G.; Alavi S.; Woo T. K. Science 2010, 330, 650. 10.1126/science.1194237. [DOI] [PubMed] [Google Scholar]
- a Lin X.; Telepeni I.; Blake A. J.; Dailly A.; Brown C. M.; Simmons J. M.; Zoppi M.; Walker G. S.; Thomas K. M.; Mays T. J.; Hubberstey P.; Champness N. R.; Schröder M. J. Am. Chem. Soc. 2009, 131, 2159. 10.1021/ja806624j. [DOI] [PubMed] [Google Scholar]; b Yan Y.; Yang S.; Blake A. J.; Schröder M. Acc. Chem. Res. 2014, 47, 296. 10.1021/ar400049h. [DOI] [PubMed] [Google Scholar]
- Yang S.; Ramirez-Cuesta A. J.; Newby R.; Garcia-Sakai V.; Manuel P.; Callear S. K.; Campbell S. I.; Tang C. C.; Schröder M. Nat. Chem. 2014, 7, 121. 10.1038/nchem.2114. [DOI] [PubMed] [Google Scholar]
- a Li B.; Zhang Z.; Li Y.; Yao K.; Zhu Y.; Deng Z.; Yang F.; Zhou X.; Li G.; Wu H.; Nijem N.; Chabal Y. J.; Lai Z.; Han Y.; Shi Z.; Feng S.; Li J. Angew. Chem., Int. Ed. 2012, 51, 1412. 10.1002/anie.201105966. [DOI] [PubMed] [Google Scholar]; b Du L.; Yang S.; Xu L.; Min H.; Zheng B. CrystEngComm 2014, 16, 5520. 10.1039/c4ce00530a. [DOI] [Google Scholar]; c Lee C. H.; Huang H. Y.; Liu Y. H.; Luo T. T.; Lee G. H.; Peng S. M.; Jiang J. C.; Chao I.; Lu K. L. Inorg. Chem. 2013, 52, 3962. 10.1021/ic302758g. [DOI] [PubMed] [Google Scholar]; d Vaidhyanathan R.; Iremonger S. S.; Shimizu G. K. H.; Boyd P. G.; Alavi S.; Woo T. K. Angew. Chem., Int. Ed. 2012, 51, 1826. 10.1002/anie.201105109. [DOI] [PubMed] [Google Scholar]
- a Alsmail N. H.; Suyetin M.; Yan Y.; Cabot R.; Krap C. P.; Lu J.; Easun T. L.; Bichoutskaia E.; Lewis W.; Blake A. J.; Schröder M. Chem. - Eur. J. 2014, 20, 7317. 10.1002/chem.201304005. [DOI] [PubMed] [Google Scholar]; b Zheng B.; Bai J.; Duan J.; Wojtas L.; Zaworotko M. J. J. Am. Chem. Soc. 2011, 133, 748. 10.1021/ja110042b. [DOI] [PubMed] [Google Scholar]; c Zheng B.; Yang Z.; Bai J.; Li Y.; Li S. Chem. Commun. 2012, 48, 7025. 10.1039/c2cc17593b. [DOI] [PubMed] [Google Scholar]; d Xiong S.; He Y.; Krishna R.; Chen B.; Wang Z. Cryst. Growth Des. 2013, 13, 2670. 10.1021/cg4004438. [DOI] [Google Scholar]; e Chen Y.-Q.; Qu Y.-K.; Li G.-R.; Zhuang Z.-Z.; Chang Z.; Hu T.-L.; Xu J.; Bu X.-H. Inorg. Chem. 2014, 53, 8842. 10.1021/ic500788z. [DOI] [PubMed] [Google Scholar]; f Chen Z.; Adil K.; Weselinski L. J.; Belmabkhout Y.; Eddaoudi M. J. Mater. Chem. A 2015, 3, 6276. 10.1039/C4TA07115H. [DOI] [Google Scholar]; g Lu Z.; Bai J.; Hang C.; Meng F.; Liu W.; Pan Y.; You X. Chem. - Eur. J. 2016, 22, 6277. 10.1002/chem.201504907. [DOI] [PubMed] [Google Scholar]
- Duan J.; Yang Z.; Bai J.; Zheng B.; Li Y.; Li S. Chem. Commun. 2012, 48, 3058. 10.1039/c2cc16231h. [DOI] [PubMed] [Google Scholar]
- Wei Y.-S.; Lin R.-B.; Wang P.; Liao P.-Q.; He C.-T.; Xue W.; Hou L.; Zhang W.-X.; Zhang J.-P.; Chen X.-M. CrystEngComm 2014, 16, 6325. 10.1039/C3CE42219D. [DOI] [Google Scholar]
- a Nandi S.; De Luna P.; Daff T. D.; Rother J.; Liu M.; Buchanan W.; Hawari A. I.; Woo T. K.; Vaidhyanathan R. Sci. Adv. 2015, 1, e1500421. 10.1126/sciadv.1500421. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nugent P.; Belmabkhout Y.; Burd S. D.; Cairns A. J.; Luebke R.; Forrest K.; Pham T.; Ma S.; Space B.; Wojtas L.; Eddaoudi M.; Zaworotko M. J. Nature 2013, 495, 80. 10.1038/nature11893. [DOI] [PubMed] [Google Scholar]
- Wang J.; Huang L.; Yang R.; Zhang Z.; Wu J.; Gao Y.; Wang Q.; O’Hare D.; Zhong Z. Energy Environ. Sci. 2014, 7, 3478. 10.1039/C4EE01647E. [DOI] [Google Scholar]
- Singh M. P.; Dhumal N. R.; Kim H. J.; Kiefer J.; Anderson J. A. J. Phys. Chem. C 2016, 120, 17323. 10.1021/acs.jpcc.6b02906. [DOI] [Google Scholar]
- a Keceli E.; Hemgesberg M.; Grunker R.; Bon V.; Wilhelm C.; Philippi T.; Schoch R.; Sun Y.; Bauer M.; Ernst S.; Kaskel S.; Thiel W. R. Microporous Mesoporous Mater. 2014, 194, 115. 10.1016/j.micromeso.2014.03.022. [DOI] [Google Scholar]; b Debatin F.; Thomas A.; Kelling A.; Hedin N.; Bacsik Z.; Senkovska I.; Kaskel S.; Junginger M.; Mueller H.; Schilde U.; Jaeger C.; Friedrich A.; Holdt H.-J. Angew. Chem., Int. Ed. 2010, 49, 1258. 10.1002/anie.200906188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.