

Abstract
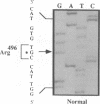
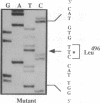
Although the A and B subtypes of Niemann-Pick disease (NPD) both result from the deficient activity of acid sphingomyelinase (ASM; sphingomyelin cholinephosphohydrolase, EC 3.1.4.12) and the lysosomal accumulation of sphingomyelin, they have remarkably distinct phenotypes. Type A disease is a fatal neurodegenerative disorder of infancy, whereas type B disease has no neurologic manifestations and is characterized primarily by reticuloendothelial involvement and survival into adulthood. Both disorders are more frequent among individual of Ashkenazi Jewish ancestry than in the general population. The recent isolation and characterization of cDNA and genomic sequences encoding ASM has facilitated investigation of the molecular lesions causing the NPD subtypes. Total RNA was reverse-transcribed, and the ASM cDNA from an Ashkenazi Jewish type A patient was specifically amplified by the polymerase chain reaction (PCR). Molecular analysis of the PCR products revealed a G----T transversion of nucleotide 1487, which occurred at a CpG dinucleotide and predicted an Arg----Leu substitution in residue 496. Hybridization of PCR-amplified genomic DNA with allele-specific oligonucleotides indicated that the proband was homoallelic for the Arg----Leu substitution and that both parents and several other relatives were heterozygous. This mutation was detected in 32% (10 of 31) of the Ashkenazi Jewish NPD type A alleles studied and occurred in only 5.6% (2 of 36) of ASM alleles from non-Jewish type A patients. Of interest, the Arg----Leu substitution occurred in one of the ASM alleles from the two Ashkenazi Jewish NPD type B patients studied and in none of the ASM alleles of 15 non-Jewish type B patients. In contrast, the mutation was not present in 180 ASM alleles from normal individuals of Ashkenazi Jewish descent. These findings identify a frequent missense mutation among NPD patients of Ashkenazi Jewish ancestry that results in neuronopathic type A disease when homoallelic and can result in the nonneuronopathic type B phenotype when heteroallelic. The identification of this ASM mutation in Ashkenazi Jewish patients should facilitate the prevention of NPD in this population by carrier detection with molecular diagnostic techniques.
Full text
PDF





Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Bernstein H. S., Bishop D. F., Astrin K. H., Kornreich R., Eng C. M., Sakuraba H., Desnick R. J. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J Clin Invest. 1989 Apr;83(4):1390–1399. doi: 10.1172/JCI114027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D. F., Desnick R. J. Affinity purification of alpha-galactosidase A from human spleen, placenta, and plasma with elimination of pyrogen contamination. Properties of the purified splenic enzyme compared to other forms. J Biol Chem. 1981 Feb 10;256(3):1307–1316. [PubMed] [Google Scholar]
- Brady R. O., Kanfer J. N., Mock M. B., Fredrickson D. S. The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae. Proc Natl Acad Sci U S A. 1966 Feb;55(2):366–369. doi: 10.1073/pnas.55.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase G. A., McKusick V. A. Controversy in human genetics: founder effect in Tay-Sachs disease. Am J Hum Genet. 1972 May;24(3):339–340. [PMC free article] [PubMed] [Google Scholar]
- Coulondre C., Miller J. H., Farabaugh P. J., Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978 Aug 24;274(5673):775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- Fraikor A. L. Tay-Sachs disease: genetic drift among the Ashkenazim Jews. Soc Biol. 1977 Summer;24(2):117–134. doi: 10.1080/19485565.1977.9988272. [DOI] [PubMed] [Google Scholar]
- Itakura K., Rossi J. J., Wallace R. B. Synthesis and use of synthetic oligonucleotides. Annu Rev Biochem. 1984;53:323–356. doi: 10.1146/annurev.bi.53.070184.001543. [DOI] [PubMed] [Google Scholar]
- Kaback M. M. Tay-Sachs disease: from clinical description to prospective control. Prog Clin Biol Res. 1977;18:1–7. [PubMed] [Google Scholar]
- Klar R., Levade T., Gatt S. Synthesis of pyrenesulfonylamido-sphingomyelin and its use as substrate for determining sphingomyelinase activity and diagnosing Niemann-Pick disease. Clin Chim Acta. 1988 Sep 15;176(3):259–267. doi: 10.1016/0009-8981(88)90185-4. [DOI] [PubMed] [Google Scholar]
- Myerowitz R., Costigan F. C. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J Biol Chem. 1988 Dec 15;263(35):18587–18589. [PubMed] [Google Scholar]
- Myerowitz R. Splice junction mutation in some Ashkenazi Jews with Tay-Sachs disease: evidence against a single defect within this ethnic group. Proc Natl Acad Sci U S A. 1988 Jun;85(11):3955–3959. doi: 10.1073/pnas.85.11.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrianthopoulos N. C., Melnick M. Tay-Sachs disease: a genetic-historical view of selective advantage. Prog Clin Biol Res. 1977;18:95–106. [PubMed] [Google Scholar]
- Myrianthopoulos N. C., Naylor A. F., Aronson S. M. Founder effect in Tay-Sachs disease unlikely. Am J Hum Genet. 1972 May;24(3):341–342. [PMC free article] [PubMed] [Google Scholar]
- Navon R., Proia R. L. The mutations in Ashkenazi Jews with adult GM2 gangliosidosis, the adult form of Tay-Sachs disease. Science. 1989 Mar 17;243(4897):1471–1474. doi: 10.1126/science.2522679. [DOI] [PubMed] [Google Scholar]
- Quintern L. E., Schuchman E. H., Levran O., Suchi M., Ferlinz K., Reinke H., Sandhoff K., Desnick R. J. Isolation of cDNA clones encoding human acid sphingomyelinase: occurrence of alternatively processed transcripts. EMBO J. 1989 Sep;8(9):2469–2473. doi: 10.1002/j.1460-2075.1989.tb08382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki R. K., Gelfand D. H., Stoffel S., Scharf S. J., Higuchi R., Horn G. T., Mullis K. B., Erlich H. A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988 Jan 29;239(4839):487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A. R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977 Dec;74(12):5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triggs-Raine B. L., Feigenbaum A. S., Natowicz M., Skomorowski M. A., Schuster S. M., Clarke J. T., Mahuran D. J., Kolodny E. H., Gravel R. A. Screening for carriers of Tay-Sachs disease among Ashkenazi Jews. A comparison of DNA-based and enzyme-based tests. N Engl J Med. 1990 Jul 5;323(1):6–12. doi: 10.1056/NEJM199007053230102. [DOI] [PubMed] [Google Scholar]
- Tsuji S., Choudary P. V., Martin B. M., Stubblefield B. K., Mayor J. A., Barranger J. A., Ginns E. I. A mutation in the human glucocerebrosidase gene in neuronopathic Gaucher's disease. N Engl J Med. 1987 Mar 5;316(10):570–575. doi: 10.1056/NEJM198703053161002. [DOI] [PubMed] [Google Scholar]
- da Veiga Pereira L., Desnick R. J., Adler D. A., Disteche C. M., Schuchman E. H. Regional assignment of the human acid sphingomyelinase gene (SMPD1) by PCR analysis of somatic cell hybrids and in situ hybridization to 11p15.1----p15.4. Genomics. 1991 Feb;9(2):229–234. doi: 10.1016/0888-7543(91)90246-b. [DOI] [PubMed] [Google Scholar]