

Abstract
The constraint-based analysis of genome-scale metabolic and regulatory networks has been successful in predicting phenotypes and useful for analyzing high-throughput data sets. Within this modeling framework, linear optimization has been used to study genome-scale metabolic models, resulting in the enumeration of single optimal solutions describing the best use of the network to support growth. Here mixed-integer linear programming was used to calculate and study a subset of the alternate optimal solutions for a genome-scale metabolic model of Escherichia coli (iJR904) under a wide variety of environmental conditions. Analysis of the calculated sets of optimal solutions found that: (1) only a small subset of reactions in the network have variable fluxes across optima; (2) sets of reactions that are always used together in optimal solutions, correlated reaction sets, showed moderate agreement with the currently known transcriptional regulatory structure in E. coli and available expression data, and (3) reactions that are used under certain environmental conditions can provide clues about network regulatory needs. In addition, calculation of suboptimal flux distributions, using flux variability analysis, identified reactions which are used under significantly more environmental conditions suboptimally than optimally. Together these results demonstrate the utilization of reactions in genome-scale models under a variety of different growth conditions.
Constraint-based modeling of reconstructed genome-scale metabolic networks has proven useful for understanding and predicting the genotype-phenotype relationship in microbes (Ibarra et al. 2002; Segre et al. 2002; Stelling et al. 2002; Fong et al. 2003; Forster et al. 2003; Price et al. 2003; Reed and Palsson 2003). Within this analysis framework, a variety of methods have been developed to characterize the metabolic steady-state flux solution space and select solutions within that space that might be physiologically relevant, including elementary mode analysis and extreme pathway analysis (Papin et al. 2003), flux balance analysis (Kauffman et al. 2003), MOMA (Segre et al. 2002), OptKnock (Burgard et al. 2003), flux-coupling (Burgard et al. 2004), random sampling (Almaas et al. 2004; Price et al. 2004; Wiback et al. 2004), and flux variability analysis (Mahadevan and Schilling 2003). Flux balance analysis uses linear optimization to find a flux distribution that maximizes a particular objective function (e.g., growth rate or ATP production; Varma and Palsson 1994; Kauffman et al. 2003). However, there are often multiple flux distributions that are equally optimal (value of the objective function is the same) giving rise to the concept of multiple alternate optima (Lee et al. 2000; Mahadevan and Schilling 2003). The existence of such multiple optima would correspond to the biological notion of silent phenotypes (Raamsdonk et al. 2001), that is, the same observed overall cellular function is achieved with different uses of underlying reaction networks (Fong et al. 2003).
Mixed-integer linear programming (MILP) has been used to study optimal solutions by identifying the minimum number of reactions needed for optimal growth (minimum reaction sets; Burgard and Maranas 2001; Burgard et al. 2001), as well as enumerating alternate basic optima for small metabolic networks, where the objective function takes on the same value but the flux distributions through the metabolic network are different (Lee et al. 2000; Phalakornkule et al. 2001). The alternate optimal flux distributions calculated using the MILP algorithm differ in that they all use a different set of reactions. For the small-scale networks previously studied (Lee et al. 2000; Phalakornkule et al. 2001), the number of alternate optima was on the order of 10 solutions. In genome-scale networks, a large number of network redundancies exist (Mahadevan and Schilling 2003) creating computational challenges in calculating all alternative optima. One approach to explore the range of optimal solutions is to fix the growth rate and then calculate the minimum and maximum flux values through each reaction in the network (referred to in this paper as `flux variability analysis'). The approach was recently used to analyze the initial genome-scale network for Escherichia coli (iJE660a; Edwards and Palsson 2000) for a select number of environmental conditions including aerobic growth on glucose, acetate, and D-lactate (Mahadevan and Schilling 2003).
Here we applied the recursive MILP algorithm (Lee et al. 2000) to calculate a subset of the alternate optimal solutions in an expanded genome-scale model of E. coli (iJR904; Reed et al. 2003) for a large number of media conditions (136). Comparisons between the optimal solutions characterized by the flux variability analysis and those calculated with an MILP approach found that the first 500 solutions identify all of the variable fluxes in the set of alternate optima, but do not fully capture the magnitude of the flux variability through the individual reactions. Looking at the alternate optima across the 136 simulated growth environments, a number of reactions were found to be utilized in all of the calculated alternative optima, whereas others were used under only a few environmental conditions. This collection of optimal solutions was studied to provide insights into the candidate regulatory mechanisms and strategies that would allow E. coli to grow optimally. Flux variability analysis was also used to study how fluxes are used in suboptimal solutions, and this approach identified reactions that if used would lead to suboptimal growth by the cell. Thus, the present study is the most comprehensive evaluation of alternative optima in genome-scale models to date.
RESULTS
We first computed a sample of 56,756 optimal solutions under 136 growth environments. We then assessed the properties of these solutions and found the correlated reaction subsets in these optimal solutions. The correlated reaction subsets were then compared to known regulon structures and expression profiling data sets. For aerobic and anaerobic growth on glucose, we compared the utilization of reactions in the alternative optima to described transcriptional regulation of the genes that are associated with some of these fluxes. Finally, we compared the reaction usage in optimal growth solutions to those in suboptimal growth solutions.
Comparing Results From MILP and Flux Variability Analysis
Previous metabolic networks studied using MILP have enumerated a finite number of flux distributions (Lee et al. 2000). A modified version of the algorithm (see Methods section) was implemented here and used to calculate basic optimal solutions for 88 aerobic and 48 anaerobic growth conditions, where different carbon sources were used (see Table 1 for a list of simulated carbon sources). The set of optima for a given environment will be referred to in this paper as the `condition-specific optima'. Application of the modified MILP algorithm to a genome-scale metabolic network showed that for many carbon sources the number of alternate optima was large, making it computationally difficult to enumerate all of the optimal solutions. As such, only the first 500 optimal solutions were calculated for each of the 136 (= 88 + 48) environmental conditions. For some simulated environments, aerobic growth with glycine as the sole carbon source and for all 48 anaerobic conditions, it was possible to enumerate all of the alternate optima.
Table 1.
Allowable Carbon Sources
| Abbr. | Metabolite name | Abbr. | Metabolite name |
|---|---|---|---|
| 2ddglcn | 2-Dehydro-3-deoxy-D-gluconate | tre | Trehalose |
| acgam | N-acetyl-D-glucosamine | uri | Uridine |
| acmana | N-Acetyl-D-mannosamine | xtsn | Xanthosine |
| acnam | N-Acetylneuraminate | xyl-D | D-Xylose |
| adn | Adenosine | 12ppd-S | (S)-Propane-1,2-diol |
| arab-L | L-Arabinose | 3hcinnm | 3-hydroxycinnamic acid |
| cytd | Cytidine | 3hpppn | 3-(3-hydroxy-phenyl)propionate |
| dad-2 | Deoxyadenosine | 4abut | 4-Aminobutanoate |
| dcyt | Deoxycytidine | ac | Acetate |
| dgsn | Deoxyguanosine | acac | Acetoacetate |
| dha | Dihydroxyacetone | acald | Acetaldehyde |
| din | Deoxyinosine | akg | 2-Oxoglutarate |
| duri | Deoxyuridine | ala-D | D-Alanine |
| fru | D-Fructose | ala-L | L-Alanine |
| fuc-L | L-Fucose | arg-L | L-Arginine |
| g6p | D-Glucose 6-phosphate | asn-L | L-Asparagine |
| gal | D-Galactose | asp-L | L-Asparate |
| galct-D | D-Galactarate | but | Butyrate (n-C4:0) |
| galctn-D | D-Galactonate | cit | Citrate |
| galt | Galactitol | etoh | Ethanol |
| galur | D-Galacturonate | fum | Fumarate |
| gam | D-Glucosamine | gin-L | L-Glutamine |
| glc-D | D-Glucose | glu-L | L-Glutamate |
| glcn | D-Gluconate | gly | Glycine |
| glcr | D-Glucarate | glyc | Glycerol |
| glcur | D-Glucuronate | glyclt | Glycolate |
| glyald | D-Glyceraldehyde | hdca | Hexadecanoate (n-C16:0) |
| glyc3p | Glycerol 3-phosphate | lac-D | D-Lactate |
| gsn | Guanosine | lac-L | L-Lactate |
| idon-L | L-idonate | mal-L | L-Malate |
| ins | Inosine | mnl | D-Mannitol |
| lcts | Lactose | ocdca | Octadecanoate (n-C18:0) |
| malt | Maltose | orn | Ornithine |
| malthx | Maltohexaose | pppn | Phenylpropanoate |
| maltpt | Maltopentaose | pro-L | L-Proline |
| malttr | Maltotriose | ptrc | Putrescine |
| maltttr | Maltotetraose | pyr | Pyruvate |
| man | D-Mannose | ser-D | D-Serine |
| man6p | D-Mannose 6-phosphate | ser-L | L-Serine |
| melib | Melibiose | succ | Succinate |
| rib-D | D-Ribose | tartr-L | L-tartrate |
| rmn | L-Rhamnose | thr-L | L-Threonine |
| sbt-D | D-Sorbitol | trp-L | L-Tryptophan |
| sucr | Sucrose | ttdca | Tetradecanoate (n-C14:0) |
Shaded metabolites can be used aerobically and anaerobically as sole carbon sources; unshaded metabolites can only be used as sole carbon sources under aerobic conditions.
To investigate how well the calculated condition-specific optima spanned or represented the actual range of optimal solutions, the number of varying fluxes and the range of those fluxes were calculated from the different sets of condition-specific optima and compared to the results using flux variability analysis. Figure 1 shows that for the 88 aerobic conditions, the first 150 optimal solutions are sufficient to identify all of the variable fluxes among the set of condition-specific optima, whereas determining the numerical range of these fluxes in the set sometimes required the calculation of more optima. For some carbon sources, the magnitude of the flux ranges when looking at all 500 optimal solutions was smaller than the actual flux ranges calculated using flux variability analysis. These results imply that the first 500 optimal solutions are adequate for getting a sample of the full set of condition-specific optima, while still remaining computationally tractable. With adequate sampling, the set of condition-specific optima can be further analyzed.
Figure 1.

Comparisons of properties for sampled optima with all optima. The number of variable fluxes and the allowable ranges for these fluxes across all optima were calculated using a flux variability analysis. Each line is for one of the 88 carbon sources capable of supporting aerobic growth. (A) shows that as the number of calculated optima increases, the number of variable fluxes found in these sampled optimal solutions approaches the total number of variable fluxes. (B) shows how the magnitude of the flux variations is represented by the sampled optima relative to the actual flux variability across all optima.
Properties of Alternate Optima
The average optimal flux distribution, found among the 136 growth conditions considered, used 294 of the 931 internal reactions in iJR904, and for a given growth condition the average number of variable fluxes was 49 (with the number of variable fluxes typically being higher under aerobic conditions). Interestingly, with the exception of anaerobic growth on adenosine and deoxyadenosine, none of the exchange fluxes with the environment varied across alternate optima. This result indicates that the external state or phenotype of the cell is normally unique for a set of condition-specific optima, whereas flexibility in the internal fluxes accounts for the different optima. In all, there were 140 internal fluxes and five exchange fluxes that were variable under at least one of the 136 tested environmental conditions (see Supplemental material). Most of the flexibility in the network resides with reactions using different electron carriers, nucleotide salvage reactions, and central metabolic reactions. There were quite a few instances in central metabolism where the flux through a reaction was variable across the different optima (most of glycolysis, TCA cycle, anaplerotic reactions, and oxidative phosphorylation reactions).
The complete set of alternate optima, both aerobic and anaerobic, are a set of 56,756 flux distributions that were further studied—grouped together they are referred to here as the `mixed optima'. For each reaction in the network, the fraction of the optimal solutions, in the set of mixed optima, which use that particular reaction (fopt) was calculated (Fig. 2A, also available in Supplemental material). Reactions in the iJR904 model were previously assigned to different metabolic subsystems based on metabolic function, and Figure 2B shows for each subsystem how many reactions are assigned to that subsystem and the distribution of fractional usage within this subsystem. For example, there are 40 oxidative phosphorylation reactions: 65% of these are never used in any of the mixed optima, 20% have a fopt between 0 and 0.25, 3% have a fopt between 0.25 and 0.5, 8% have a fopt between 0.75 and 1.0, and 5% are used in all optima.
Figure 2.

Reaction usage in optimal flux distributions. (A) shows for each reaction in the metabolic network, what fraction of the optimal flux distributions utilize this reaction (fopt). The reactions are then rank-ordered by frequency of use in optimal flux distributions. Each reaction in the model was previously classified into one of 30 subsystems. (B) shows for each subsystem how many reactions belong to that subsystem (No. Rxns) and what fraction of these reactions are: never used (fopt = 0), used in less than 25% of the solutions (0 < fopt < 0.25), used in between 25% and 50% of the solutions (0.25 < fopt < 0.5), used in between 50% and 75% of the solutions (0.5 < fopt < 0.75), used in between 75% and 100% of the solutions (0.75 < fopt < 1), and used in all of the solutions (fopt = 1). The individual fractions are shaded according to value: less than 0.1 is white, between 0.1 and 0.5 is light gray, and larger than 0.5 is dark gray.
A total of 201 reactions were used in all optimal solutions across the different environmental conditions; these include reactions involved in amino acid metabolism (cys, his, ile, leu, lys, met, phe, and tyr—the amino acids that cannot serve as carbon sources), folate metabolism, and membrane lipid biosynthesis. These 201 reactions are needed for optimal growth across all 136 simulated growth environments, and are related to the intersection of minimal reactions sets (Burgard and Maranas 2001; Burgard et al. 2001) for the different growth environments. Comparisons to experimental gene essentiality data for growth on rich media (Gerdes et al. 2003) shows that of the 201 reactions, 81 are associated with genes essential for growth on rich media, 20 have multiple isozymes explaining why single knockouts might not have been essential experimentally, and another 20 do not have any ORFs associated with it in iJR904. For the remaining 80 reactions, the rich media might contain metabolites that cannot be used as sole carbon sources, allowing for some of these reactions to be unessential. All mixed optima use histidine biosynthesis reactions, but if histidine was present in the growth medium these associated genes would be unessential. These 201 reactions are needed for optimal growth across the tested environmental conditions, but not necessarily for suboptimal growth, further explaining why not all of the reactions were associated with lethal genes.
In contrast to the 201 reactions that are used across all of the mixed optima, a number of reactions (351) were never utilized by any of the optimal solutions for any of the tested environments. These include 185 blocked reactions—reactions that will never be used by the network even if all exchange fluxes are free (Burgard et al. 2004). In addition to these blocked reactions are reactions needed for unsimulated environments (e.g., anaerobic growth with alternate electron acceptors such as dimethylsulfoxide or nitrate) and reactions that are less efficient than others (e.g., oxidative phosphorylation reactions that transfer different amounts of protons across the membrane). This first-pass analysis of the mixed optima can be advanced by a more detailed investigation of how reactions are used relative to each other and with respect to specific environmental conditions.
Correlated Reaction Sets
Further examination of the mixed optima identified sets of reactions that are always used together in a flux distribution; these reaction sets have been previously referred to as `correlated reactions,' `fully coupled reactions,' and `reaction/enzyme subsets' (Papin et al. 2002; Schilling et al. 2002; Schuster et al. 2002; Burgard et al. 2004). The 66 correlated reaction sets calculated in the present study arise from flux distributions which maximize biomass yields, leading to the hypothesis that the reactions in a correlated reaction set would have similar regulation if a cell uses its metabolic network to maximize biomass production. The number of reactions in a set of correlated reactions varied between two and nine, with a set size of two being the most frequent (Fig. 3).
Figure 3.

Distribution of correlated reaction sets. The 66 correlated reaction sets can be categorized by the number of reactions in a set as well as the set's metabolic purpose (metabolite biosynthesis or degradation or other). Using known regulation from databases and available literature, 45 out of 66 sets involved genes where at least half of the genes belonged to the same regulon (strong regulation). For six of the two reaction sets, one or both of the reactions were not associated with any genes, so no comparison could be made.
The regulatory structure in E. coli—taken from EcoCyc (Karp et al. 2002), RegulonDB (Salgado et al. 2004), and primary literature data—was used to test whether the genes associated with correlated reaction sets have been found to be coregulated. For 9% of the correlated reaction sets no comparisons could be made, because there were not at least two reactions in the set with associated genes. Comparisons between regulon information and correlated reaction sets showed that 68% of the correlated reaction sets were consistent with established regulatory structure (where more than half of the associated genes belonged to the same regulon), and for 23% of the correlated reaction sets there was only weak evidence of conserved regulatory structure (less than half of the associated genes belonged to the same regulon). The transcriptional regulatory network in E. coli, however, has not been completely elucidated. Recent analysis of expression data in the context of a regulatory model indicates that roughly only 25% of the transcriptional regulatory mechanisms have been described in the primary literature (Covert et al. 2004).
As a further comparison, expression data were used to determine whether genes in a correlated reaction set have similar expression patterns under different conditions. Using publicly available AffyMetrix data, from the ASAP database, (Allen et al. 2003), the average pairwise correlation coefficient between genes involved in a reaction set was calculated. P-values, indicating whether the correlation coefficient is significant, were calculated by computing the average correlation coefficients from a random set of genes (see Methods). The resulting average correlation coefficients and associated P-values for the correlated reaction sets are shown in Figure 4A (also available in Supplemental materials). For comparison the average correlation coefficients and P-values for genes belonging to the same transcription unit (Karp et al. 2002) were calculated (Fig. 4B). Only transcription units with at least two genes with measured expression data were included in the analysis.
Figure 4.

Correlation of genes in correlated reaction sets and transcription units based on expression data. The average correlation coefficients between genes associated with reactions in correlated reaction sets was calculated using a set of publicly available (ASAP) expression data from 20 different conditions. The calculated average correlation coefficients and their corresponding P-values are plotted in A (six of the two reaction sets were omitted from the graph because at least one of the reactions had no associated genes). Circles are used to denote small sets (only two genes with expression data), and triangles are used to denote larger sets (greater than two genes with expression data); open shapes are used for sets that are used in less than 10,000 optimal solutions, and filled shapes are used for sets used in more than 10,000 optimal solutions. (B) shows average correlation coefficients between genes on the same transcription unit (Karp et al. 2002) using the same set of expression data. Open circles are used to denote small sets (only two genes with expression data) and solid triangles to denote larger sets (greater than two genes with expression data).
The transcription units appear to have more significant correlation than the correlated reaction sets: 18 out of 66 (27.3%) correlated reaction sets have P-values less than 0.05, whereas 159 of 321 (49.5%) transcription units have P-values less than 0.05. Almost all of the correlated reaction sets with significant correlation were classified as having evidence of being part of the same regulon (17). The number of times a correlated reaction set is used in the mixed optima does not seem to affect how well the set correlates with the expression data.
Condition-Dependent Fluxes
The condition-specific optimal solutions for glucose aerobic and glucose anaerobic growth were further analyzed to investigate whether the transcriptional regulatory network controlling metabolic enzymes pushes the cell towards an optimal state. The fraction of solutions that use a particular reaction under glucose aerobic conditions was compared to the fraction for glucose anaerobic conditions (Fig. 5, also available in Supplemental materials). Each point in Figure 5 represents a different metabolic reaction. One may expect that reactions that fall below the line (meaning that they are used more highly in glucose aerobic optima) would have their genes up-regulated under aerobic conditions and down-regulated under anaerobic conditions; the opposite would be true for reactions falling above the line.
Figure 5.

Preferential usage of fluxes under aerobic glucose vs. anaerobic glucose optimal growth. Each point in the graph represents one of the 931 metabolic reactions; the x-axis plots the fraction of optimal solutions that utilize that reaction under glucose aerobic conditions (500 optima), and the y-axis plots the fraction of optimal solutions that utilize that reaction under glucose anaerobic conditions (204 optima). Using a regulatory model (iMC1010v2) that accounts for known regulation and hypothesized regulation based on expression data, some reactions were predicted to have more isozymes present aerobically (black) or more isozymes present anaerobically (white). Discrepancies between regulation and usage of fluxes in alternate optima are circled and labeled in the figure, see text for further details.
A previously developed regulatory model, iMC1010v2, was used to compare these predictions with the transcriptional regulatory structure in E. coli. The iMC1010v2 model was constructed based on established regulatory mechanisms and went through one iteration of improvement using transcription factor knockout strains whose expression was measured under glucose aerobic and glucose anaerobic conditions (Covert et al. 2004). The transcriptional regulatory model predicted for 61 reactions that more isozymes would be expressed under aerobic glucose conditions (aerobic reaction set), and for 53 reactions that more isozymes would be expressed under anaerobic glucose conditions (anaerobic reaction set). Black squares in Figure 5 are used to indicate reactions belonging to the aerobic reaction set, white squares for reactions in the anaerobic reaction set, and gray squares for the remaining reactions in the model. Assuming that the bacteria operate optimally, it would be expected that the white points would fall above the line and the black points below the line.
Most of the reactions that have predicted changes in the expression of associated genes, based on iMC1010v2, are not used in any of the condition-specific optimal solutions for glucose aerobic or glucose anaerobic conditions. For both the aerobic and anaerobic reaction sets, nine reactions in each set fell on the predicted half of the graph. Discrepancies occurred in both reaction sets, where more isozymes are expressed under the condition that utilizes a reaction the least under optimal conditions.
One of the two discrepancies in the aerobic reaction set is used only slightly more in the anaerobic case (fanaerobic = 0.681 vs. faerobic = 0.676); further sampling of the alternate optima could result in higher aerobic usage (faerobic). The other aerobic reaction set discrepancy is for the glycolate transport reaction (GLYCLT2r) which is used in all glucose anaerobic optimal solutions and in no glucose aerobic optimal solutions. Deletion of GLYCLT2r from the network only drops the maximal anaerobic growth rate by 0.1%, so even if the transporter is not expressed there would be negligible differences in observed growth rate. It is also important to note that the model predicts that glycolate cannot serve as a carbon source under anaerobic conditions, explaining why this transporter might be expressed aerobically.
There are also six discrepancies for reactions in the aerobic reaction set: hydrogenase reactions (HYD1, HYD2, HYD3), fumarate reductase (FRD2), formate-hydrogen lyase (FHL), and formate dehydrogenase (FDH2). Like the glycolate transporter, all six reactions can be deleted simultaneously with negligible effect on the predicted maximal growth rate (drops only by 0.1%). The fumarate reductase enzyme is responsible for two reactions, FRD2 and FRD3 (where different quinones are used as electron donors in the two reactions). The FRD3 reaction is used in all glucose anaerobic optima, explaining why the fumarate reductase enzyme is expressed under anaerobic conditions.
Suboptimal Fluxes
The previous sections focused on properties of optimal solutions; this section examines properties of suboptimal solutions. Flux variability analysis rather than MILP was used to calculate the variability of fluxes for different fixed growth rates (99% maximal growth, 90% maximal growth, 50% maximal growth, and 25% maximal growth) under the 88 aerobic environments. For each of the aerobic growth conditions (see Table 1) the set of suboptimal reactions (those reactions that can be used in a suboptimal flux distribution) for the different suboptimal growth rate levels was identical. The fraction of environmental conditions, fenv, (out of a total of 88 considered) in which a reaction can be used in an optimal solution (gray curve) or suboptimal solution (black curve) was determined (Fig. 6; note that the 185 blocked reactions are left out of this figure). The black curve in Figure 6 is always greater than or equal to the gray curve, indicating that the set of optimal reactions (those reactions that can be used in an optimal flux distribution) is always a subset of the suboptimal reactions. Again reactions can be categorized according to their usage: 380 reactions are used under the same number of conditions optimally and suboptimally (ignoring blocked reactions), and 366 reactions are used under more growth conditions suboptimally than optimally (see Supplemental materials).
Figure 6.

Reaction usage in optimal and suboptimal flux distributions. For each of the tested 88 aerobic environmental conditions, reactions were classified as being used in optimal solutions or used only in suboptimal solutions. The black line in the figure shows for each reaction the fraction of environments (fenv) that can use this reaction suboptimally. The gray line in the figure shows for each reaction the fraction of environments that can use this reaction in optimal solutions. The 185 blocked reactions in the network are not shown in the figure.
Looking at the usage patterns of exchange fluxes in optimal and suboptimal aerobic solutions (these are not shown in Fig. 6) gives insights into how certain metabolites are used by the cell (Table 2). Some metabolites can only serve as carbon sources and will never be secreted either optimally or suboptimally; this is true for 52 extracellular metabolites. Thirteen extracellular metabolites can be secreted suboptimally and cannot be used as carbon sources (they can only serve as by-products). Another set of 30 metabolites can be used as a sole carbon source and can be secreted during suboptimal growth on any of the other carbon sources, and additionally six carbon sources can be secreted during suboptimal growth on only a limited number of other carbon sources. Finally, the metabolites urea, xanthine, uracil, and indole cannot serve as carbon sources and are secreted optimally under aerobic growth conditions on a select number of carbon sources.
Table 2.
Characterization of Extracellular Metabolites by Their Roles in Various Single Carbon Aerobic Environments
| CS | BP | Metabolite names |
|---|---|---|
| O | N | (52) 3-hydroxycinnamic acid; 3-(3-hydroxyphenyl)propionate: Acetoacetate; N-Acetyl-D-glucosamine; N-Acetyl-D-mannosamine; N-Acetylneuraminate; L-Arabinose; L-Aspartate; Butyrate (n-C4:0); Citrate; Deoxyadenosine; Deoxycytidine; Deoxyguanosine; Deoxyinosine; Deoxyuridine; D-Fructose; L-Fucose; D-Glucose 6-phosphate; D-Galactose; D-Galactarate; D-Galactonate; Galactitol; D-Galacturonate; D-Glucarate; D-Glucuronate; L-Glutamine; Glycerol 3-phosphate; Guanosine; Hexadecanoate (n-C16:0); Lactose L-Malate; Maltose; Maltohexaose; Maltopentaose; Maltotriose; Maltotetraose; D-Mannose; D-Mannose 6-phosphate; Melibiose; D-Mannitol; octadecanoate (n-C18:0); Phenylpropanoate; D-Ribose; L-Rhamnose; D-Sorbitol; D-Serine; Sucrose; L-tartrate; Trehalose; tetradecanoate (n-C14:0); D-Xylose |
| O | S | (36) 4-Aminobutanoate; Acetate; Acetaldehyde; Adenosine; 2-Oxoglutarate; D-Alanine; L-Alanine; L-Arginine; L-Asparagine; Cytidine; Dihydroxyacetone; Ethanol; Fumarate; L-Glutamate; Glycine; D-Glyceraldehyde; Glycerol; Glycolate; Inosine; D-Lactate; Ornithine; L-Proline; Putrescine; Pyruvate; L-Serine; Succinate; L-Threonine; L-Tryptophan; Uridine; Xanthosine; D-Glucose; 2-Dehydro-3-deoxy-D-gluconate; (S)-Propane-1,2-diol; D-Gluconate; L-Idonate; L-Lactate |
| N | S | (13) 1,5-Diaminopentane; Adenine; Formate; Guanine; L-Histidine; Hypoxanthine; L-Isoleucine; L-Leucine; L-Lysine; L-Phenylalanine; Thymidine; L-Tyrosine; L-Valine |
| N | O,S | (4) urea, xanthine, uracil, indole |
| N | N | (31) meso-2,6-Diaminoheptanedioate; Allantoin; AMP; Cob(l)alamin; Choline; L-Carnitine; Cytosine; Cyanate; L-Cysteine; Dimethyl sulfide; Dimethyl sulfoxide; Fe2+; L-Fucose 1-phosphate; gamma-butyrobetaine; Glycine betaine; K+; D-Methionine; L-Methionine; Sodium; Nicotinate; Nicotinamide adenine dinucleotide; NMN; Nitrite; Nitrate; (R)-Pantothenate; Spermidine; Taurine; Thiamin; Trimethylamine; Trimethylamine N-oxide; Thiosulfate |
| N | O | (7) CO2, H2O, h, ammonium, phosphate, O2, sulfate |
Columns correspond to carbon source (CS) and by-product (BP). O, both optimal and suboptimal; N, neither optimal nor suboptimal; S, only suboptimal.
Interestingly, a set of 147 metabolic reactions are used suboptimally but never optimally (Fig. 6), so the presence of the responsible enzymes under the tested conditions would drive the cell towards a less optimal state. These reactions include ABC transporters, when alternate transport mechanisms are available; the first step in the Entner Dourdoroff pathway; phospholipid recycling; nucleotide degradation; transporters associated with by-products that cannot be utilized as carbon sources; and oxidative phosphorylation reactions.
DISCUSSION
Alternate optimal growth solutions exist in genome-scale models, and these solutions need to be studied. Herein, application of an MILP algorithm to a genome-scale metabolic model of E. coli revealed that the number of alternate optima for genome-scale models is often large, making it computationally challenging to enumerate all of the optima for some conditions. Analysis of the calculated sets of optimal solutions showed that: (1) only a small subset of reactions in the network have variable fluxes across optima, (2) correlated reaction sets showed moderate agreement with the regulatory structure elucidated to date using classical methods and with expression data, and (3) condition-dependent reactions help provide clues about network regulatory needs. The additional calculation of suboptimal flux distributions identified reactions which are used under more environmental conditions suboptimally than optimally.
There are many more alternate optima than the number of fluxes that can vary across the set of alternate optimal solutions. Only a relatively small subset of reactions in the network (140 of 931 internal reactions) has variable fluxes across optimal solutions. For only a few environmental conditions (2 of 136) will the exchange fluxes vary across optima, indicating that the internal state of the cell is where the variability lies. In some cases it will be difficult to exactly determine which optima a cell might use based on expression data or protein levels, as some variable reactions are carried out by the same enzymes. For example, there are 45 nucleotide salvage reactions whose fluxes can vary under at least one of the tested environmental conditions; however, there are only 19 different enzymes associated with these reactions. For some variable reactions, such as those involved in central metabolism, expression data, proteomic data, and flux data could be used to help identify which reaction a cell is utilizing. Another level of complication in identifying the physiological solution is that an affine convex combination of the basic alternate optima is also an alternate optima; it will also be a valid flux distribution with the same objective value. For these reasons it may prove too difficult to determine exactly what solution a living cell utilizes.
The first 500 alternate optima for each condition were used to identify sets of correlated reactions. The majority of these correlated reaction sets (45 of 66) were consistent with established regulatory structure. However, comparisons made using established transcriptional regulatory structure are limited by the fact that a large portion of the transcriptional regulatory network has not been elucidated. A better agreement with regulon structure and correlated reaction sets in genome-scale models could emerge as the transcriptional regulatory network is further characterized. It was recently shown that the consistency between transcriptional regulatory network structure and expression data can vary depending on the structural features of network elements and functional classes of genes (Herrgard et al. 2003). Analysis of expression data showed that only a quarter of the correlated reaction sets (27.3%) showed significant correlation (P < 0.05) across different conditions. However, 49.5% of the genes belonging to the same transcription unit showed significant correlation across expression data sets. In contrast, comparisons between correlated reaction sets and expression data in yeast found that the correlated reaction sets were highly correlated with expression data (Schuster et al. 2002). The yeast network studied, however, contained only central metabolism, and the study looked at expression data for only a small number of genes under two different environmental conditions.
Unraveling the transcriptional regulatory network in E. coli is currently of great interest to many researchers. Comparing condition-specific optima provides useful insights into why the bacteria choose to express certain enzymes under certain conditions. Comparisons between glucose anaerobic and glucose aerobic reaction usage in optimal solutions generated testable hypotheses, some of which have already been proven experimentally. It will be important to investigate multiple sets of conditional optima, as the reasons behind enzyme regulation might not be apparent by studying only two environmental conditions.
By looking at optimal and suboptimal flux distributions, reactions which are used only in suboptimal solutions can be identified. Why would E. coli over the course of evolution retain these enzymes, since these reactions are never used optimally? These enzymes might be useful for reasons not captured based on the assumed condition of optimal growth. Network topology cannot capture the importance of enzymes that might catalyze a less efficient overall reaction; these enzymes could have higher turnover rates, better kinetics, allosteric regulation, or other such reasons making them beneficial. For example, to convert pyruvate into acetyl-CoA, many of the aerobic optimal solutions use pyruvate formate lyase (PFL) rather than pyruvate dehydrogenase (PDH), because formate, a product of PFL reaction, can be converted to hydrogen gas whose electrons are then carried down the electron transport chain. Using PFL the cell can make slightly more ATP than if it used PDH (with NADH transferring electrons to the electron transport chain). The network topology alone cannot predict possible loss of hydrogen gas making PFL more efficient. As another example, E. coli has two cytochrome oxidases, one capable of translocating two protons and the other capable of translocating 2.5 protons per electron pair donated to oxygen. The enzymes have different affinities for oxygen, making one better than the other at different oxygenation levels (Gennis and Stewart 1996).
For the analysis presented in this paper, further sampling of the alternate optima would not significantly affect the results. From the flux variability analysis results, the set of fluxes that are always used or never used across the mixed alternate optima would not change; however, deviations could occur in the fraction of mixed optima (fopt) that use a particular reaction for those reactions in Figure 2 with fopt between 0 and 1. Investigation of the correlated reaction sets, in conjunction with the flux variability analysis results, also indicated that these sets would not change if more sampling of the optima took place. It should be noted, however, that biases in the sampling could affect other types of analysis of the resulting flux distributions, such as the distribution of flux values through individual reactions (Wiback et al. 2004).
Taken together, these in silico results indicate that an optimal E. coli growth phenotype might be achievable by a large number of internal flux distributions; distinguishing the differences between these optima experimentally might prove difficult. In addition, studying the optimal and suboptimal utilization of reactions in the network could lead to understanding why enzymes are expressed under different conditions.
METHODS
Metabolic Network
A recently reconstructed E. coli metabolic network was used in this study (Reed et al. 2003). Type III extreme pathways are thermodynamically infeasible combinations (Beard et al. 2002; Price et al. 2002) of reactions with no net production or consumption of metabolites. To avoid having flux distributions utilizing type III pathways, 17 reactions in the network were removed by constraining the flux through these reactions to zero: CYTDt2, ABUTt2, ACCOAL, GALUi, GLUt4, INSt2, LCADi, ADK1, ADNt2, PROt4, SERt4, ALARi, THMDt2, THRt4, URAt2, URIt2, and VPAMT [reaction abbreviations match those previously reported (Reed et al. 2003) and can be found in the Supplemental materials].
Environmental Conditions Tested
All external metabolites were tested in silico for their ability to support aerobic and/or anaerobic growth in minimal medium. Exchange fluxes for ammonium, phosphate, sulfate, CO2, Fe2+, K+, Na+, water, and protons were allowed to be free. Uptake rates for oxygen were set to a maximum of 1000 and 0 mmole/g DW-h for aerobic and anaerobic simulations, respectively. Substrate uptake rates for the tested carbon sources were set to 10 mmole/g DW-h. Of the 143 external metabolites in the model, 88 supported aerobic growth as the sole carbon source, and only 48 of these also supported anaerobic growth with a biomass yield greater than 0.005 (Table 1). A total of 136 conditions (88 aerobic and 48 anaerobic) were then used to calculate sets of alternative optima.
MILP Algorithm
A recursive algorithm for calculating alternate optima using MILP has been published (Lee et al. 2000). These alternate optima utilize different sets of reactions. This algorithm was used to study a genome-scale metabolic network of E. coli. Some minor alterations to the algorithm were implemented and are described in more detail. The LP for this network is of the following form:
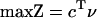 |
1a |
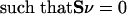 |
1b |
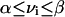 |
1c |
where S is the stoichiometric matrix, v is the steady-state flux vector, and α and β are the upper and lower limits for the individual flux values. In the prior algorithm (Lee et al. 2000), inequality constraints were transformed to standard form by introduction of slack variables. This initial transformation was not done in the present study, because the only inequality constraints were for the individual fluxes; instead the problem remained in the form stated above. The following additional constraints were adopted to ensure that different solutions are calculated (the ranges on the last set of constraints (2d) were modified from the original version of the algorithm):
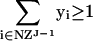 |
2a |
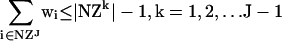 |
2b |
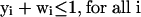 |
2c |
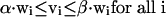 |
2d |
At each iteration, J, at least one of the non-zero fluxes from the previous solution (NZJ-1) must be set to zero, where the binary variable yi is 1 if that flux is selected to be removed from the basis at iteration J (equation 2a). The binary variable wi is subsequently forced to zero if yi is one (equation 2c), and the upper and lower bounds for that particular flux are then constrained to zero (equation 2d). Equation 2b ensures that alternate bases are not revisited by eliminating at least one non-zero variable found in previous iterations. The cplex solver in GAMS (GAMS Development) was used to enumerate the first 500 optima for each environmental condition.
Correlated Reactions Sets
A binary matrix (B) was formed from the set of mixed optimal solutions, where rows correspond to different reactions and columns are different optima. Non-zero fluxes have an entry of 1 in the binary matrix, and zero fluxes have an entry of zero. The correlated reaction sets could be determined by studying the resulting reaction participation matrix (BBT; Papin et al. 2002).
Transcriptional Regulatory Network Predictions
A previously developed transcriptional regulatory model (iMC1010v2; Covert et al. 2004) was used to simulate what genes are expressed under glucose minimal media conditions in the presence and absence of oxygen. The model uses Boolean rules to determine whether a gene is expressed or not expressed under given environmental conditions (Covert et al. 2001).
Analysis of Expression Data
AffyMetrix expression data sets (20) were collected from the ASAP database (Allen et al. 2003); replicates were averaged. The estimated transcript copy number was used in the calculations (for a description see https://asap.ahabs.wisc.edu/~glasner/Protocols/DataDefinitionDefinitions.txt). Gene-protein-reaction (GPR) associations from the iJR904 model (Reed et al. 2003) were used to translate reaction sets into gene sets. MATLAB (Math-works) was used to calculate all pairwise correlation coefficients between associated genes across different data sets. The individual pairwise correlation coefficients for a given reaction set were then averaged, yielding the reported average correlation coefficient. When isozymes were available for a given reaction, the average correlation coefficients were calculated separately using the different isozymes, and only the highest correlation coefficient was reported for the set. P-values were calculated using 100,000 randomly selected gene sets from the 904 genes included in the model. The transcription units were downloaded from EcoCyc (Karp et al. 2002); only the 321 transcription units containing at least two genes with measured expression data were used in the analysis. Average correlation coefficients and P-values for transcription units were calculated as described above.
Acknowledgments
This work was funded by research grants from NIH (GM57089) and NSF (BES-01-20363). The authors and UCSD disclose potential conflicting financial interests.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2546004.
References
- Allen, T.E., Herrgard, M.J., Liu, M., Qiu, Y., Glasner, J.D., Blattner, F.R., and Palsson, B.Ø. 2003. Genome-scale analysis of the uses of the Escherichia coli genome: Model-driven analysis of heterogeneous data sets. J. Bacteriol. 185: 6392-6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almaas, E., Kovacs, B., Vicsek, T., Oltvai, Z.N., and Barabasi, A.L. 2004. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427: 839-843. [DOI] [PubMed] [Google Scholar]
- Beard, D.A., Liang, S.D., and Qian, H. 2002. Energy balance for analysis of complex metabolic networks. Biophys. J. 83: 79-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard, A.P. and Maranas, C.D. 2001. Probing the performance limits of the Escherichia coli metabolic network subject to gene additions or deletions. Biotechnol. Bioeng. 74: 364-375. [DOI] [PubMed] [Google Scholar]
- Burgard, A.P., Vaidyaraman, S., and Maranas, C.D. 2001. Minimal reaction sets for Escherichia coli metabolism under different growth requirements and uptake environments. Biotechnol. Prog. 17: 791-797. [DOI] [PubMed] [Google Scholar]
- Burgard, A.P., Pharkya, P., and Maranas, C.D. 2003. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 84: 647-657. [DOI] [PubMed] [Google Scholar]
- Burgard, A.P., Nikolaev, E.V., Schilling, C.H., and Maranas, C.D. 2004. Flux coupling analysis of genome-scale metabolic network reconstructions. Genome Res. 14: 301-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covert, M.W., Schilling, C.H., and Palsson, B. 2001. Regulation of gene expression in flux balance models of metabolism. J. Theoret. Biol. 213: 73-88. [DOI] [PubMed] [Google Scholar]
- Covert, M.W., Knight, E.M., Reed, J.L., Herrgard, M.J., and Palsson, B.Ø. 2004. Integrating high-throughput and computational data elucidates bacterial networks. Nature 429: 92-96. [DOI] [PubMed] [Google Scholar]
- Edwards, J.S. and Palsson, B.Ø. 2000. The Escherichia coli MG1655 in silico metabolic genotype: Its definition, characteristics, and capabilities. Proc. Natl. Acad. Sci. 97: 5528-5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, S.S., Marciniak, J.Y., and Palsson, B.Ø. 2003. Description and interpretation of adaptive evolution of Escherichia coli K-12 MG1655 using a genome-scale in silico metabolic model. J. Bacteriol. 185: 6400-6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster, J., Famili, I., Palsson, B.Ø., and Nielsen, J. 2003. Large-scale evaluation of in silico gene knockouts in Saccharomyces cerevisiae. OMICS 7: 193-202. [DOI] [PubMed] [Google Scholar]
- Gennis, R.B. and Stewart, V. 1996. Respiration. In Escherichia coli and salmonella (ed. F.C. Neidhardt), pp. 217-261. ASM Press, Washington, DC.
- Gerdes, S.Y., Scholle, M.D., Campbell, J.W., Balazsi, G., Ravasz, E., Daugherty, M.D., Somera, A.L., Kyrpides, N.C., Anderson, I., Gelfand, M.S., et al. 2003. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J. Bacteriol. 185: 5673-5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrgard, M.J., Covert, M.W., and Palsson, B.Ø. 2003. Reconciling gene expression data with known genome-scale regulatory network structures. Genome Res. 13: 2423-2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra, R.U., Edwards, J.S., and Palsson, B.Ø. 2002. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420: 186-189. [DOI] [PubMed] [Google Scholar]
- Karp, P.D., Riley, M., Saier, M., Paulsen, I.T., Collado-Vides, J., Paley, S.M., Pellegrini-Toole, A., Bonavides, C., and Gama-Castro, S. 2002. The EcoCyc Database. Nucleic Acids Res. 30: 56-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, K.J., Prakash, P., and Edwards, J.S. 2003. Advances in flux balance analysis. Curr. Opin. Biotechnol. 14: 491-496. [DOI] [PubMed] [Google Scholar]
- Lee, S., Phalakornkule, C., Domach, M.M., and Grossmann, I.E. 2000. Recursive MILP model for finding all the alternate optima in LP models for metabolic networks. Comp. Chem. Eng. 24: 711-716. [Google Scholar]
- Mahadevan, R. and Schilling, C.H. 2003. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 5: 264-276. [DOI] [PubMed] [Google Scholar]
- Papin, J.A., Price, N.D., and Palsson, B.Ø. 2002. Extreme pathway lengths and reaction participation in genome-scale metabolic networks. Genome Res. 12: 1889-1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papin, J.A., Price, N.D., Wiback, S.J., Fell, D.A., and Palsson, B.Ø. 2003. Metabolic pathways in the post-genome era. Trends Biochem. Sci. 28: 250-258. [DOI] [PubMed] [Google Scholar]
- Phalakornkule, C., Lee, S., Zhu, T., Koepsel, R., Ataai, M.M., Grossmann, I.E., and Domach, M.M. 2001. A MILP-based flux alternative generation and NMR experimental design strategy for metabolic engineering. Metab. Eng. 3: 124-137. [DOI] [PubMed] [Google Scholar]
- Price, N.D., Famili, I., Beard, D.A., and Palsson, B.Ø. 2002. Extreme pathways and Kirchhoff's second law. Biophys. J. 83: 2879-2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, N.D., Papin, J.A., Schilling, C.H., and Palsson, B. 2003. Genome-scale microbial in silico models: The constraints-based approach. Trends Biotechnol. 21: 162-169. [DOI] [PubMed] [Google Scholar]
- Price, N.D., Schellenberger, J., and Palsson, B.Ø. 2004. Uniform sampling of steady state flux spaces: Means to design experiments and to interpret enzymopathies. J. Biol. Chem. (in press). [DOI] [PMC free article] [PubMed]
- Raamsdonk, L.M., Teusink, B., Broadhurst, D., Zhang, N., Hayes, A., Walsh, M.C., Berden, J.A., Brindle, K.M., Kell, D.B., Rowland, J.J., et al. 2001. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 19: 45-50. [DOI] [PubMed] [Google Scholar]
- Reed, J.L. and Palsson, B.Ø. 2003. Thirteen years of building constraint-based in silico models of Escherichia coli. J. Bacteriol. 185: 2692-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed, J.L., Vo, T.D., Schilling, C.H., and Palsson, B.Ø. 2003. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 4: R54.51-R54.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado, H., Gama-Castro, S., Martinez-Antonio, A., Diaz-Peredo, E., Sanchez-Solano, F., Peralta-Gil, M., Garcia-Alonso, D., Jimenez-Jacinto, V., Santos-Zavaleta, A., Bonavides-Martinez, C., et al. 2004. RegulonDB (version 4.0): Transcriptional regulation, operon organization and growth conditions in Escherichia coli K-12. Nucleic Acids Res. 32: D303-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, C.H., Covert, M.W., Famili, I., Church, G.M., Edwards, J.S., and Palsson, B.Ø. 2002. Genome-scale metabolic model of Helicobacter pylori 26695. J. Bacteriol. 184: 4582-4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster, S., Klamt, S., Weckwerth, W., Moldenhauer, F., and Pfeiffer, T. 2002. Use of network analysis of metabolic systems in bioengineering. Bioprocess Biosyst. Eng. 24: 363-372. [Google Scholar]
- Segre, D., Vitkup, D., and Church, G.M. 2002. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. 99: 15112-15117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelling, J., Klamt, S., Bettenbrock, K., Schuster, S., and Gilles, E.D. 2002. Metabolic network structure determines key aspects of functionality and regulation. Nature 420: 190-193. [DOI] [PubMed] [Google Scholar]
- Varma, A. and Palsson, B.Ø. 1994. Metabolic flux balancing: Basic concepts, scientific and practical use. Bio/Technology 12: 994-998. [Google Scholar]
- Wiback, S.J., Famili, I., Greenberg, H.J., and Palsson, B.Ø. 2004. Monte Carlo sampling can be used to determine the size and shape of the steady state flux space. J. Theor. Biol. 228: 437-447. [DOI] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- https://asap.ahabs.wisc.edu/~glasner/Protocols/DataDefinitionDefinitions.txt; Web site describes how the estimated transcript copy number is calculated from the gene expression data.