

Abstract
Dense coverage of the rice genome with polymorphic DNA markers is an invaluable tool for DNA marker-assisted breeding, positional cloning, and a wide range of evolutionary studies. We have aligned drafts of two rice subspecies, indica and japonica, and analyzed levels and patterns of genetic diversity. After filtering multiple copy and low quality sequence, 408,898 candidate DNA polymorphisms (SNPs/INDELs) were discerned between the two subspecies. These filters have the consequence that our data set includes only a subset of the available SNPs (in particular excluding large numbers of SNPs that may occur between repetitive DNA alleles) but increase the likelihood that this subset is useful: Direct sequencing suggests that 79.8% ± 7.5% of the in silico SNPs are real. The SNP sample in our database is not randomly distributed across the genome. In fact, 566 rice genomic regions had unusually high (328 contigs/48.6 Mb/13.6% of genome) or low (237 contigs/64.7 Mb/18.1% of genome) polymorphism rates. Many SNP-poor regions were substantially longer than most SNP-rich regions, covering up to 4 Mb, and possibly reflecting introgression between the respective gene pools that may have occurred hundreds of years ago. Although 46.2% ± 8.3% of the SNPs differentiate other pairs of japonica and indica genotypes, SNP rates in rice were not predictive of evolutionary rates for corresponding genes in another grass species, sorghum. The data set is freely available at http://www.plantgenome.uga.edu/snp.
The discovery of large numbers of single nucleotide polymorphisms (SNPs) in genome-scale sequencing initiatives opens new doors into the study of the genome-wide distribution of diversity and its significance. SNP markers open the possibility of “variation maps” at 100-fold higher resolution than current collections of DNA polymorphisms. This potential is being realized in humans (Sachidanandam et al. 2001) and Arabidopsis (Jander et al. 2002; Schmid et al. 2003; Torjek et al. 2003), in which SNP marker density is measured on a kilobase scale, whereas current genetic maps in major crops only begin to break the megabase barrier. Owing to this high density and advanced genotyping technologies, it is possible to apply these markers to genome-scale linkage disequilibrium and association studies as well as provide a tool for applications such as DNA marker-assisted breeding.
The recent sequencing of representatives from two diverse “subspecies” of rice provides an early glimpse into the genomic structure of variation in a monocot plant. After Arabidopsis (a dicot), the rice (Oryza sativa) genome is the most completely sequenced plant genome. Fortuitously, rice genomes have been sequenced from two rice subspecies (indica and japonica), which are thought to have diverged more than 1 million years ago (Bennetzen 2000). The International Rice Genome Sequencing Project (IRGSP) has used a BAC-based strategy to sequence rice ssp. japonica cv. Nipponbare (Sasaki and Burr 2000), and assembled sequences have been published for rice Chromosomes 1 (Sasaki et al. 2002), 4 (Feng et al. 2002), and 10 (Rice Chromosome 10 Sequencing Consortium 2003). Recently, The Institute for Genomic Research (TIGR; http://www.tigr.org) released a tentative assembly of all 12 chromosomes. Syngenta has sequenced the same japonica cultivar using a shotgun approach (Goff et al. 2002). The Beijing Genomics Institute (BGI) has sequenced an indica subspecies using a shotgun approach (ssp. indica cv. 93-11; Yu et al. 2002). The availability of extensive sequence for each of the two rice subspecies offers a unique and rich comparative genomics opportunity.
In this study, we have aligned indica contigs to the japonica genome assembly for the purpose of providing an SNP resource useful for rice breeding and genetics. This provides about a 100-fold increase relative to prior SNP studies (Nasu et al 2002), in the sampling of loci and SNP alleles available to study genome-wide patterns of genetic and physical distributions of SNP variation in rice. We investigate the predictive value of levels of SNP variation in the sequenced strains to other rice genotypes, and to other monocots (e.g., Sorghum). This data set provides a generalized framework useful to perform high-density marker studies involving indica/japonica crosses, and also has application to many evolutionary and/or functional studies.
RESULTS
Polymorphism Discovery and General Statistics
We used BLAST (Altschul et al. 1990) to align the BGI indica contigs with the TIGR japonica pseudomolecule assembly. Prior to alignment, all indica shotgun contigs were masked for repetitive elements using a comprehensive rice repetitive element database (see Methods). This step served to minimize the data set to low-copy DNA in which SNPs were more readily distinguished from paralogs, and reduced the uncompressed BLAST output from 180 GB to 4 GB, a more computationally manageable size. SNPs and single-base INDELs were then extracted from the BLAST alignments after a filtering schema that (1) excluded SNPs/INDELs with more than one polymorphism over a 40-bp window, (2) excluded polymorphisms with redundant hits to the pseudomolecule (i.e., unmasked repetitive/paralogous DNA), and (3) corrected for polymorphisms of low quality (excluded SNPs with a high quality score, Q < 20, in the indica contig). It should be noted that quality information was only available for the indica contigs, but the high depth of coverage from the sequenced japonica BACs should ensure high-quality sequence at the individual nucleotide level in the pseudomolecules. The results of this analysis are shown in Table 1.
Table 1.
Indica-Japonica Polymorphisms
| Chromosome | Lengtha | POLY | POLY/kbb | SNP | SNP/kbb | INDEL | INDEL/kbb | cMc | cM/100 kb |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 42,904,173 | 56,815 | 1.32 (1.97) | 53,652 | 1.25 (1.86) | 3163 | 0.07 (0.11) | 182 | 0.42 |
| 2 | 35,514,782 | 48,937 | 1.38 (2.02) | 46,202 | 1.30 (1.91) | 2735 | 0.08 (0.11) | 158 | 0.44 |
| 3 | 35,023,388 | 43,674 | 1.25 (1.78) | 41,172 | 1.18 (1.68) | 2502 | 0.07 (0.10) | 166 | 0.48 |
| 4 | 34,446,165 | 31,759 | 0.92 (1.58) | 29,659 | 0.86 (1.48) | 2100 | 0.06 (0.10) | 130 | 0.38 |
| 5 | 28,498,211 | 30,361 | 1.07 (1.73) | 28,508 | 1.00 (1.62) | 1853 | 0.07 (0.11) | 122 | 0.43 |
| 6 | 29,923,250 | 31,613 | 1.06 (1.66) | 29,792 | 1.00 (1.57) | 1821 | 0.06 (0.10) | 124 | 0.42 |
| 7 | 29,275,636 | 33,200 | 1.13 (1.88) | 30,326 | 1.04 (1.71) | 2874 | 0.10 (0.16) | 119 | 0.41 |
| 8 | 27,648,541 | 29,884 | 1.08 (1.81) | 28,178 | 1.02 (1.71) | 1706 | 0.06 (0.10) | 121 | 0.44 |
| 9 | 21,127,180 | 25,312 | 1.20 (1.93) | 24,011 | 1.14 (1.84) | 1301 | 0.06 (0.10) | 94 | 0.44 |
| 10 | 22,432,522 | 25,383 | 1.13 (1.94) | 23,962 | 1.07 (1.83) | 1421 | 0.06 (0.11) | 84 | 0.37 |
| 11 | 24,871,670 | 26,747 | 1.08 (1.76) | 25,069 | 1.01 (1.65) | 1678 | 0.07 (0.11) | 118 | 0.47 |
| 12 | 26,881,443 | 25,213 | 0.94 (1.64) | 23,810 | 0.89 (1.55) | 1403 | 0.05 (0.09) | 110 | 0.41 |
| Total | 358,546,961 | 408,898 | 1.13 (1.81) | 384,431 | 1.06 (1.70) | 24,557 | 0.07 (0.11) | 1527 | 3.82 |
TIGR pseudomolecule assembly v1.0.
First number is polymorphisms (POLY) per total pseudomolecule length. The number in parentheses is the number of polymorphisms per low-copy pseudomolecule length.
Recombination distances determined using RGP genetic markers.
After applying the filters described, the total number of polymorphisms remaining was 408,898 (384,341 SNPs and 24,557 single-base INDELs). On average, this resulted in a polymorphism rate of 1.70 SNPs/kb and 0.11 INDEL/kb of unmasked (nonrepetitive) pseudomolecule length. There was not a dramatic difference in SNP frequency at the level of whole chromosomes (Table 1). However, there does appear to be a somewhat higher SNP frequency in Chromosomes 1 and 2 and a lower SNP frequency in Chromosomes 4 and 12. Although others have suggested that changes in the recombination frequency per unit of physical distance affect nucleotide diversity (Begun and Aquadro 1992; Stephan and Langley 1998), such a trend was not apparent in our data. Most of the polymorphisms were transitions (61.8%), with transversions making up 32.8%, and INDELs accounting for 6.0%. Because of the stringent filtering methods used and exclusion of repetitive DNA, this is surely an underestimate of the actual polymorphism frequency at corresponding nucleotides among the two rice subspecies, which we consider to be best estimated at 4.31 SNPs/kb based on direct sequencing of orthologous loci (Nasu et al. 2002).
To test empirically the quality of the sample of SNPs between the two rice subspecies that are included in our data set, we sequenced random loci containing SNPs from each chromosome. PCR primers were designed from japonica sequence and used to amplify japonica (cv. Nipponbare) and indica (cv. 93-11) DNA. Of the 109 high-quality reads that aligned between the two subspecies, 87 contained SNPs that matched the database, and 22 matched the flanking sequence but did not contain a polymorphism. Based on this sample, we can assert with 95% confidence (based on binomial statistics) that the fraction of true SNPs in this data set falls within the range of 79.8% ± 7.5%.
SNP Variation Across the Rice Genome
To explore the genomic distribution of the patterns of DNA polymorphism between indica and japonica subspecies, SNP (excluding INDELs) frequency based on our sample was plotted at 100-kb intervals along each pseudomolecule (Fig. 1). SNP frequency (Fig. 1, solid blue line) was defined as the number of SNPs divided by the amount of unmasked (i.e., low-copy) DNA within the 100-kb interval. The amount of repeat-masked DNA was also shown (Fig. 1, dotted black line). Each interval was compared with the average SNP frequency for its chromosome, to identify the subset that showed nonrandom SNP frequencies (at p < 0.001 after Bonferroni correction). These intervals were then assembled together into contigs if they were immediately adjacent to, or separated by, intervals that showed statistically significant local positive spatial autocorrelation (Anselin's Local I, p < 0.01), a measurement of the tendency of like values to cluster in space.
Figure 1.

SNP variation across the rice genome. The x-axis shows the pseudomolecule position in which each tick-mark is a megabase. The y-axis shows the number of SNPs (solid blue line) per 100 kb of total DNA after subtraction of repetitive DNA. The right y-axis shows the percent of a 100-kb DNA stretch that is masked for repetitive DNA (dotted black line). Regions >300 kb that showed lower than expected SNP frequencies are noted with green blocks, and regions of higher than expected SNP frequencies are noted with red blocks.
Nonrandom patterns of SNP distribution are strongly supported by the highly significant levels of global spatial autocorrelation (Moran's I, p < 0.001) detected on all chromosomes (Table 2). These data support the findings of Nasu et al. (2002) that polymorphism in the rice genome (from the indica-japonica perspective) appears to be nonrandomly distributed. However, the present results go beyond the data of Nasu et al. in sampling a far higher number of loci (albeit in only two genotypes), thus offering the opportunity to investigate nonrandom patterns on a much finer scale. Among a total of 3585 intervals tested, 328 contigs (48.6 Mb; 486 intervals) of higher than expected SNP frequency and 237 contigs (64.7 Mb; 647 intervals) of low SNP frequency were found (Supplemental Table V). Contigs that were 300 kb or larger are indicated in Figure 1. Together, these contigs make up 31.6% (Table 2) of the total genome and range from 12.5% (Chromosome 10) to 43.5% (Chromosomes 5 and 12).
Table 2.
Nonrandom Distribution of Indica-Japonica Polymorphic Regions
| High polymorphism regions
|
Low polymorphism regions
|
All polymorphism regions
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr | Lengtha | Intervalsb | Contigs | Mb | Intervalsb | Contigs | Mb | Intervalsb | Contigs | Mb | % Chr | Moran's /c |
| 1 | 42.9 | 23 | 20 | 2.3 | 59 | 26 | 5.9 | 82 | 46 | 8.2 | 19.1% | 0.372*** |
| 2 | 35.5 | 23 | 22 | 2.3 | 48 | 19 | 4.8 | 71 | 41 | 7.1 | 20.0% | 0.370*** |
| 3 | 35.0 | 37 | 25 | 3.7 | 61 | 23 | 6.1 | 98 | 48 | 9.8 | 28.0% | 0.499*** |
| 4 | 34.4 | 70 | 38 | 7 | 79 | 27 | 7.9 | 149 | 65 | 14.9 | 43.3% | 0.474*** |
| 5 | 28.5 | 45 | 33 | 4.5 | 79 | 16 | 7.9 | 124 | 49 | 12.4 | 43.5% | 0.489*** |
| 6 | 29.9 | 68 | 33 | 6.8 | 58 | 18 | 5.8 | 126 | 51 | 12.6 | 42.1% | 0.554*** |
| 7 | 29.3 | 50 | 38 | 5 | 62 | 16 | 6.2 | 112 | 54 | 11.2 | 38.3% | 0.433*** |
| 8 | 27.6 | 41 | 30 | 4.1 | 45 | 14 | 4.5 | 86 | 44 | 8.6 | 31.1% | 0.421*** |
| 9 | 21.1 | 19 | 16 | 1.9 | 32 | 16 | 3.2 | 51 | 32 | 5.1 | 24.1% | 0.307*** |
| 10 | 22.4 | 12 | 11 | 1.2 | 16 | 8 | 1.6 | 28 | 19 | 2.8 | 12.5% | 0.220*** |
| 11 | 24.9 | 39 | 28 | 3.9 | 50 | 30 | 5 | 89 | 58 | 8.9 | 35.8% | 0.294*** |
| 12 | 26.9 | 59 | 34 | 5.9 | 58 | 24 | 5.8 | 117 | 58 | 11.7 | 43.5% | 0.284*** |
| All | 358.5 | 486 | 328 | 48.6 | 647 | 237 | 64.7 | 1133 | 565 | 113.3 | 31.6% | |
Megabase.
100 kb.
Measurement of global spatial autocorrelation; ***indicates significance (P < 0.001).
Regions of unusually high SNP density are present on all chromosomes. On average, these 328 regions include slightly less than average repetitive DNA (34.3% vs. genome-wide coverage of 37.2%). Although most (298 contigs) of the regions are 100-200 kb in length, 30 of the regions are >300 kb long, with the longest being 1.3 Mb and 1.0 Mb on Chromosomes 6 and 4, respectively.
Regions of low SNP frequency are considerably longer and cover more of the genome than the regions of high SNP density (Table 2). In the Discussion, we elaborate on the possibility that this asymmetry (Fig. 2) may reflect introgression of chromosomal segments between the two subspecies. Most of the SNP-poor regions (171 contigs) are 200 kb or shorter, but 66 are 300 kb or longer, with 11 regions being >1 Mb. The longest is found on Chromosome 5 (4 Mb) followed by Chromosome 8 (2.3 Mb) and Chromosome 7 (2.1 Mb). These regions contain the genome-wide average of repetitive DNA, thus the lack of polymorphisms cannot be due to masking effects.
Figure 2.

Asymmetry in contigs length for regions of high and low SNP frequency. The x-axis shows the contig length in 100 kb bins for regions of high (diamond) and low (square) SNP frequency. The y-axis shows the cumulative percent of total contigs that fall into a size range.
Genomic regions with low recombination rates have been shown to correlate with low DNA sequence polymorphism in other organisms (Begun and Aquadro 1992; Stephan and Langley 1998). To investigate whether this correlation holds true or not in rice, we plotted the SNP polymorphism rate against the recombination rate (Supplemental Fig. 3) for genetic markers mapped between the japonica variety Nipponbare and the indica variety Kasalath (Harushima et al. 1998). Plateaus in the recombination rates correlate with centromere positions (data not shown), but these plateaus do not appear to correlate with altered SNP frequency. An exception to this is Chromosome 5, but this reduced SNP frequency can be explained by the high frequency of repetitive DNA (Fig. 1). Therefore, there does not appear to be a gross reduction in low-copy SNP frequency per unit of low-copy DNA in regions of reduced recombination rates in the rice genome.
Comparison of Rice Versus Sorghum SNPs
The importance of rice as a model for a wide range of cereal grains and grasses makes it of interest to determine the value of the present data in predicting levels of variation on a per-locus basis in other taxa. This is of interest not only from the practical standpoint of efficient DNA marker identification, but also from the perspective of identifying genes that are rapidly evolving in different Poaceae lineages and thus may account for a disproportionately large share of divergence among the grains and grasses. To investigate this, we created a “unigene” set of sorghum loci from public EST databases, determined the number of polymorphisms between two sorghum species (Sorghum propinquum and Sorghum bicolor), and compared the number of indica-japonica polymorphisms at homologous rice loci. On a superficial level, the intersubspecific rice and interspecific sorghum comparisons are analogous. The two rice subspecies are largely interfertile, although some reproductive barriers exist, as is true of the two sorghum species. Briefly, 17,714 contigs were constructed from 106,100 sorghum EST traces. Of these, 2236 contigs were found to be polymorphic between the two sorghum genotypes. These were then aligned to 56,056 rice CDS (coding DNA minus 5′-UTR/3′-UTR/introns) sequences to find orthologous rice loci. Only sorghum contigs that hit the rice genome exactly once were considered to increase the chance of identifying true orthologs. This resulted in 575 single-hit rice-sorghum homologs, and the number of rice and sorghum SNPs was determined for each locus. A total of 7131 rice SNPs were found in 2,503,374 bp, and 2217 sorghum SNPs were found in 787,419 bp. Therefore the average SNP frequency at these loci was 2.8 SNPs/kb for both rice and sorghum.
To investigate whether the levels of polymorphism found in a rice gene were related to those found in the corresponding sorghum gene, we determined the correlation between SNP frequencies in the 575 genes for rice and sorghum. The correlation coefficient (-0.074) was not significant (p = 0.078). As a first effort toward the identification of genes that are highly polymorphic in both crops, we found 73 genes that demonstrated a polymorphism rate ≥3.0 SNPs/kb in both rice and sorghum. These highly polymorphic genes are listed in Supplemental Table III. We note that this is a biased sample in that it represents only the subset of sorghum genes that were (1) identified in EST libraries and (2) matched only one locus in rice. Other genes that have eluded EST analysis and/or have more complex family structure may show different patterns.
DISCUSSION
Utility of the SNP Data Set
We have identified a sample of 408,898 SNP/INDEL polymorphisms between two subspecies of rice, using a stringent filtering approach that favors high SNP quality (i.e., avoiding false positives) over exhaustive SNP sampling (i.e., avoiding false negatives), to provide a resource of immediate value for crop improvement. By sequencing a random sampling of the SNP data set, we estimate that a minimum of 295,633 (79.8%-7.5%) of the SNPs are real, leaving 0.82 SNPs/kb based on the japonica pseudomolecule size (358,546,961 bp). This is an intentional underestimate of the exact rate of polymorphism that excludes many true SNPs between corresponding nucleotides across the genome, in particular excluding virtually all SNPs in allelic copies of repetitive DNA elements. However, this large sample of variation in low-copy DNA is useful not only for DNA marker-assisted improvement, but also to provide new information about how variation is distributed across the genome.
There are several possible explanations for the ∼20% rate of false positives remaining in the SNP data set. (1) The indica genomic DNA amplified during verification was from the 93-11 cultivar, but from a different source than that sequenced by BGI. (2) Although every effort was made to eliminate low-quality base pairs, sequencing errors surely still exist in both genomic and validation sequences. (3) Occasional misassembled contigs surely also exist in both data sets that lead to improper PCR primer design and amplification of a different region that does not contain the predicted SNP. In addition, SNPs could not be detected in physical gaps between indica contigs and pseudomolecules, which leave portions of the genome without SNP coverage at present. Although the quality of the data set presented here is surprisingly high given the draft nature of the genomes, this quality should improve with later genome releases. In its current state, the data set provides SNP variation at a large sampling of polymorphic low-copy loci collectively offering high-resolution DNA marker coverage for most of the rice genome.
This SNP/INDEL data set (available at http://www.plantgenome.uga.edu/snp) is a valuable resource for genetic experiments involving indica and japonica parents. Some possible applications include enhancing current genetic maps (Nasu et al. 2002), global linkage disequilibrium mapping (Ching et al. 2002; Flint-Garcia et al. 2003), association studies (Botstein and Risch 2003), and marker-assisted selection (Koebner and Summers 2003). A researcher could select from our database a small number of SNPs in a candidate region (Thornsberry et al. 2001; Whitt et al. 2002) or perform a genome-wide LD probe (Hansen et al. 2001). Initial estimates suggest that LD blocks may be as large as 100 kb in rice (Garris et al. 2003). The data set presented here contains more than enough SNPs for a low-resolution scan of the genome, followed by further high-resolution scans in selected LD blocks.
One important consideration in the application of our results is the fact that this SNP data set is based on (albeit large numbers of) differences between only one japonica and one indica genotype. Only a subset of the SNPs identified herein will be applicable to other combinations of indica-japonica cultivars. An initial estimate of the extent to which these data extrapolate to other cultivar combinations can be made using the SNP study by Nasu et al. (2002). In this work, three japonica cultivars, two indica cultivars, and one wild rice genotype were scanned for SNPs at 417 loci by direct sequencing. This group successfully geno-typed 213 SNPs in the six rice genotypes. We examined a subset of these (86 SNPs) for overlap with our data set, in which all three japonica genotypes shared the same nucleotide and both indica genotypes shared a different nucleotide. Of these, 47.7% (41/86) showed exact overlap with our indica-japonica data set. This is a conservative estimate of overlap because our SNP set was constructed with a high degree of stringency and the genome sequence we aligned may not be represented in the Nasu data set. Similarly, we have sequenced samples from two indica and two japonica varieties (data not shown), and found 43.9% (25/57) of the SNPs in our database. Based on an overall success rate of 66/143, we can assert with 95% confidence (based on binomial statistics) that the fraction of SNPs found in any indica-japonica cross that overlap our data set is 46.2% ± 8.3%. Therefore, assuming a 72.3% low-end accuracy of our data set and a low-end 37.9% extrapolation to other indica-japonica crosses, we roughly estimate that a minimum of 26.6% (102,234) of the SNPs that we have identified would be informative in other indica-japonica crosses. This lower limit represents an informative indica-japonica SNP frequency of 0.26 SNP/kb for the complete 400-Mb rice genome. In practical terms, if a plant breeder needed two markers flanking an important gene or QTL in a novel indica-japonica cross, eight candidate SNPs from the database would need to be evaluated on the parents prior to genotyping in a large number of progeny.
Genome-Scale Observations
Although the exclusion of repetitive DNA and other stringent filters applied to our data have the consequence that we have only identified a sampling of the true SNPs that exist between the two strains studied, this is still a far larger sampling that had previously been described in detail and thus brings new resolution to the study of how polymorphism is distributed across the genome. Spatial autocorrelation analysis suggests that the SNP rate is not randomly distributed across the rice genome, consistent with findings in other genomes (Waterston et al. 2002). Our rigorous filters to maximize the number of reproducible polymorphisms may reduce our ability to detect features such as localized “hot-spots” of variation (because we exclude SNPs that are <20 nt apart). Furthermore, alternate classes of polymorphism (e.g., simple sequence repeats and >1-bp INDELs) in other genomic areas are not included in the global polymorphic frequency maps.
The exclusion of most repetitive DNA would tend to reduce our SNP rate relative to other studies (Feng et al. 2002; Nasu et al. 2002; Zhao et al. 2004), because genes and low-copy DNA generally evolve more slowly than repetitive DNA. We do not exclude the idea that repetitive DNA could generate new alleles that in principle might be used as SNPs. However, the widespread tendency of repetitive DNA in plants to exist in large families of recent origin (e.g., Bennetzen 2000) makes it a less-promising reagent than in organisms in which many repetitive DNA families are of ancient origin and individual family members are highly divergent. Although we emphasize once again that our overall SNP rate is an underestimate of the true rate for these taxa, such a large sampling of high-quality SNPs nonetheless provides a more detailed resource than previously was available for the examination of indica-japonica diversity.
Some of the SNP-rich and SNP-poor regions may reflect local variation in underlying mutation rates caused by differences in DNA metabolism or chromosome physiology (Waterston et al. 2002), but other regions show distinctive patterns that suggest other explanations. About 13.9% of SNP-poor contigs, accounting for 9.2% of the genome, are 500 kb or more in length, versus only 2.1% of SNP-rich contigs accounting for 1.5% of the genome (Fig. 2). The relatively greater length of SNP-poor regions than SNP-rich regions is consistent with an a priori expectation that there may have been historical introgression of chromosomal segments between the subspecies. Indica and japonica rice subspecies are thought to have diverged more than 1 million years ago (Bennetzen 2000), and both remain highly bred and widely cultivated. “Wide crosses” between the ssp. japonica and indica gene pools are occasionally made in breeding programs, and it is no surprise that introgression would be found. In fact, japonica strains were specifically known to have been present in the ancestry of 93-11 (the indica strain that was sequenced: Yu et al. 2002). Introgression is a very plausible explanation of why centimorgan-sized stretches of chromosome could be SNP-poor, and would also explain why many SNP-poor regions were longer than most SNP-rich regions.
If introgression does, indeed, account for the greater length of SNP-poor than SNP-rich regions, a first approximation of its antiquity might be obtained based on the length of SNP-poor chromosome segments. Spatial autocorrelation (described above) was used to circumscribe chromosomal segments that contained lower SNP diversity than could be explained by a random distribution of the SNP sample contained in our database. A total of 33 intervals showed low SNP diversity over stretches of >500 kb, thus representing a population of intervals that are significantly longer than the average length of the SNP-rich regions (Fig. 2). A total of 22 such regions contain two or more genetic markers, permitting estimation of their recombinational length. The distances between the terminal markers in these regions average 852 kb and 3.07 cM, but this is naturally an underestimate of the length of the intervals (because markers do not lie at the breakpoints). Based on the average physical length of these 22 intervals (1.13 Mb), and assuming that recombination is evenly distributed across the intervals, one would estimate their true recombinational length to average ∼4.07 cM. By using formulas derived by Hansen (1959), one can estimate that it would require ∼25 consecutive generations of backcrossing to a recurrent geno-type to reduce introgressed chromosome segments from a donor to this average length. Because rice is bred largely by self-pollinating methods with outcrosses typically only at intervals of six to eight generations, the putatively introgressed segments may be hundreds of years old. We note that this probably occurred not as a single event but instead as multiple introgression events of different antiquities, contributing to the varying lengths of introgressed chromosome segments.
These SNP-poor regions raise many interesting questions for future research. Determination of whether they truly represent introgression or not will require resequencing of a large sampling of SNPs in populations of genotypes that adequately sample the japonica and indica gene pools, respectively. This will also reveal in which direction the introgression occurred for each segment. Investigation of the patterns of introgression in other genotypes may shed light on whether the persistence of such chromosomal segments is random, or has some selective basis. If the latter, comparison of introgressed segments in diverse cultivars may help to pin down the specific genes in the segment(s) on which selection is acting.
Regions of low polymorphism found herein do not appear to correlate closely with regions of low recombination. Several studies have shown that regions of the genome that experience low crossing-over rates during meiosis have lower levels of nucleotide diversity that the rest of the genome. This has been demonstrated in species such as fruit fly (Begun and Aquadro 1992), human (Yu et al. 2001), and tomato (Stephan and Langley 1998). Whereas most SNP-rich regions do occur on the longer rice chromosomes, our data do not clearly show this trend across the genome. The RGP japonica-indica (1527 cM) genetic map (Harushima et al. 1998) shows an overall recombination rate of 3.82 cM/Mb assuming a rice genome size of 400 Mb. Deviations from this are seen on the pseudomolecule at the 100-kb scale with severe recombination restrictions surrounding the centromeres. The regions of reduced polymorphism that we identified do not correlate with these centromeric regions.
Regions of high polymorphism are also a fascinating topic for future research. One can envision many hypotheses that may explain subsets of these regions, including the presence of genes that account for partial reproductive isolation between the subspecies, linkage drag associated with strong selection for particular alleles that are ancestral to one of the subspecies, variations in the amount of low-copy noncoding DNA that is free to evolve rapidly, or other hypotheses. We hope that the SNP resources described herein will foster these and other new avenues of inquiry.
METHODS
Plant Materials and Sequence Data Sets
We downloaded 12 rice pseudomolecules (ssp. japonica cv. Nipponbare) from TIGR (ver1.0; www.tigr.org/tdb/e2k1/osa1) and 127,551 rice shotgun contigs (ssp. indica cv 93-11) and corresponding quality files from BGI (http://btn.genomics.org.cn:8080/rice). Nipponbare and 93-11 genomic DNA was kindly provided by Susan McCouch at Cornell University.
Polymorphism Discovery
Computations were performed on a 32-node Linux Cluster running Red Hat 9.0. The 127,551 indica contigs were masked with a rice repetitive element database using cross_match (-minmatch 10 -minscore 20; http://www.phrap.org). The sequences in the repeat data set were initially recovered from 400 Mb of publicly available Nipponbare genomic sequences (downloaded from http://rgp.dna.affrc.go.jp on Aug. 23, 2002) using RECON (Bao and Eddy 2002), a program for the de novo identification of repeat families. Thereafter, the sequences were individually cu-rated based on the putative identity of each repeat (N. Jiang, Z. Bao, S. Eddy, and S. Wessler, in prep.). Each of the masked indica contigs were aligned against the 12 japonica pseudomolecules using NCBI-BLAST (ver2.2.5; E < 1 × 10-10). A Perl script was written that pulled out high-scoring segment pairs (HSP) that have an identity between 95% and 100% and a length exceeding 100 bp. SNPs and single-base INDELs were extracted from these alignments with the criteria that there was 100% identical sequence of 20 bp on either side of the polymorphism resulting in 729,819 SNP/INDEL polymorphisms. These were deposited into a MySQL (http://www.mysql.com) database and indexed by their pseudomolecule position. Polymorphisms that had an “N” in either genotype were removed (1979 filtered). Polymorphisms from multiple contigs that perfectly matched the same pseudomolecule position were removed (88,879 filtered). SNPs with low indica contig quality scores (Q < 20) were removed (46,030 filtered). This left 592,931 SNP/INDEL polymorphisms. Then 20 bases up and downstream of the polymorphism (41 bp total) were BLAST aligned (E < 1 × 10-6) to the pseudomolecules, and the number of hits to the genome were determined. Of the 41 bp sequences, 408,898 hit the rice genome exactly once. This was the data set used in subsequent analyses. The entire 592,931 polymorphism set is available online at http://www.plantgenome.uga.edu/snp.
SNP Verification by Direct Sequencing
For each SNP in the database, 500 bp was extracted from either side of the SNP. These 1001-bp fragments were mixed by chromosome, and 20 SNP-flanking PCR primers (Supplemental Table IV) were designed per chromosome from pseudomolecule sequence using Fast PCR (http://www.biocenter.helsinki.fi/bi/bare-1_html/download.htm; ver2.7.20). Primer design parameters were 18-22 bp in length, 50%-60% GC, and 58°-62C° Tm. Nipponbare or 93-11 genomic DNA (50 ng) was PCR-amplified (1× cloned Pfu buffer [Stratagene], 0.2 mM dNTP, 1 μM primer mix, 2.5 units of Taq [Promega], and 0.2 mU of Pfu [Stratagene]) for 35 cycles under the appropriate annealing temperatures using an MJ Research PTC-100 thermocycler. PCR products were cleaned as follows: 5 μL of the PCR products were mixed with 2 μL of exonuclease (1 U/μL) and shrimp alkaline phosphatase (1 U/μL), heated for 15 min at 37°C, and heat-inactivated for 15 min at 80°C. Both enzymes were from USB. Cycle sequencing reactions were performed on 2 μL of cleaned PCR product using the BigDye Terminator Cycle Sequencing Kit Version 3.1 (Applied Biosystems) and a PTC-100 thermocycler. Finished cycle sequencing reactions were filtered through Sephadex filter plates (Krakowski et al. 1995) directly into Perkin-Elmer MicroAmp Optical 96-well reaction plates. Sequencing was performed using an ABI 3700. ABI sequencer trace data were manually evaluated for the presence of an SNP using PHRED/CROSS_MATCH/PHRAP/CONSED software (Ewing et al. 1998; Gordon et al. 1998). Only clones with a Ph/Pr value >16 over 100 continuous base pairs and sequences >50 bp in length were used in sequence analyses. Sequences were deposited in GenBank (accession nos. CL299954-CL300320).
Identification of Regions of High and Low Variability
All 100-kb intervals were analyzed for significant deviations from the average SNP frequency using binomial statistics on a per chromosome basis. The significance threshold was p < 0.001 after Bonferroni correction, which was calculated individually for each chromosome. Individual 100-kb outliers were then assembled into contigs if they were immediately adjacent or were considered to be single occurrences if they were found to occur in regions of significant local spatial autocorrelation (p < 0.01). Local spatial autocorrelation was determined using Anselin's Local I with significance determined by a conditional randomization test (Anselin 1995). In this randomization approach, the value for SNP diversity at a given bin is held constant while the remaining values are randomly distributed across all other bins. The observed Local I value for that bin may then be compared with the distribution of the permuted values to determine the significance level. To be scored as significantly autocorrelated at the p < 0.01 level, the observed value of the bin had to be greater than the maximum of the permuted values for 99 permutations.
Global Spatial Autocorrelation The spatial clustering of SNP diversity for each chromosome was quantified using a modified form of the Moran's I (Moran 1948, 1950) value for global spatial autocorrelation. The Moran's I for global autocorrelation of SNP richness for a chromosome was calculated as
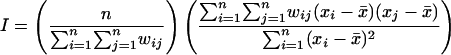 |
where n is the total number of intervals in the chromosome, xi is the SNP richness of interval i, xj is the SNP richness of interval j, x̄ is the mean SNP richness for the chromosome, and wij is the weighted distance between intervals i and j. This weighted distance was determined by a simple binary weight matrix in which the intervals within 200 kb of i were assigned a weight of 1 and all other intervals were assigned a weight of 0. Thus, only the four nearest neighbors of any interval were considered in the global Moran's I calculation.
Possible values for Moran's I range between 1 and -1. The expected value for a data set that shows no spatial autocorrelation is a negative value very near 0. A Moran's I approaching 1 represents a situation in which similar values are clustered together, and a value near -1 represents a situation in which similar values repel one another.
The significance of I was tested using a randomization approach in which the values for SNP diversity were randomized 999 times for each chromosome, and a Moran's I value was determined for each randomization. The observed value of Moran's I was compared with this set of randomized values. An observed value that was greater than any of the I values resulting from these 999 permutations was declared significant at the p < 0.001 level.
Determination of Recombination Rate for Rice
We downloaded 3267 genetic marker sequences and their cM positions from the Rice Genome Program (RGP) Web site (http://rgp.dna.affrc.go.jp/publicdata/geneticmap2000/). These markers were aligned to the 12 japonica pseudomolecules using BLAST (E < 1 × 10-20). All BLAST hits that matched the chromosome on which it was genetically mapped were plotted along the corresponding pseudomolecule.
Comparative Analysis of Sorghum SNPs
We downloaded 106,100 sorghum EST trace files from two S. propinquum (RHIZ2, FM1) and nine S. bicolor EST libraries (DG1, IP1, EM1, PIC1, PI1, WS1, LG1, OV1, OV2; http://cagt03.agtec.uga.edu/traceSequence/). Bases were called with phred (-trim) and vector sequence masked with cross_match.manyreads (-minmatch 12 -penalty -2 -minscore 20). Contigs were built with phrap.manyreads (-minscore 75). This resulted in 17,714 contigs and 7482 singlets. Each contig (“unigene”) was examined for polymorphisms using Polybayes (Marth et al. 1999) software (-priorPoly .001). SNP/INDEL information was parsed with a custom Perl script and dumped into an MySQL database. Contigs containing polymorphisms between S. propinquum and S. bicolor were identified using SQL statements resulting in 2236 polymorphic sorghum contigs. We downloaded 56,056 rice CDS sequences from TIGR (ftp.tigr.org/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_1.0). Each of the 2236 polymorphic sorghum loci was aligned to a CDS data set with NCBI-BLAST (E < 1e-20). The coordinates of sorghum-rice corresponding CDS loci were determined. The indica-japonica SNP database was searched with these coordinates to determine the relative number of polymorphisms between rice and sorghum at corresponding loci. In all, 575 sorghum contigs were found that hit the rice genome only once. The SNP frequency between rice and sorghum were determined for these 575 loci. Highly polymorphic loci (73 total) were defined as having an SNP polymorphism rate ≥3.0 SNP/kb in both rice and sorghum.
Acknowledgments
This work was supported by the Rockefeller Foundation initiative on “Resilient Crops for Water-Limited Environments,” USAID Comparative Cereal Genomics Initiative, and the US National Science Foundation Plant Genome Research Program. Special thanks go to John Bowers, Sue Wessler, and Jon Robertson for comments and suggestions. We also thank the rice sequencing community for depositing much data in public databases for further study by a wide range of scientists.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to GenBank under accession nos. CL299954-CL300320. The following individuals kindly provided reagents, samples, or unpublished information as indicated in the paper: S. McCouch.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2479404.
References
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. 1990. Basic local alignment search tool. J. Mol. Biol. 215: 403-410. [DOI] [PubMed] [Google Scholar]
- Anselin, L. 1995. Local Indicators of Spatial Association—LISA. Geograph. Anal. 27: 93-115. [Google Scholar]
- Bao, Z. and Eddy, S.R. 2002. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12: 1269-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D.J. and Aquadro, C.F. 1992. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 356: 519-520. [DOI] [PubMed] [Google Scholar]
- Bennetzen, J.L. 2000. Comparative sequence analysis of plant nuclear genomes: Microcolinearity and its many exceptions. Plant Cell 12: 1021-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein, D. and Risch, N. 2003. Discovering genotypes underlying human phenotypes: Past successes for Mendelian disease, future approaches for complex disease. Nat. Genet. 33 Suppl: 228-237. [DOI] [PubMed] [Google Scholar]
- Ching, A., Caldwell, K.S., Jung, M., Dolan, M., Smith, O.S., Tingey, S., Morgante, M., and Rafalski, A.J. 2002. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing, B., Hillier, L., Wendl, M.C., and Green, P. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8: 175-185. [DOI] [PubMed] [Google Scholar]
- Feng, Q., Zhang, Y., Hao, P., Wang, S., Fu, G., Huang, Y., Li, Y., Zhu, J., Liu, Y., Hu, X., et al. 2002. Sequence and analysis of rice chromosome 4. Nature 420: 316-320. [DOI] [PubMed] [Google Scholar]
- Flint-Garcia, S.A., Thornsberry, J.M., and Buckler, E.S.t. 2003. Structure of linkage disequilibrium in plants. Annu. Rev. Plant Biol. 54: 357-374. [DOI] [PubMed] [Google Scholar]
- Garris, A.J., McCouch, S.R., and Kresovich, S. 2003. Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165: 759-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff, S.A., Ricke, D., Lan, T.H., Presting, G., Wang, R., Dunn, M., Glazebrook, J., Sessions, A., Oeller, P., Varma, H., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92-100. [DOI] [PubMed] [Google Scholar]
- Gordon, D., Abajian, C., and Green, P. 1998. Consed: A graphical tool for sequence finishing. Genome Res. 8: 195-202. [DOI] [PubMed] [Google Scholar]
- Hansen, M., Kraft, T., Ganestam, S., Sall, T., and Nilsson, N.O. 2001. Linkage disequilibrium mapping of the bolting gene in sea beet using AFLP markers. Genet. Res. 77: 61-66. [DOI] [PubMed] [Google Scholar]
- Hansen, W.D. 1959. Early generation analysis of lengths of heterozygous chromosome segments around a locus held heterozygous with backcrossing or selfing. Genetics 44: 833-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harushima, Y., Yano, M., Shomura, A., Sato, M., Shimano, T., Kuboki, Y., Yamamoto, T., Lin, S.Y., Antonio, B.A., Parco, A., et al. 1998. A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148: 479-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander, G., Norris, S.R., Rounsley, S.D., Bush, D.F., Levin, I.M., and Last, R.L. 2002. Arabidopsis map-based cloning in the post-genome era. Plant Physiol. 129: 440-450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koebner, R.M. and Summers, R.W. 2003. 21st century wheat breeding: Plot selection or plate detection? Trends Biotechnol. 21: 59-63. [DOI] [PubMed] [Google Scholar]
- Krakowski, K., Bunville, J., Seto, J., Baskin, D., and Seto, D. 1995. Rapid purification of fluorescent dye-labeled products in a 96-well format for high-throughput automated DNA sequencing. Nucleic Acids Res. 23: 4930-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth, G.T., Korf, I., Yandell, M.D., Yeh, R.T., Gu, Z., Zakeri, H., Stitziel, N.O., Hillier, L., Kwok, P.Y., and Gish, W.R. 1999. A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 23: 452-456. [DOI] [PubMed] [Google Scholar]
- Moran, P.A.P. 1948. The interpretation of statistical maps. J. Royal Stat. Soc. 10: 243-251. [Google Scholar]
- ____. 1950. Notes on continuous stochastic phenomena. Biometrika 37. [PubMed]
- Nasu, S., Suzuki, J., Ohta, R., Hasegawa, K., Yui, R., Kitazawa, N., Monna, L., and Minobe, Y. 2002. Search for and analysis of single nucleotide polymorphisms (SNPs) in rice (Oryza sativa, Oryza rufipogon) and establishment of SNP markers. DNA Res. 9: 163-171. [DOI] [PubMed] [Google Scholar]
- Rice Chromosome 10 Sequencing Consortium. 2003. In-depth view of structure, activity, and evolution of rice chromosome 10. Science 300: 1566-1569. [DOI] [PubMed] [Google Scholar]
- Sachidanandam, R., Weissman, D., Schmidt, S.C., Kakol, J.M., Stein, L.D., Marth, G., Sherry, S., Mullikin, J.C., Mortimore, B.J., Willey, D.L., et al. 2001. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409: 928-933. [DOI] [PubMed] [Google Scholar]
- Sasaki, T. and Burr, B. 2000. International Rice Genome Sequencing Project: The effort to completely sequence the rice genome. Curr. Opin. Plant Biol. 3: 138-141. [DOI] [PubMed] [Google Scholar]
- Sasaki, T., Matsumoto, T., Yamamoto, K., Sakata, K., Baba, T., Katayose, Y., Wu, J., Niimura, Y., Cheng, Z., Nagamura, Y., et al. 2002. The genome sequence and structure of rice chromosome 1. Nature 420: 312-316. [DOI] [PubMed] [Google Scholar]
- Schmid, K.J., Sorensen, T.R., Stracke, R., Torjek, O., Altmann, T., Mitchell-Olds, T., and Weisshaar, B. 2003. Large-scale identification and analysis of genome-wide single-nucleotide polymorphisms for mapping in Arabidopsis thaliana. Genome Res. 13: 1250-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan, W. and Langley, C.H. 1998. DNA polymorphism in lycopersicon and crossing-over per physical length. Genetics 150: 1585-1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornsberry, J.M., Goodman, M.M., Doebley, J., Kresovich, S., Nielsen, D., and Buckler, E.S.t. 2001. Dwarf8 polymorphisms associate with variation in flowering time. Nat. Genet. 28: 286-289. [DOI] [PubMed] [Google Scholar]
- Torjek, O., Berger, D., Meyer, R.C., Mussig, C., Schmid, K.J., Rosleff Sorensen, T., Weisshaar, B., Mitchell-Olds, T., and Altmann, T. 2003. Establishment of a high-efficiency SNP-based framework marker set for Arabidopsis. Plant J. 36: 122-140. [DOI] [PubMed] [Google Scholar]
- Waterston, R.H., Lindblad-Toh, K., Birney, E., Rogers, J., Abril, J.F., Agarwal, P., Agarwala, R., Ainscough, R., Alexandersson, M., An, P., et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520-562. [DOI] [PubMed] [Google Scholar]
- Whitt, S.R., Wilson, L.M., Tenaillon, M.I., Gaut, B.S., and Buckler, E.S.t. 2002. Genetic diversity and selection in the maize starch pathway. Proc. Natl. Acad. Sci. 99: 12959-12962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, A., Zhao, C., Fan, Y., Jang, W., Mungall, A.J., Deloukas, P., Olsen, A., Doggett, N.A., Ghebranious, N., Broman, K.W., et al. 2001. Comparison of human genetic and sequence-based physical maps. Nature 409: 951-953. [DOI] [PubMed] [Google Scholar]
- Yu, J., Hu, S., Wang, J., Wong, G.K., Li, S., Liu, B., Deng, Y., Dai, L., Zhou, Y., Zhang, X., et al. 2002. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79-92. [DOI] [PubMed] [Google Scholar]
- Zhao, W., Wang, J., He, X., Huang, X., Jiao, Y., Dai, M., Wei, S., Fu, J., Chen, Y., Ren, X., et al. 2004. BGI-RIS: An integrated information resource and comparative analysis workbench for rice genomics. Nucleic Acids Res. 32 Database issue: D377-D382. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp.tigr.org/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_1.0; TIGR.
- http://btn.genomics.org.cn:8080/rice/; Rice GD.
- http://cagt03.agtec.uga.edu/; Comparative Grass Genomics Center.
- http://rgp.dna.affrc.go.jp/; Rice Genome Research Program.
- http://www.biocenter.helsinki.fi/bi/bare-1_html/download.htm; FAST-PCR.
- http://www.mysql.com; MYSQL.
- http://www.phrap.org; Phred/Phrap/Consed System.
- http://www.plantgenome.uga.edu/snp; Plant Genome Mapping Lab.
- http://www.tigr.org/tdb/e2k1/osa1; The TIGR Rice Genome Project.