

Abstract
Estrogen receptor α (ERα) mediates the biological actions of estrogens and also contributes to the development and progression of breast cancer. To gain a more comprehensive understanding of ERα-mediated transcription, we used chromatin immunoprecipitation and promoter focused microarrays (ChIP-chip) to identify ERα binding sites in T-47D human breast cancer cells. Transcription factor binding site analysis revealed that the estrogen response element (ERE) was significantly over-represented and was found in 50% of the 243 ERα-bound regions identified. Interestingly, multiple ERα-bound regions were detected in the upstream regulatory sequences of the CYP2B gene cluster. Because ERα has been reported to regulate the expression of other cytochrome P450 enzymes and CYP2B6 is highly expressed in ERα-positive breast tumors, we focused on characterizing the ERα-dependent regulation of CYP2B6. Reporter gene assays revealed that ERα and ERβ increased CYP2B6-regulated gene expression through a functional ERE located at −1669 to −1657 in the upstream regulatory region of CYP2B6. E2 increased ERα and nuclear receptor coactivator 3 (NCoA3) recruitment to the 5′-flanking region of CYP2B6, and increased CYP2B6 mRNA levels in T-47D but not in MCF-7 human breast cancer cells. RNAi-mediated knockdown of ERα in the T-47D cells resulted in a significant decrease in CYP2B6 mRNA levels. Taken together, our study provides evidence for cell-type specific transcriptional regulation of the CYP2B6 gene by ERs.
Keywords: Chromatin immunoprecipitation and microarray chip (ChIP-chip), Estrogen receptor, Estrogen, Transcription factor binding analysis, Gene regulation, Cytochrome P450 2B6
1. Introduction
Estrogens act through their nuclear receptors, estrogen receptor α (ERα; NR2A1) and ERβ (NR3A2) to regulate diverse transcriptional responses that include both stimulation and repression of gene expression [1]. ERs share the evolutionarily conserved functional domains typical of other nuclear receptor family members [2]. These include the amino-terminal activation function-1 (AF1) domain, the centrally located DNA-binding domain (DBD) and the carboxy-terminal ligand-binding domain (LBD), which also contains the ligand-dependent activation function-2 (AF2) region that is important for interaction with coactivators [3]. Ligand binding induces conformational changes in the receptors resulting in homodimerization and subsequent DNA binding. ER homodimers bind to perfect and imperfect palindromic DNA sequences termed estrogen responsive elements (EREs) located in the regulatory region of their target genes. Once bound to DNA, ERs act as nucleation sites for the recruitment of chromatin remodeling complexes and co-regulators (i.e. nuclear receptor coactivator 3 (NCoA3), p300/Creb binding protein), and the assembly of basal transcriptional machinery [4]. ERs also regulate transcription via half-site EREs and by a tethering mechanism that involves protein:protein interactions with other DNA-bound transcription factors, including activating protein 1 and stimulating protein 1 [4].
Estrogens are critical for the growth, development and differentiation of the mammary gland. ER expression in breast cancer is associated with improved responsiveness to endocrine targeted therapies, such as treatment with the anti-estrogen tamoxifen to reduce ER activity or treatment with aromatase inhibitors to deplete estrogen levels [5]. In addition to increasing the expression of genes that regulate cell proliferation and cancer development, ERα has also been reported to regulate the expression of many drug metabolizing enzymes, including cytochrome P450 1A1 (CYP1A1), CYP1B1, and CYP2A6 [6–8], many of which are involved in the metabolism of estrogens. Two recent studies have reported higher CYP2B6 expression in ERα-positive compared to ERα-negative breast tumors [9,10], implicating CYP2B6 as an ERα target gene. ER-dependent regulation of the aforementioned drug metabolizing enzymes may contribute to alterations in endogenous steroid levels as well as increased metabolism or bioactivation of therapeutic agents and xenobiotics.
Hepatic CYP2B genes are the most inducible CYP isoforms by phenobarbital (PB)-type inducers. CYP2B6 is expressed in the human liver, where it constitutes approximately 6% of total microsomal P450s [11], but it is also expressed in extra-hepatic tissues including the intestine, kidney, lung, skin, brain and mammary gland [12–14]. CYP2B6 metabolizes a wide variety of pharmaceutical agents including cyclophosphamide, buproprion and tamoxifen; environmental contaminants such as aflatoxin B and dibenzanthracene; nicotine and methylenedioxymethamphetamine (MDMA “ecstasy”) [15,16]. CYP2B6 expression is regulated through the constitutive androstane receptor (CAR; NR1I3), a member of the nuclear receptor superfamily of transcription factors [17]. Upon nuclear translocation, CAR associates with its dimerization partner retinoid X receptor (RXR; NR2B1). The CAR-RXR complex binds the phenobarbital-responsive enhancer module (PBREM) and recruits co-regulator proteins to modulate target gene expression, such as CYP2B6 [18]. Although CAR has long been recognized as a key regulator of CYP2B, emerging evidence suggests that other nuclear receptors such as pregnane X receptor (PXR; NR1I2) [19] and glucocorticoid receptor (GR; NR3C1) [20], and liver enriched transcription factors are also involved in the regulation of this gene [21]. Interestingly, many of the potent inducers of CYP2B6 expression are ligands for PXR but not CAR [22]. Putative glucocorticoid response elements (GREs) present in the upstream regulatory regions of mouse Cyp2b10 and rat CYP2B1/2 are also responsive to the synthetic glucocorticoid, dexamethasone (DEX) [23,24]. DEX treatment has been reported to be required for maximum induction of Cyp2b10 expression by activators of CAR [23]. It is unclear if GR is involved in the regulation of CYP2B6, since the CYP2B6 promoter region does not contain a GRE. However, activation of GR has been reported to increase the expression of PXR and CAR in human hepatocytes [25]. Estrogens have also been implicated in the regulation of Cyp2b10 with one study reporting that high doses (μM) of 17β-estradiol (E2) activate mouse but not human CAR [26]. However, direct regulation of CYP2B6 by ERs has not been determined.
Recent studies using chromatin immunoprecipitation (ChIP) combined with microarrays (ChIP-chip) have identified several ERα-bound regions across the genome [27,28]. A number of breast cancer cell lines have been used in in vitro studies of estrogen responses, but most of the data, including genome-wide analyses of ERα binding sites, have come from experiments using MCF-7 cells [27–30]. MCF-7 cells express higher ERα levels than T-47D human breast cancer cells, while both cell lines expressing low, but similar, levels of ERβ[31,32]. Although MCF-7 and T-47D cells exhibit similar global gene expression profiles in response to E2 treatment [33], significant differences in the expression of many genes have been reported [10]. Determining ERα binding profiles in other ERα-positive breast cancer cell lines, such as T-47D, will ensure that any cell line-specific differences in ERα action are not overlooked. In the present study, we performed ChIP-chip to identify ERα-bound genomic regions in T-47D human breast cancer cells. One of the ERα-bound regions identified was located in the 5′-regulatory region of CYP2B6. Reporter gene assays, mRNA expression and protein expression analysis provide evidence for ER-dependent regulation of CYP2B6.
2. Material and methods
2.1. Chemicals
Dimethyl sulfoxide (DMSO), 17β-estradiol (E2) and ICI 182,780 were purchased from Sigma (St. Louis, MO). Primers for quantitative real-time polymerase chain reaction (qPCR) were purchased from Integrated DNA Technology (Coralville, Iowa, USA). Cell culture media, fetal bovine serum (FBS) and trypsin were purchased from Wisent (St. Bruno, Canada). All other chemicals and biochemicals were of the highest quality available from commercial vendors.
2.2. Plasmids
To generate pGL3p-2B7P, the ChIP-chip isolated region_6 (chr19: 46,118,736 to 46,119,920) was amplified from T-47D genomic DNA and cloned into the KpnI and BglII sites of pGL3 promoter vector using primers: 5′-CAAAGGTACCCCACTGGTGCTTCACCCTGG-3′ and 5′-CAAAAGATCTTCCTTTATCAGTCCTTCTGTGAC-3′. The restriction enzyme sites used in the cloning are underlined. Region_6 was approximately 68-kb upstream from CYP2B6, but was also approximately 10-kb upstream from CYP2B7P, so we named the plasmid pGL3p-2B7P. The pGL3p-2B7P plasmid contained region_6 upstream of the SV40 promoter, allowing us to study the E2-dependent regulation of the isolated ERα-bound region. To generate pGL3-2B6, we amplified approximately −1.8 kb of the 5′-flanking region of CYP2B6 promoter (chr19: 46,187,266 to 46,189,032) from T-47D genomic DNA using primers: 5′-CAAAGGTACCGTGTGTAAAGCACTTCACGCCT-3′ and 5′-CAAAAGATCTCTGCACCCTGCTGCAGCCT-3′. The restriction enzyme sites used in the cloning are underlined. This sequence included region_42 (chr19: 46,187,186 to 46,187,511) identified from our ChIP-chip experiments. The −1.8 kb 5′-flanking region was cloned into the KpnI and BglII sites of pGL3 basic vector to create pGL3-2B6, which included 60-bp downstream of the TATA-box promoter of CYP2B6 generating a plasmid that is similar to −1.6 k/PBREM plasmid described by others [34]. PGL3-2B6_EREmut was created by site-directed mutagenesis using the following primers: 5′-GCTCCTCCTGTTTCAAAGTAAC-3′ and 5′-GTTACTTTGAAACAGGAGGAGC-3′ in region_42. For the plasmid pGL3-2B6_ΔERE, a KpnI site was introduced downstream of the ERE using the following primers: 5′-CAGGTCCTGGTACCAGCAAAGG-3′ and 5′-CCTTTGCTGGTACCAGGACCTG-3′. KpnI digestion removed a 156 bp DNA fragment containing both the PBREM and putative ERE. The linear plasmid was then ligated creating pGL3-2B6_ΔERE. pSG5 ERα and pSG5 ERβ were generous gifts from Prof. Jan-Åke Gustafsson (University of Houston, TX, USA). For the plasmid pSG5 ERα DBD, E203A and G204A mutations were introduced to the DNA-binding domain of ERα using the following primers: 5′-GTCTGGTCCTGTGCGGCCTGCAAGGCCTTC-3′ and 5′-GAAGGCCTTGCAGGCCGCACAGGACCAGAC-3′. All mutations were verified by DNA sequencing.
2.3. Cell culture
T-47D human breast carcinoma cells were cultured in Dulbecco’s modified Eagle’s medium and F12 medium in a 1:1 mixture ratio (DMEM:F12), supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (PEST). HuH-7 human hepatoma cells were cultured in high glucose DMEM supplemented with 10% FBS and 1% PEST. MCF-7 human breast carcinoma cells were cultured in DMEM, supplemented with 10% FBS and 1% PEST. In experiments investigating the effect of E2, cells were plated in phenol red free media, supplemented with 5% dextran-coated charcoal (DCC) treated FBS for 72 h before transient transfection. All cells were maintained at 37 °C in a 5% CO2 environment and subcultured at 80% confluency.
2.4. Chromatin immunoprecipitation and ChIP-chip
ChIP assays were performed as described by Matthews et al. [7]. Chromatin was sonicated to an average size of 500 bp. T-47D and MCF-7 cells were grown in phenol red free media supplemented with 5% DCC-FBS and 1% PEST for 3 days and treated with either 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780 for 1 h or 4 h. The isolated chromatin was incubated with 0.8 μg of either normal rabbit IgG, anti-ERα (HC-20; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-NCoA3 (M-397; Santa Cruz). Promoter occupancy was quantified using quantitative real-time PCR (qPCR). Primer sequences are provided in Supplementary File 1.
For the ChIP-chip experiments, T-47D cells were plated in 10-cm dishes in DMEM:F12 supplemented with 10% FBS and 1% PEST. After 48 h, ChIP assays were performed as previously described [7]. Immunoprecipitated DNA from 10 cm/antibody was linearly amplified using Primer A: GTTTCCCAGTCACGGTC(N)9 and Primer B: GTTTCCCAGTCACGGTC according to the manufacturer’s instructions (Affymetrix, Santa Clara, CA). Linearly amplified DNA (7.5 μg) was fragmented by limited DNAseI digestion and hybridized to Affymetrix human promoter tiling arrays 1.0R. Hybridization and washing steps were performed according to the manufacturer’s protocol. Data were normalized and analyzed using CisGenome [35]. Enriched peaks were determined by comparing triplicate anti-ERα immunoprecipitated samples to IgG using a moving average approach and the default settings in CisGenome using hg18 [36]. Regions were merged if the gap between them was <300 bp and the number of probes failing to reach the cut-off was <5. Regions were discarded if they were <120 bp or did not contain at least 5 continuous probes above the cut-off.
2.5. cis-Regulatory element search
To calculate the enrichment of individual cis-regulatory elements, we performed a position weight matrix (PWM) search across the ChIP-enriched sequences using an in-house cis-regulatory element search application. Our application was written in Ruby (v 1.8.6) and used the RinRuby package for connectivity to the R statistical interpreter (v 2.8.1). We first searched all of the 243 enriched sequences at 1% FDR using all of the JASPAR (http://jaspar.cgb.ki.se/) PWMs for human, mouse, rat or undeclared species (i.e., denoted as “−”). Our algorithm defined putative binding sites as those within a ChIP-chip site that exceeded our matrix similarity score cut-off of 0.80. For comparison purposes, the application also counted the average number of times a putative cis-regulatory element occurred within 100 randomly generated sequences based on the same nucleotide frequencies as the ChIP-chip site. Site-based and whole dataset enrichment probability scores were calculated using a Bayesian probabilistic method. For the site-based enrichment probability, we used the cumulative rather than regular probability for ease of biological interpretation. For the whole dataset enrichment, we calculated the total counts from all of the ChIP-chip sites and the total random counts from all of the sites, and performed Poisson-based probability calculations.
2.6. Transient transfections and reporter gene assays
HuH-7 cells were plated in 12-well dishes in DMEM medium containing either 10% FBS or phenol red free DMEM containing 5% DCC-FBS. Twenty-four hours after plating, the cells were transfected with pSG5-ERα, pSG5-ERβ, and luciferase reporter vectors using Lipofectamine 2000 (Invitrogen Corp., Burlington, Canada). The cells were dosed 24 h post-transfection with either 0.1% DMSO (solvent), 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780. The following day, cells were lysed and luciferase activity was determined according to the manufacturer’s instructions (Promega, Madison, WI). The firefly luciferase activity was normalized to that of the renilla luciferase (Promega) and the normalized data were presented relative to empty vector control.
2.7. RNA isolation, cDNA synthesis, and quantitative real-time PCR
T-47D and MCF-7 cells were dosed for 6 h with either 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780. RNA was isolated using RNeasy Mini Kit as described by the manufacturer (Qiagen, Mississauga, Canada). Half a microgram of the isolated RNA was reversed transcribed using random hexamer primers and Super-ScriptII Reverse Transcriptase (Invitrogen). The cDNA was amplified with the appropriate primers (Primer sequences are provided in Supplementary File 1), and quantified using SYBR green (Bio-Rad Laboratories, Mississauga, Canada).
2.8. Western blot analysis
Western blots were performed as described previously [37], using anti-ERα (HC-20; Santa Cruz), anti-β-actin antibody (Sigma) or anti-CYP2B6 antibody (AB1283; Chemicon International, Temecula, CA). Enhanced chemiluminescence (ECL) reagents from GE Healthcare (Mississauga, Canada) and film from Denville Scientific (Saint-Laurent, Canada) were used to detect the specified proteins.
2.9. Small interfering iRNA
ERα knockdown experiments were performed as described previously [37], using ERα (J-003401-11-0020 and J-003401-14-0020), non-targeting #2 (D-001810-02-20; targeting luciferase) ON-TARGETplus siRNAs and DharmaFECT1 transfection reagent were purchased from Dharmacon (Lafayette, CO). Briefly, T-47D cells were seeded 300,000 per well in six-well plates containing 2 ml of medium. After 24 h, 100 nM of each siRNA against ERα or luciferase was transfected into T-47D cells using 4 μl of DharmaFECT1. Changes in mRNA and protein levels were assessed 48 h post-transfection.
2.10. Statistical analysis
All results are expressed as means±standard error of the means (S.E.M.). Statistical analysis was calculated using GraphPad Prism 5 statistical software (San Diego, CA). One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison tests and the Student’s two tailed t-tests were used when appropriate. Statistical significance was assessed at p<0.05.
3. Results
3.1. Estrogen receptor binding sites in T-47D cells
To identify the ERα binding sites in T-47D human breast cancer cells, we performed ChIP-chip assays on cells plated in medium containing 10% FBS and cultured for 48 h to reach 85% confluency. ERα was active under these conditions due to residual levels of estrogens in the serum [38]. ELISA assays showed that the E2 concentration in our medium was approximately 0.1 nM (data not shown). Therefore due to the level of residual estrogens in the serum-containing medium, exogenous E2 was not added to the medium prior to performing the ChIP-chip assays. Chromatin was isolated using either an anti-ERα antibody or normal rabbit IgG and linearly amplified DNA was hybridized to Affymetrix Human tiling 1.0R microarrays. We performed three biological replicates and data were normalized and analyzed as described in the Materials and methods. We identified 243 promoter regions bound by ERα using a false detection rate (FDR) of 1%. The identified regions were assigned a relative rank by CisGenome where the region number correlates with statistical significance such that region 1 represents the highest ranked region within the data set [35]. The identified regions corresponded to 217 putative ERα target genes, since in some cases a gene contained multiple ERα-bound regions in its 5′-flanking region. A BED file containing genomic coordinates based on hg18 for the identified regions and the corresponding genes is provided in Supplementary File 2.
We then compared our ERα-bound regions in T-47D cells to those identified in a genome-wide analysis of ERα-binding sites in MCF-7 cells that were cultured in estrogen-reduced serum for 72 h before treating cells with 100 nM E2 [27]. Only 88 regions (36%) overlapped with at least 50% sequence identity between the two data sets (Fig. 1). This relatively low level of overlap could be due to differences in data analysis software used or culture conditions between the two studies. However, the low level of overlap in identified regions might also reflect cell-specific differences in ERα-action between the two breast cancer cell lines.
Fig. 1.
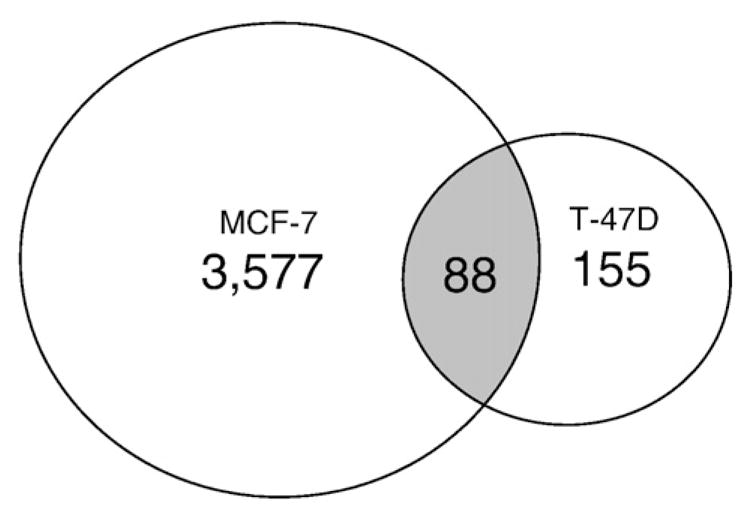
ERα-bound regions in T-47D cells. Comparison of the ERα-bound regions identified in the current study (T-47D) with those reported in a genome-wide analysis in MCF-7 cells [27]. As shown in the Venn diagram 88 of the 243 regions identified in our study overlapped with at least 50% sequence identity to those reported previously [27].
To determine which cis-regulatory elements were significantly over-represented in the isolated ChIP-chip regions, we performed a position weight matrix (PWM) search across the ChIP-enriched sequences. Our analysis identified 36 transcription factor motifs that were significant at p<0.01 (Table 1 summarizes the top ranking transcription factor motifs; see Supplementary File 3 for the complete list). The ERE, or ESR1 binding motif as defined in the JASPAR database (http://jaspar.cgb.ki.se/), was significantly over-represented in the ERα-bound regions. Overall, approximately 50% of the 243 regions contained an ERE.
Table 1.
Transcription factor motifs that were over-represented in the ERα-bound regions identified in T-47D. Table shows the top ranking transcription factor motifs. For the complete list, see Supplementary File 3.
| Binding motifa | Sum of real count | Sum of random count | Over representation | Poisson p-values |
|---|---|---|---|---|
| ESR1 | 162 | 25.23 | 6.42 | 0 |
| Myf | 427 | 192.85 | 2.21 | 0 |
| NHLH1 | 305 | 143.07 | 2.13 | 0 |
| IRF2 | 110 | 51.69 | 2.13 | 5.50E−13 |
| Hand1-Tcfe2a | 2355 | 1228.77 | 1.92 | 0 |
| Foxd3 | 814 | 433.79 | 1.88 | 0 |
| Roaz | 376 | 203.67 | 1.85 | 0 |
| NR2F1 | 138 | 76.91 | 1.79 | 1.28E−10 |
| HNF4A | 378 | 212.63 | 1.78 | 0 |
| FOXI1 | 1830 | 1035.8 | 1.77 | 0 |
Refer to JASPAR (http://jaspar.cgb.ki.se/) for detail descriptions of the transcription factor motifs.
3.2. Confirmation of ChIP-chip results
To verify the results obtained from the ChIP-chip analysis and determine the E2-dependent recruitment of ERα to the regions identified from the analysis, we performed conventional ChIP on T-47D cells grown for 3 days in a steroid-reduced medium before treatment with E2 for 1 h. We selected 13 ERα-bound regions to further investigate. The binding regions were chosen to cover a range of enrichment values but also included known ERα-regulated genes. Gene names and region numbers shown in bold font indicate that these sequences contained at least one ERE. E2-dependent recruitment of ERα was confirmed in all 13 regions examined, although the fold enrichments varied among them (Fig. 2A). Some of the identified regions included known estrogen target genes such as gene regulated in breast cancer 1 (GREB1), RAS-like estrogen-regulated growth inhibitor (RERG) and estrogen receptor α (ESR1) (Fig. 2B). Surprisingly, ERα binding to trefoil factor 1 (TFF1), a gene routinely used to study ERα-mediated transcription [39], was not detected under our assay conditions. Several of the identified ERα-bound regions have not been previously reported in ChIP-chip studies using MCF-7 cells [27–29], including region_6 and region_42 that map to the 5′-regulatory regions of CYP2B7P and CYP2B6, respectively (Fig. 2B). Since CYP2B6 is involved in the metabolism of several clinically important drugs and it has been reported to correlate with ERα expression [10], we studied the ER-dependent regulation of CYP2B6.
Fig. 2.

Confirmation of ERα-bound regions identified in the ChIP-chip study. (A) Conventional ChIP was performed to confirm 13 ERα-bound regions in T-47D plated in DCC-FBS containing medium and treated with E2 for 1 h. These 13 regions were chosen to represent a range of enriched values from the array. ERα recruitment level significantly (p<0.05 Student’s t-test) different from DMSO is indicated by an asterisk for each region. Regions shown in bold font indicate the presence of at least one estrogen response element in the sequence. (B) Selected ERα-bound regions relative to their closest genes are shown using USCS Genome Browser (http://genome.ucsc.edu/). The black blocks represent ERα-bound regions identified from our ChIP-chip study, whereas the arrows indicate the transcriptional direction of the closest annotated genes. (C) The diagram shows the genomic coordinates and the sequences of the putative EREs are boxed in CYP2B7P (6) and CYP2B6 (42) and the phenobarbital-responsive enhancer module (PBREM). Nuclear receptor 1 (NR1) and NR2 represent two direct repeat 4 (DR4) nuclear receptor motifs, which are boxed, while NF1 denotes a putative nuclear factor 1 binding site as previously described [18]. The perfect palindromic ERE from vitellogenin A2 is also shown for comparison. A colon indicates homology among the sequences, where an underline indicates nucleotide differences among the sequences at that position.
3.3. ER expression positively correlates with CYP2B6 levels
To investigate the ER-dependent regulation of CYP2B6, we first cloned a ~1.8 kb fragment upstream from the CYP2B6 start site, which included region_42. Transcription factor binding site analysis identified a putative ERE located approximately −1669 to −1657 bp upstream of the CYP2B6 start site, which is also located 40 bp downstream of the CAR and PXR responsive PBREM. The putative ERE is an imperfect palindromic sequence (GGTCAnnnTAACT) compared to the vitellogenin A2 ERE (GGTCAnnnTGACC) [40]. Region_6 contained three putative EREs with the same sequences as the ERE in region_42 (Fig. 2C). Comparative genomic analysis revealed that the ERE present in the regulatory region of CYP2B6 was not conserved in the 5′-regulatory region of the mouse homolog Cyp2b10 nor in the rat homolog Cyp2B1 (data not shown). This suggests that the direct regulation of CYP2B6 by ERs might exhibit species-specificity and be observed in humans but not in mice or rat models.
To investigate the transcriptional activation of CYP2B6 by ERs, luciferase reporter assays were performed (pGL3-2B6) with increasing amounts of ERα or ERβ in ER-negative HuH-7 human hepatoma cells. The reporter activity of pGL3-2B6 (Fig. 3A and C) or pGL3p-2B7P (Fig. 3B and D) increased with increasing amounts of ERα or ERβ. To determine the E2-dependent regulation of CYP2B6, HuH-7 cells were plated in a steroid-deprived medium and pGL3-2B6 and pGL3p-2B7P were transiently transfected with fixed amounts of ERα or ERβ. In HuH-7 cells transfected with either ERα or ERβ, treatment with 10 nM E2 caused a significant increase in luciferase activity for both pGL3-2B6 (Fig. 3E and G) and pGL3p-2B7P (Fig 3F and H), which was inhibited by co-treatment with the ER antagonist, ICI 182,780.
Fig. 3.

ER-dependent transcriptional activation of pGL3-2B6 and pGL3p-2B7P in HuH-7 cells. HuH-7 cells were transfected with (A) 200 ng of pGL3-2B6 or (B) pGL3p-2B7P and increasing amounts of the plasmid pSG5 ERα for 24 h prior to luminescence detection. pGL3-2B6 activity is regulated by the natural promoter of CYP2B6, whereas pGL3p-2B7P, by the regulatory region located 69 kb upstream of CYP2B6, which also maps to the 5′ regulatory region of CYP2B7P. Results shown are means±S.E.M. for three independent experiments. Similar results were obtained when increasing amounts of the plasmid pSG5 ERβ were transfected with the two reporter gene plasmids (C and D). In order to evaluate the estrogen responsiveness of CYP2B6, HuH-7 cells were plated in DCC-FBS containing medium for 24 h. Cells were co-transfected with (E) 200 ng of pGL3-2B6 and 50 ng of pSG5 ERα or (F) 200 ng of pGL3p-2B7P and 5 ng of pSG5 ERα for 24 h. The cells were treated with either 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780 or 10 nM E2+100 nM ICI 182,780 for an additional 24 h prior to luminescence detection. Similar results were obtained when (G) 200 ng of pGL3-2B6 and 100 ng of pSG5 ERβ or (H) 200 ng of pGL3p-2B7P and 25 ng of pSG5 ERβ were transfected into the HuH-7 cells. Luciferase activity significantly (p<0.05 one-way ANOVA) different from DMSO is indicated by an asterisk.
We then introduced two point mutations in the ERE to create pGL3-2B6_EREmut to determine the role of the ERE in the CYP2B6 promoter at mediating the ERα-dependent induction of CYP2B6. We also created a promoter truncation, pGL3-2B6_ΔERE where the putative ERE was removed. E2-induced and ERα-dependent luciferase activity was not observed in cells transfected with pGL3-2B6_EREmut and pGL3-2B6_ΔERE (Fig. 4A). In agreement with these findings, the introduction of two mutations, E203A and G204A, in the ERα DNA-binding domain (DBD) which prevented ERα-dependent regulation of ERE-mediated responses [41], reduced the ERα-dependent regulation of pGL3-2B6 (Fig. 4B). Western blots showed that wild-type ERα and DBD mutant were expressed at similar levels, indicating that differences in reporter gene expression were not due to reduced protein levels (Fig. 5C).
Fig. 4.

ERα-dependent regulation of pGL3-2B6 occurs through the ERE site. (A) HuH-7 cells were transfected with 200 ng of pGL3-2B6, pGL3-2B6_EREmut or pGL3-2B6_ΔERE and 50 ng of pSG5 ERα for 24 h. Cells were treated with either 0.1% DMSO or 10 nM E2 for 24 h prior to luminescence detection. Results shown are means±S.E.M. for five independent experiments. Luciferase activity that is statistically significant (p<0.05 one-way ANOVA) from DMSO pGL3-2B6 is indicated by an asterisk. (B) HuH-7 cells were plated in full serum and transfected with 200 ng of pGL3-2B6 ERE and 100 ng of either pSG5 ERα or pSG5 ERα DBD. Results shown are means±S.E.M. for three independent experiments. Luciferase activity that was significantly different (p<0.05 one-way ANOVA) from pSG5 (vector control) or wild-type ERα are indicated by an asterisk or dagger, respectively. (C) Western blot analysis shows that the wild-type and the mutant receptors were expressed at similar levels.
Fig. 5.

E2-dependent regulation of CYP2B6 expression in T-47D cells. T-47D cells were plated in DCC-FBS containing medium for 72 h before treatment. (A) After 6 h treatment with 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780, RNA was isolated and CYP2B6 expression level was analyzed by qPCR as described in Materials and methods. Expression level significantly (p<0.05 one-way ANOVA) different from DMSO is indicated by an asterisk. Results shown are means±S.E.M. for three independent experiments. (B) T-47D cells were treated with 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780 for 24 h before western analysis. Images shown are representative of two independent experiments. (C) ChIP assays were performed in T-47D cells treated with 0.1% DMSO, 10 nM E2, 100 nM ICI 182,780, or 10 nM E2+100 nM ICI 182,780 for the time points indicated. Results shown are means±S.E.M. for three independent experiments. Recruitment levels are presented as a percentage of a 5% total chromatin input. Recruitment level significantly (p<0.05 one-way ANOVA) different from time-matched DMSO is indicated by an asterisk.
3.4. ERα regulates CYP2B6 expression in an E2-dependent manner in T-47D cells
To investigate the effect of E2 and ICI 182,780 on CYP2B6 mRNA expression, we isolated RNA from T-47D cells treated with E2 alone, ICI 182,780 alone, or in combination. A modest but statistically significant increase in CYP2B6 mRNA levels was observed in cells treated with 10 nM E2 for 6 h (Fig. 5A) and 24 h (data not shown). Co-treatment with 100 nM ICI 182,780 reduced the E2-dependent increase in CYP2B6 mRNA levels. ICI 182,780 treatment alone had no significant effect on CYP2B6 mRNA expression. We did not observe any increases in CYP2B7P mRNA expression level (data not shown). Western blot analysis revealed a modest E2-dependent increase in CYP2B6 protein expression at 24 h (Fig. 5B). ChIP assays showed an E2-dependent increase in recruitment of ERα to the ERE in the 5′-regulatory region of CYP2B6 at 1 h; however, detectable ERα occupancy was evident in DMSO samples (Fig. 5C). ERα recruitment returned to a basal level after 4 h of E2 treatment (Fig. 5C). Although the inhibitory effect of ICI 182,780 on ERα recruitment to the 5′-regulatory region of CYP2B6 was not apparent after 1 h of treatment, ICI 182,780 significantly reduced E2-dependent recruitment of ERα after 4 h (Fig. 5C). RNAi-mediated knockdown of ERα using two different siRNAs targeting ERα resulted in an approximately 30% reduction in CYP2B6 mRNA expression levels (Fig. 6A). This finding demonstrates that ERα plays a role in regulating CYP2B6.
Fig. 6.

RNAi-mediated knockdown of ERα in T-47D cells. (A) T-47D cells were transfected with either siRNA targeting luciferase (siLuc) or two different siRNAs targeting ERα (siERα-11 or siERα-14) for 48 h. RNA was isolated and CYP2B6 and ERα expression levels were analyzed by qPCR as described in Materials and methods. CYP2B6 mRNA levels were normalized to non-transfected T-47D cells (control), which was set to 1. Statistical significance (p<0.05 Student’s t-test) compared to non-targeting pool is indicated by an asterisk. Results shown represent the means±S.E.M. for three independent experiments. (B) T-47D cells were transfected with either non-targeting pool reagent or siRNA against ERα for 48 h. ChIP assays were performed and recruitment levels are presented as a percentage of a 5% total chromatin input. Statistical significance (p<0.05 Student’s t-test) compared to non-targeting pool is indicated by an asterisk. Results shown are the means± S.E.M. for three independent experiments.
3.5. Differential recruitment of ERα and NCoA3 is associated with altered expression of TFF1 and CYP2B6 in MCF-7 and T-47D cells
The estrogen responsive TFF1 gene was not identified in our ChIP-chip study in T-47D cells, whereas previous studies in MCF-7 cells have not reported ERα occupancy at CYP2B6 [27,28]. To investigate these discrepancies between our study and previous studies [27,28], we evaluated the regulation of TFF1, CYP2B6 and the estrogen responsive GREB1 in T-47D and MCF-7 cells. In agreement with another report [10], we observed increased levels of CYP2B6 mRNA expression in T-47D compared to MCF-7 cells (Fig. 7A). Moreover, T-47D cells exhibited an E2-dependent increase in CYP2B6 mRNA expression that was not observed in MCF-7 cells. Although E2-induced a two-fold increase in TFF1 mRNA levels in both cell lines, the basal level of TFF1 mRNA was three orders of magnitude higher in MCF-7 cells compared to T-47D cells (Fig. 7B). Similar E2-dependent increases in GREB1 mRNA levels were observed in both T-47D and MCF-7 cells, but the expression levels of GREB1 were higher in MCF-7 compared to T-47D cells (Fig. 7C). ChIP assays were performed to examine if the observed differences in TFF1 and CYP2B6 expression levels were due to altered recruitment of ERα and/or nuclear coactivator 3 (NCoA3/SRC3/pCIP/AIB1). NCoA3 was chosen over the related coactivators, NCoA1 and NCoA2, since it is frequently amplified and overexpressed in human breast cancer and has been shown to enhance estrogen-dependent transactivation [42]. MCF-7 cells are known to express higher relative levels of NCoA3 compared to T-47D with both cell lines expressing similar levels of NCoA1 and NCoA2 [42]. We observed an increased E2-dependent recruitment of ERα and NCoA3 to the TFF1 promoter region in MCF-7 cells but not in T-47D cells (Fig. 8A and D). In contrast, E2-dependent increases in ERα and NCoA3 recruitment to the CYP2B6 ERE region were observed in T-47D cells but not in MCF-7 cells (Fig. 8B and E). E2-dependent increases in recruitment of ERα and NCoA3 to GREB1 Enh3 were observed in both T-47D and MCF-7 cells (Fig. 8C and F). The fold recruitment levels of ERα and NCoA3 at GREB1 Enh3 were similar between the two cell lines, but the absolute magnitude of ERα and NCoA3 occupancy was higher in MCF-7 than in T-47D cells.
Fig. 7.

Differential expression of TFF1 and CYP2B6 in T-47D and MCF-7 after E2 stimulation. T-47D and MCF-7 cells were plated in DCC-FBS containing medium for 72 h before treatment. After 6 h treatment with 0.1% DMSO or 10 nM E2, RNA was isolated and the expression of (A) CYP2B6, (B) TFF1, and (C) GREB1 was analyzed by qPCR as described in Materials and methods. Statistical significance (p<0.05 Student’s t-test) between treatment groups from the same cell line is indicated by an asterisk. Statistical significance (p<0.05 Student’s t-test) between cell lines with the same treatment is indicated by a dagger. Results shown are means±S.E.M. for three independent experiments.
Fig. 8.

Differential recruitment of ERα and NCoA3 to TFF1, CYP2B6 and GREB1 in T-47D and MCF-7 cells after E2 stimulation. T-47D and MCF-7 cells were plated in DCC-FBS containing medium for 72 h before treatment. ChIP assays were then performed on cells treated with 0.1% DMSO or 10 nM E2 for 1 h and the recruitment of ERα (A–C) or NCoA3 (D–F) to TFF1, CYP2B6 and GREB1 Enh 3. Results shown are means±S.E.M. for three independent experiments. Recruitment levels are presented as a percentage of a 5% total chromatin input. Recruitment level significantly (p<0.05 Student’s t-test) different from cell line-matched DMSO is indicated by an asterisk.
4. Discussion
The regulation of CYP2B6 is important in drug metabolism and xenochemical toxicity [15]. Most studies on CYP2B6 regulation have focused on the transcriptional induction mediated by xenobiotics, where the main focus of the studies was the transcriptional regulation of CYP2B enzymes by CAR [16]. However CAR does not operate alone in the transactivation of P450 genes, but rather works in concert with several transcription factors, nuclear receptors, and coactivators to control signaling pathways that regulate lipids, bile acids, and hormone homeostasis [43]. Moreover, there are few reports assessing the regulation of CYP2B6 in extra-hepatic tissues.
In this study, we demonstrate the ER-dependent regulation of CYP2B6 in different human cancer cell lines. Because we detected ERα binding sites in the upstream regulatory region of CYP2B6 gene using ChIP-chip, we characterized the ER-dependent regulation of CYP2B6 gene. qPCR and Western blots revealed that the CYP2B6 mRNA levels were increased in an ER-dependent manner. After analysis of truncated promoters and mutant construct reporters for the gene, we found that ERs increased CYP2B6 promoter activity through an ERE site in the upstream regulatory in that gene. ChIP assays confirmed the E2-dependent recruitment of ERα to the 5′-flanking region of CYP2B6 and a distal estrogen responsive region upstream of CYP2B7P; although no changes in CYP2B7P mRNA levels were observed. A significant level of ERα occupancy to both the 5′-regulatory regions of CYP2B6 and CYP2B7P were apparent in the absence of E2. Previous studies have also reported significant ERα binding to different E2-responsive target promoters in the absence of ligand [27,33]. The high level of basal binding of ERα is unclear, but it might be due to very low levels of estrogens or growth factors present in our estrogen-reduced serum-containing medium. In support of this notion, prolonged treated with ICI 182,780, an anti-estrogen that promotes proteolytic degradation of ERα, reduced the level of ERα occupancy below basal levels at both CYP2B6 and CYP2B7P.
Previous studies have reported that high doses of E2 (10 μM) activate mouse CAR, but not human CAR assayed under the same experimental conditions [26]. The authors concluded that the E2-dependent increase in Cyp2b10 protein levels was independent of ER, since the potent synthetic ER agonist, diethylstilbestrol had no effect. However, this might not be the case for the regulation of CYP2B6. Here we show that physiological levels of E2 induce CYP2B6 mRNA and protein levels in an ERα-dependent manner, but also in a cell line selective manner. The ER action was mediated through an ERE located at −1669 to −1657 bp upstream of the CYP2B6 start site and with 40 bp of the PBREM sequence. The ERE site in human CYP2B6 is not conserved in mouse Cyp2b10, which may explain that the reported E2-dependent changes in Cyp2b10 expression levels might be ER-independent in mice. The proximity of the ERE to the PBREM site suggests that crosstalk between CAR and ERα at the CYP2B6 promoter may occur in tissues where both receptors are expressed. Ongoing reporter gene assays revealed that ERα potentiates CAR-mediated regulation of CYP2B6 expression in HuH-7 hepatoma cells (Raymond Lo and Jason Matthews unpublished results). Activating transcription factor (ATF) 5 and members of the CCAAT/enhancer-binding protein have also potentiate CAR-dependent regulation of CYP2B6 in human hepatocytes and hepatoma cells [21]. ERα is expressed in liver tissue, and has important regulatory roles in hepatic signaling [44], thus ER might serve as another hepatic regulator of CYP2B6 expression. Furthermore, given that ERs are expressed in several extra-hepatic tissues, it is possible that ERs might also serve as an extra-hepatic regulator of CYP2B6. Further investigation is needed to understand the specific transcriptional environment that determines the role of ERs in tissue-specific regulation of CYP2B6.
CYP2B6 metabolizes a wide range of xenobiotics and endogenous compounds and is an important regulator of hormone homeostasis [45]. CYP2B6 catalyzes the hydroxylation of testosterone [46,47] and thus indirectly influences E2 levels. Since, ERs are also known to regulate the expression of CYP1A1 and CYP1B1, both of which metabolize endogenous estrogens [8,48–50], the ER-dependent regulation of CYPs may be part of a regulatory mechanism to control estrogen and/or other hormone levels.
A comparison of the ChIP-chip data from T-47D cells with similar studies using MCF-7 cells [27,28] revealed that only about a third of the identified ERα-bound regions overlapped with ERα-bound regions following E2-treatment of MCF-7 cells [27]. The cell line-specific differences in ERα-bound regions might be due to differences in assay conditions, array platforms and data analysis strategies between the studies [27,28]. In terms of CYP2B6, E2-dependent regulation of this gene was evident in T-47D but not MCF-7 cells. Conversely, the recruitment level of ERα to the promoter region of TFF1 was not observed in T-47D cells after 1 h of E2 treatment. These differences were due to cell line-specific differences in E2-dependent occupancy of ERα and NCoA3. Despite the lack of E2-induced recruitment of ERα to TFF1 in T-47D cells, we observed a 2-fold increase in TFF1 mRNA levels. This may be due to differences in sensitivity between ChIP assays and mRNA expression determination by qPCR. Nevertheless, the identification of different ERα-bound regions between the two cell lines highlights the importance of studying ERα activity in multiple breast cancer cell lines to obtain a complete understanding of ERα-dependent transcription.
In the studies reported here, we have established that ERs regulate CYP2B6 expression through direct binding to an ERE located in the CYP2B6 promoter, but in a breast cancer cell line selective manner. Our data add ERs to the growing list of transcription factors involved in the regulation of CYP2B6 expression. We have also demonstrated differential ERα-mediated gene expression in two commonly used ERα-positive breast cancer cell lines, indicating that it is necessary to use multiple cancer, immortalized, and/or primary cell lines to gain a more accurate understanding of ER-mediated transcription.
Supplementary Material
Acknowledgments
We would like to thank Dr. Rachel Tyndale (University of Toronto, Toronto, ON) and Prof. Jan-Åke Gustafsson (University of Houston, TX, USA) for providing the materials used in this study. JM is the recipient of a Canadian Institute of Health Research New Investigator Award. This work was supported by the CIHR operating grant MOP-82715 to JM.
Abbreviations
- ER
estrogen receptor
- ChIP
chromatin immunoprecipitation
- ChIP-chip
chromatin immunoprecipitation on microarray chip
- NCoA3
nuclear coactivator 3
- ERE
estrogen response element
- CYP
cytochrome P450
- CAR
constitutive androstane receptor
- RXR
retinoid X receptor
- PBREM
phenobarbital response enhancer module
- PXR
pregnane X receptor
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- DEX
dexamethasone
- TFF1
trefoil factor 1
- E2
17β-estradiol
- ICI 182,780
fulvestrant
- PWM
position weight matrix
- FDR
false detection rate
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbagrm.2010.01.005.
References
- 1.Nilsson S, Gustafsson JA. Biological role of estrogen and estrogen receptors. Crit Rev Biochem Mol Biol. 2002;37:1–28. doi: 10.1080/10409230290771438. [DOI] [PubMed] [Google Scholar]
- 2.Ruff M, Gangloff M, Wurtz JM, Moras D. Estrogen receptor transcription and transactivation: structure–function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. 2000;2:353–359. doi: 10.1186/bcr80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, Enmark E, Pettersson K, Warner M, Gustafsson JA. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- 4.Sanchez R, Nguyen D, Rocha W, White JH, Mader S. Diversity in the mechanisms of gene regulation by estrogen receptors. BioEssays. 2002;24:244–254. doi: 10.1002/bies.10066. [DOI] [PubMed] [Google Scholar]
- 5.Dutta U, Pant K. Aromatase inhibitors: past, present and future in breast cancer therapy. Med Oncol. 2008;25:113–124. doi: 10.1007/s12032-007-9019-x. [DOI] [PubMed] [Google Scholar]
- 6.Higashi E, Fukami T, Itoh M, Kyo S, Inoue M, Yokoi T, Nakajima M. Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab Dispos. 2007;35:1935–1941. doi: 10.1124/dmd.107.016568. [DOI] [PubMed] [Google Scholar]
- 7.Matthews J, Wihlen B, Thomsen J, Gustafsson JA. Aryl hydrocarbon receptor-mediated transcription: ligand-dependent recruitment of estrogen receptor alpha to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin-responsive promoters. Mol Cell Biol. 2005;25:5317–5328. doi: 10.1128/MCB.25.13.5317-5328.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsuchiya Y, Nakajima M, Kyo S, Kanaya T, Inoue M, Yokoi T. Human CYP1B1 is regulated by estradiol via estrogen receptor. Cancer Res. 2004;64:3119–3125. doi: 10.1158/0008-5472.can-04-0166. [DOI] [PubMed] [Google Scholar]
- 9.Bieche I, Girault I, Urbain E, Tozlu S, Lidereau R. Relationship between intratumoral expression of genes coding for xenobiotic-metabolizing enzymes and benefit from adjuvant tamoxifen in estrogen receptor alpha-positive postmenopausal breast carcinoma. Breast Cancer Res. 2004;6:R252–R263. doi: 10.1186/bcr784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tozlu S, Girault I, Vacher S, Vendrell J, Andrieu C, Spyratos F, Cohen P, Lidereau R, Bieche I. Identification of novel genes that co-cluster with estrogen receptor alpha in breast tumor biopsy specimens, using a large-scale real-time reverse transcription-PCR approach. Endocr Relat Cancer. 2006;13:1109–1120. doi: 10.1677/erc.1.01120. [DOI] [PubMed] [Google Scholar]
- 11.Stresser DM, Kupfer D. Monospecific antipeptide antibody to cytochrome P-450 2B6. Drug Metab Dispos. 1999;27:517–525. [PubMed] [Google Scholar]
- 12.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43:149–173. doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 13.Janmohamed A, Dolphin CT, Phillips IR, Shephard EA. Quantification and cellular localization of expression in human skin of genes encoding flavin-containing monooxygenases and cytochromes P450. Biochem Pharmacol. 2001;62:777–786. doi: 10.1016/s0006-2952(01)00718-3. [DOI] [PubMed] [Google Scholar]
- 14.Miksys S, Lerman C, Shields PG, Mash DC, Tyndale RF. Smoking, alcoholism and genetic polymorphisms alter CYP2B6 levels in human brain. Neuropharmacology. 2003;45:122–132. doi: 10.1016/s0028-3908(03)00136-9. [DOI] [PubMed] [Google Scholar]
- 15.Hodgson E, Rose RL. The importance of cytochrome P450 2B6 in the human metabolism of environmental chemicals. Pharmacol Ther. 2007;113:420–428. doi: 10.1016/j.pharmthera.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Tompkins LM. CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Curr Drug Metab. 2008;9:598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wortham M, Czerwinski M, He L, Parkinson A, Wan YJ. Expression of constitutive androstane receptor, hepatic nuclear factor 4 alpha, and P450 oxidoreductase genes determines interindividual variability in basal expression and activity of a broad scope of xenobiotic metabolism genes in the human liver. Drug Metab Dispos. 2007;35:1700–1710. doi: 10.1124/dmd.107.016436. [DOI] [PubMed] [Google Scholar]
- 18.Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem. 1999;274:6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- 19.Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol. 2001;60:427–431. [PubMed] [Google Scholar]
- 20.Audet-Walsh E, Anderson A. Dexamethasone induction of murine CYP2B genes requires the glucocorticoid receptor. Drug Metab Dispos. 2009;37:580–588. doi: 10.1124/dmd.108.022772. [DOI] [PubMed] [Google Scholar]
- 21.Pascual M, Gomez-Lechon MJ, Castell JV, Jover R. ATF5 is a highly abundant liver-enriched transcription factor that cooperates with constitutive androstane receptor in the transactivation of CYP2B6: implications in hepatic stress responses. Drug Metab Dispos. 2008;36:1063–1072. doi: 10.1124/dmd.107.019380. [DOI] [PubMed] [Google Scholar]
- 22.Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 2007;320:72–80. doi: 10.1124/jpet.106.112136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honkakoski P, Moore R, Gynther J, Negishi M. Characterization of phenobarbital-inducible mouse Cyp2b10 gene transcription in primary hepatocytes. J Biol Chem. 1996;271:9746–9753. doi: 10.1074/jbc.271.16.9746. [DOI] [PubMed] [Google Scholar]
- 24.Stoltz C, Vachon MH, Trottier E, Dubois S, Paquet Y, Anderson A. The CYP2B2 phenobarbital response unit contains an accessory factor element and a putative glucocorticoid response element essential for conferring maximal phenobarbital responsiveness. J Biol Chem. 1998;273:8528–8536. doi: 10.1074/jbc.273.14.8528. [DOI] [PubMed] [Google Scholar]
- 25.Pascussi JM, Gerbal-Chaloin S, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone enhances constitutive androstane receptor expression in human hepatocytes: consequences on cytochrome P450 gene regulation. Mol Pharmacol. 2000;58:1441–1450. doi: 10.1124/mol.58.6.1441. [DOI] [PubMed] [Google Scholar]
- 26.Kawamoto T, Kakizaki S, Yoshinari K, Negishi M. Estrogen activation of the nuclear orphan receptor CAR (constitutive active receptor) in induction of the mouse Cyp2b10 gene. Mol Endocrinol. 2000;14:1897–1905. doi: 10.1210/mend.14.11.0547. [DOI] [PubMed] [Google Scholar]
- 27.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 28.Kwon YS, Garcia-Bassets I, Hutt KR, Cheng CS, Jin M, Liu D, Benner C, Wang D, Ye Z, Bibikova M, Fan JB, Duan L, Glass CK, Rosenfeld MG, Fu XD. Sensitive ChIP-DSL technology reveals an extensive estrogen receptor alpha-binding program on human gene promoters. Proc Natl Acad Sci USA. 2007;104:4852–4857. doi: 10.1073/pnas.0700715104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, Yeo A, George J, Kuznetsov VA, Lee YK, Charn TH, Palanisamy N, Miller LD, Cheung E, Katzenellenbogen BS, Ruan Y, Bourque G, Wei CL, Liu ET. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kininis M, Kraus WL. A global view of transcriptional regulation by nuclear receptors: gene expression, factor localization, and DNA sequence analysis. Nucl Recept Signal. 2008;6:e005. doi: 10.1621/nrs.06005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaw LE, Sadler AJ, Pugazhendhi D, Darbre PD. Changes in oestrogen receptor-alpha and -beta during progression to acquired resistance to tamoxifen and fulvestrant (Faslodex, ICI 182, 780) in MCF7 human breast cancer cells. J Steroid Biochem Mol Biol. 2006;99:19–32. doi: 10.1016/j.jsbmb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Cappelletti V, Miodini P, Di Fronzo G, Daidone MG. Modulation of estrogen receptor-beta isoforms by phytoestrogens in breast cancer cells. Int J Oncol. 2006;28:1185–1191. [PubMed] [Google Scholar]
- 33.Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, Vergara LA, Tang S, Chong A, Bajic VB, Miller LD, Gustafsson JA, Liu ET. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H, Faucette S, Sueyoshi T, Moore R, Ferguson S, Negishi M, LeCluyse EL. A novel distal enhancer module regulated by pregnane X receptor/constitutive androstane receptor is essential for the maximal induction of CYP2B6 gene expression. J Biol Chem. 2003;278:14146–14152. doi: 10.1074/jbc.M212482200. [DOI] [PubMed] [Google Scholar]
- 35.Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol. 2008;26:1293–1300. doi: 10.1038/nbt.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ji H, Wong WH. TileMap: create chromosomal map of tiling array hybridizations. Bioinformatics. 2005;21:3629–3636. doi: 10.1093/bioinformatics/bti593. [DOI] [PubMed] [Google Scholar]
- 37.Ahmed S, Valen E, Sandelin A, Matthews J. Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor alpha at human promoters. Toxicol Sci. 2009;111:254–266. doi: 10.1093/toxsci/kfp144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy CS, Meisner LF, Wu SQ, Jordan VC. Short- and long-term estrogen deprivation of T47D human breast cancer cells in culture. Eur J Cancer Clin Oncol. 1989;25:1777–1788. doi: 10.1016/0277-5379(89)90348-9. [DOI] [PubMed] [Google Scholar]
- 39.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 40.Klein-Hitpass L, Schorpp M, Wagner U, Ryffel GU. An estrogen-responsive element derived from the 5′ flanking region of the Xenopus vitellogenin A2 gene functions in transfected human cells. Cell. 1986;46:1053–1061. doi: 10.1016/0092-8674(86)90705-1. [DOI] [PubMed] [Google Scholar]
- 41.Jakacka M, Ito M, Weiss J, Chien PY, Gehm BD, Jameson JL. Estrogen receptor binding to DNA is not required for its activity through the nonclassical AP1 pathway. J Biol Chem. 2001;276:13615–13621. doi: 10.1074/jbc.M008384200. [DOI] [PubMed] [Google Scholar]
- 42.Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997;277:965–968. doi: 10.1126/science.277.5328.965. [DOI] [PubMed] [Google Scholar]
- 43.Pascussi JM, Gerbal-Chaloin S, Duret C, Daujat-Chavanieu M, Vilarem MJ, Maurel P. The tangle of nuclear receptors that controls xenobiotic metabolism and transport: crosstalk and consequences. Annu Rev Pharmacol Toxicol. 2008;48:1–32. doi: 10.1146/annurev.pharmtox.47.120505.105349. [DOI] [PubMed] [Google Scholar]
- 44.Leong GM, Moverare S, Brce J, Doyle N, Sjogren K, Dahlman-Wright K, Gustafsson JA, Ho KK, Ohlsson C, Leung KC. Estrogen up-regulates hepatic expression of suppressors of cytokine signaling-2 and -3 in vivo and in vitro. Endocrinology. 2004;145:5525–5531. doi: 10.1210/en.2004-0061. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez FJ. Human cytochrome P450: possible roles of drug-metabolizing enzymes and polymorphic drug oxidation in addiction. NIDA Res Monogr. 1991;111:202–213. [PubMed] [Google Scholar]
- 46.Imaoka S, Yamada T, Hiroi T, Hayashi K, Sakaki T, Yabusaki Y, Funae Y. Multiple forms of human P450 expressed in Saccharomyces cerevisiae. Systematic characterization and comparison with those of the rat. Biochem Pharmacol. 1996;51:1041–1050. doi: 10.1016/0006-2952(96)00052-4. [DOI] [PubMed] [Google Scholar]
- 47.Waxman DJ, Attisano C, Guengerich FP, Lapenson DP. Human liver microsomal steroid metabolism: identification of the major microsomal steroid hormone 6 beta-hydroxylase cytochrome P-450 enzyme. Arch Biochem Biophys. 1988;263:424–436. doi: 10.1016/0003-9861(88)90655-8. [DOI] [PubMed] [Google Scholar]
- 48.Frasor J, Stossi F, Danes JM, Komm B, Lyttle CR, Katzenellenbogen BS. Selective estrogen receptor modulators: discrimination of agonistic versus antagonistic activities by gene expression profiling in breast cancer cells. Cancer Res. 2004;64:1522–1533. doi: 10.1158/0008-5472.can-03-3326. [DOI] [PubMed] [Google Scholar]
- 49.Kininis M, Chen BS, Diehl AG, Isaacs GD, Zhang T, Siepel AC, Clark AG, Kraus WL. Genomic analyses of transcription factor binding, histone acetylation, and gene expression reveal mechanistically distinct classes of estrogen-regulated promoters. Mol Cell Biol. 2007;27:5090–5104. doi: 10.1128/MCB.00083-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–124. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.