

Abstract
Background
Cluster Headache (CH) is a severe primary headache, with a poorly understood pathophysiology. Complex genetic factors are likely to play a role in CH etiology; however, no confirmed gene associations have been identified. The aim of this study is to identify genetic variants influencing risk to CH and to explore the potential pathogenic mechanisms.
Methods
We have performed a genome-wide association study (GWAS) in a clinically well-defined cohort of 99 Italian patients with CH and in a control sample of 360 age-matched sigarette smoking healthy individuals, using the Infinium PsychArray (Illumina), which combines common highly-informative genome-wide tag SNPs and exonic SNPs. Genotype data were used to carry out a genome-wide single marker case-control association analysis using common SNPs, and a gene-based association analysis focussing on rare protein altering variants in 745 candidate genes with a putative role in CH.
Results
Although no single variant showed statistically significant association at the genome-wide threshold, we identified an interesting suggestive association (P = 9.1 × 10−6) with a common variant of the PACAP receptor gene (ADCYAP1R1). Furthermore, gene-based analysis provided significant evidence of association (P = 2.5 × 10−5) for a rare potentially damaging missense variant in the MME gene, encoding for the membrane metallo-endopeptidase neprilysin.
Conclusions
Our study represents the first genome-wide association study of common SNPs and rare exonic variants influencing risk for CH. The most interesting results implicate ADCYAP1R1 and MME gene variants in CH susceptibility and point to a role for genes involved in pain processing. These findings provide new insights into the pathogenesis of CH that need further investigation and replication in larger CH samples.
Electronic supplementary material
The online version of this article (doi:10.1186/s10194-016-0705-y) contains supplementary material, which is available to authorized users.
Keywords: Cluster headache, Association studies in genetics, Genome-Wide Association Study, Pituitary adenylate cyclase-activating polypeptide receptor (ADCYAP1R1), Membrane metalloendopeptidase (MME), Neprylisin, Missense mutation
Background
Cluster Headache (CH) is a primary headache with a mean prevalence of 0.1% among general population and a clear male predominance [1]. It is characterized by severe unilateral retro-orbital or fronto-temporal pain attacks, occurring even eight times a day and associated with homolateral autonomic signs [2]. Pain attacks show a circadian and seasonal rhythmicity and smoking is the most frequent life-time habit in CH patients with a prevalence over 80%.
The biology of CH is poorly understood. Current hypotheses are focused on vasomotor changes, inflammation, immune changes, autonomic system imbalance and hypothalamic dysfunction [3]. Twin and family studies have indicated the importance of genetic factors in CH [1]. Genetic predisposition seems to be complex, with the environment and genetic factors interacting together [1].
Association studies focussing on candidate genes have suggested a possible role for HCRTR2 [4–6], not confirmed in a recent meta-analysis [7]. No associations or conflicting results were found for variants in CACNA1A [8], NOS [9], MTHFR [10], PER3 [11], SERPINA [12], and ADH4 [13, 14] genes.
Given the largely unknown pathophysiology of CH, we have undertaken a whole-genome approach to identify the underlying genetic determinants in a clinically well-defined cohort of 99 Italian patients with CH. A cohort of 360 healthy Italian individuals, composed of age-matched cigarette smokers, was used as a control sample. We have used the Infinium PsychArray (Illumina) [15], which combines both common highly-informative genome-wide tag single-nucleotide polymorphisms (SNPs) and exonic SNPs, with an enrichment in genetic variants associated with common psychiatric conditions. This approach thus offers greater potential for the identification of rare or common variants with phenotypic effects in CH.
Methods
Study samples
One hundred patients with CH were enrolled at the Division of Toxicology and Clinical Pharmacology, Headache Centre, University of Modena and Reggio Emilia (Italy). CH diagnosis was made following the ICHD-III beta criteria [2]. CH patients clinical assessment included sex, age, headache history (age of onset, duration of the active phase, highest number of clusters per day and per month, triggering factors, associated autonomic symptoms, correlation with cigarette smoking in the active phase), drug treatment history and smoking status (number of cigarettes per day, CPD, and Fagerström test for nicotine dependence, FTND [16]). The control sample consisted of 360 age-matched Italian smokers, not suffering from CH or migraine, recruited at the same centre as part of a study on the genetics of nicotine dependence (RF-2009‐1549619). All participants were of self-reported Italian ancestry and provided a written informed consent to participate. This study was approved by the local Ethical Committee and took place in observation of the declaration of Helsinki (protocol number 2224/2013).
Genotyping and quality control procedures
DNA for genotyping was extracted from blood or saliva. Genotyping was performed on Illumina Infinium® PsychArray microarrays (Illumina, San Diego, California, USA) in two batches, the first using Illumina PsychArray-24 v1.0, the second using PsychArray-24 v1-1 array [15]. We limited our analysis to 566178 variants present on both versions of the array. Content for the Infinium PsychArray includes 265,000 proven tag SNPs found on the Infinium Core-24 BeadChip, 245,000 markers from the Infinium Exome-24 BeadChip, and 50,000 additional markers associated with common psychiatric disorders.
The clustering algorithm implemented in GenomeStudio was used to cluster the data. Array clustering and quality control (QC) was performed according to recommended guidelines [17] (see Additional file 1 for details). The overall data quality was high: only one control DNA sample failed QC thresholds (one additional CH sample was subsequently removed because of non-European ethnicity); 10 replicate pairs of samples undergoing genotyping were assessed for consistency and reproducibility of the assay and agreement of genotyping calls was 99.99% over all genotypes across these pairs. Principal component analysis (PCA) was performed using PLINK 1.9 [18] on SNPs with minor allele frequency (MAF) > 0.01, pruned for linkage disequilibrium (200 kb window size, r2 > 0.5). PCA plots were generated using the Genesis tool [19]. One outlying sample was identified and excluded from subsequent association analysis (Additional file 1: Figure S1). Relatedness among subjects was checked using PLINK –genome option. 458 samples (99 cases, 359 controls) and 556485 variants pass filters and QC, of these 211927 are monomorphic and were excluded from association analysis.
Sanger sequencing was used to screen the MME coding region in all CH affected individuals. Primers sequences are available from the authors upon request.
Association analysis
Single marker SNP association analysis was performed using the PLINK v 1.9 software [18]. Basic case-control analysis was performed on 290505 SNPs that passed QC and MAF > 0.01 using Fisher’s exact test. Results from the association analysis were clumped according to P-value using PLINK. Each clump is represented by an index variant (that with the lowest P-value in the clump), and contains all SNPs with P-value < 0.01 in linkage disequilibrium with it (R2 > 0.5, within 250 kb of the index SNP). Logistic regression was performed using PLINK –logistic command. To correct for multiple testing we applied the Bonferroni correction (P < 1.7 × 10−7, corresponding to 0.05/number of analyzed SNPs). Quantile-quantile (Q-Q) plot for test statistics was performed using the R package; the regional association plot was created using LocusZoom [20].
Gene-based association analysis was conducted using the Sequence Kernel Association Test (SKAT) [21], which aggregates individual score test statistics of a set of SNPs and efficiently computes gene-level p-values. Post QC, non-monomorphic SNPs were mapped to RefSeq genes (downloaded from the UCSC Table Browser, GRCh37/hg19). SKAT analysis was restricted to a comprehensive collection of candidate genes with a plausible role in CH pathogenesis. The selection of candidate genes was based on Gene Ontology (GO) biological process annotations (http://geneontology.org/page/download-annotations). We selected a group of 1038 genes annotated with sixty-six GO terms implicated in pain perception and response, blood pressure regulation and vasodilation/vasoconstriction, circadian rhythms, alcohol metabolism, neuropeptide signalling, ion channels, nicotinic acetylcholine receptors, tumour necrosis factor (TNF) signalling. The complete list of candidate genes and GO annotations is listed in Additional file 1: Table S1. Gene-based analysis was conducted on all protein altering variants (non-synonymous and splicing variants) with MAF <0.05 within these candidate genes, for a total of 2568 variants in 745 candidate genes (Additional file 1: Table S2). These included 2497 missense, 11 frameshift, 40 stopgain, 3 stoploss, and 17 splicing variants. We used a Bonferroni correction based on the number of the evaluated genes to assess the significance of the gene-based test results (p < 6.7 × 10−5). Functional annotation of variants was performed using Polyphen-2 [22] and the Combined Annotation–Dependent Depletion method (CADD) [23]. CADD is a tool for scoring the deleteriousness of variants in the human genome by incorporating a variety of algorithms in an integrated score (C score).
Results
After stringent quality control filtering and exclusion of 211927 monomorphic SNPs, 344558 high quality SNPs were retained for genetic association analysis for CH disease susceptibility. The population sample included 99 individuals with CH and 359 controls. Demographic and clinical characteristics are summarized in Table 1. We conducted genome-wide association tests for single SNPs, as well as gene-based tests focussing on a subset of rare protein altering variants (PAVs), which a priori, are more likely to impact on disease status.
Table 1.
Sample characteristics
| Cluster headache patients | Controls | |
|---|---|---|
| N | 99 | 359 |
| Gender (N,%) | ||
| Male | 83, 83.84% | 204, 56.82% |
| Female | 16, 16.16% | 155, 43.18% |
| Age, ys (mean ± sd, range) | 47.39 ± 13.12, 23–81 | 48.96 ± 11.91, 18–70 |
| CH attacks | ||
| Age at onset, ys (mean ± sd, range) | 30.04 ± 12.65, 6–80 | |
| Chronic (N, %) | 14, 14.14% | |
| N attacks/year (mean ± sd, range) | 1.29 ± 0.77, 0.17–4 | |
| N attacks/day (mean ± sd, range) | 2.85 ± 1.68, 1–9 | |
| Smoking | ||
| Ever smoking (N,%) | 88, 88.89% | 359, 100% |
| N cigarette/day (mean ± sd) | 19.17 ± 10.83 | 21.77 ± 9.13 |
| Fagerström test (mean ± sd); N | 4.60 ± 2.54 (N = 72) | 5.95 ± 2.21 |
Genome wide association analysis
Single marker case-control association analysis was performed on high quality SNPs with minor allele frequency (MAF) > 0.01 (290505 SNPs) in the study population, using the Fisher exact test (Additional file 1: Table S3). Calculation of the genomic inflation factor indicated no overall inflation of the genome-wide statistical results (λ =1), and the Q-Q plot displayed no global departure from the expected null distribution of P-values (Additional file 1: Figure S2). Both of these results indicate negligible inflation of the genome-wide association signals caused by population stratification. Overall association results are shown in the Manhattan plot (Fig. 1). Although no single variant showed statistically significant association at the genome-wide threshold (P < 1.7 × 10−7), we identified some suggestive associations (P < 10−5). Results of the top loci are shown in Table 2. A SNP cluster tagged by rs1006417 on chromosome 14 was the most significant hit (P = 1.4 × 10−6), located in a gene poor region with no known genes (Additional file 1: Figure S3). The second suggestive hit was for an intronic SNP in the ADCYAP receptor type I gene (ADCYAP1R1), encoding for pituitary adenylate cyclase-activating peptide (PACAP) receptor (Additional file 1: Figure S3). Because CH patients are more frequently males, the male:female ratio differed in the cases and controls, we thus performed a logistic regression analysis with adjustment for sex as a covariate. This analysis confirmed the chromosome 14 locus as the most significant result while the significance of the ADCYAP1R1 SNP was somewhat decreased (Additional file 1: Table S4), suggesting a possible sex effect at this locus. Adding adjustment for age and CPD to the logistic regression model provided similar results.
Fig. 1.
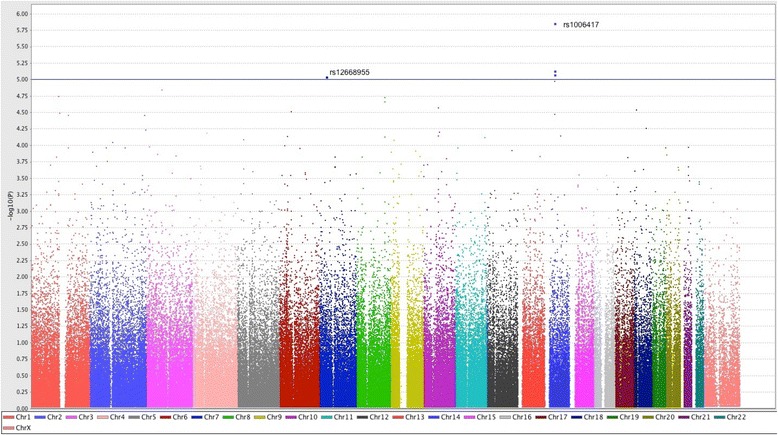
Manhattan plot of genome-wide association P-values. The y axis represents the -log10 Fisher’s test P-values; the horizontal line indicates the threshold for suggestive significance (P < 10−5)
Table 2.
Association analysis results of SNPs with suggestive genome-wide significance
| Index SNP | Position | Other SNPs in clump | Gene | A 1 | A 2 | A1-F case/controls | A1-F 1000G_all (1000G_EUR)a | P-value | OR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|
| rs1006417 | 14:41803291 | rs1782180, rs1779522, rs1778408, rs11157219, rs715334, rs12433558 | G | A | 0.11/0.26 | 0.19 (0.19) | 1.40 × 10−6 | 0.34 (0.21–0.55) | |
| rs12668955 | 7:31116168 | ADCYAP1R1 | G | A | 0.34/0.52 | 0.55 (0.50) | 9.10 × 10−6 | 0.48 (0.34–0.66) | |
| rs1495452 | 3:65834076 | MAGI1 | A | G | 0.55/0.38 | 0.39 (0.45) | 1.43 × 10−5 | 2.03 (1.48–2.79) | |
| rs2182605 | 1:117463537 | PTGFRN | G | A | 0.32/0.50 | 0.51 (0.48) | 1.80 × 10−5 | 0.49 (0.35–0.68) | |
| rs16895584 | 8:122518935 | A | G | 0.24/0.04 | 0.070 (0.054) | 1.85 × 10−5 | 3.50 (2.03–6.02) | ||
| rs6469999 | 8:122415207 | rs13255877, rs7833779 | A | G | 0.31/0.17 | 0.46 (0.16) | 2.16 × 10−5 | 2.23 (1.56–3.19) | |
| rs1509957 | 10:64610718 | rs224308, rs10822065 | G | A | 0.60/0.43 | 0.43 (0.49) | 2.66 × 10−5 | 1.99 (1.44–2.74) |
Abbreviations: A1 minor allele in study sample, A2 major allele, A1-F A1 frequency, OR odds ratio for minor allele, CI confidence interval
aAllele 1 frequency in 1000 Genomes phase 3, all populations (1000G_all) and european population (1000G_EUR)
Notably, our dataset did not provide support to previously reported putative association signals for CH in the HCRTR2 and ADH4 genes. None of the genotyped SNPs mapping to these genes, including the previously implicated SNPs rs2653349 (G1246A) in HCRTR2 and rs1126671 in ADH4, reached a nominally significant P-value (Additional file 1: Table S3).
We also investigated a possible overlap with genetic susceptibility to migraine. A recent meta-analysis identified 45 independent confirmed SNPs that map to 38 distinct loci [24], of which 29 were directly genotyped or captured by a tag SNP in our study. Only rs9349379 in PHACTR1 achieved a nominally significant P value (P = 0.034) (Additional file 1: Table S5).
Furthermore, given the relationship between CH and tobacco smoke, we hypothesized that genetic risk factors involved in smoking-related traits might also have a possible role in CH susceptibility. We inspected the CHRNA5-CHRNA3-CHRNB4 cluster, the CHRNA6-CHRNB3 cluster and CHRNA4, which are genes showing confirmed association to nicotine dependence and smoking behaviours [25–27]. However, no allelic imbalance was detected between CH cases and control smokers at any of the tested polymorphic variants (including the functional non-synonymous SNP rs16969968) (Additional file 1: Table S3). Given the availability of smoking measures in our sample, we also tested if an effect of these variants on smoking quantity (CPD) or nicotine dependence (FTND) was detectable in our sample. Linear regression analysis, with adjustment for sex, age, and CH case status, did not reveal any signal (data not shown).
Gene based analysis
To increase power to identify disease causing variants, we restricted our analysis to rare (MAF < 0.05) PAVs mapping to a comprehensive collection of candidate genes associated to cellular processes with a plausible role in CH, including pain perception and response, vasodilation and vasoconstriction, circadian rhythms, alcohol metabolism, neuropeptide signalling, ion channels, nicotinic acetylcholine receptors, TNF signalling (Additional file 1: Table S1). A total of 2568 rare PAVs belonging to 745 candidate genes were included in gene-based analysis using SKAT [21], which produces a gene-level P value that indicates the degree of enrichment of rare variant associations for SNPs within a gene (Additional file 1: Table S1). The most significant results (nominal P-value < 0.001) are shown in Table 3. The membrane metalloendopeptidase gene (MME), was the most significant gene with nominal P value of 2.5 × 10−5, which survived Bonferroni correction for multiple testing (P = 0.019). All the individual SNPs contributing to the gene-level tests (Table 3) had good genotyping cluster properties and genotyping rate > 99.9%. Interestingly, the most significant SNP (rs147564881) is a missense variant in MME (NM_000902.3:c.674G > C, p.G225A), which was found in 3.5% (7/99) CH patients and in none of 359 Italian controls. This variant is very rare in the general population (MAF = 0.1% in all 1000 Genome Project phase 3 individuals, 0.2% in 60,706 individuals of the Exome Aggregation Consortium ExAC), and has a possible damaging effect based on bioinformatic tools; the CADD PHRED-scaled C score is > 20, thus ranking in the top 1% of predicted deleteriousness of all possible single nucleotide variants [23]. The MME variants were confirmed by Sanger sequencing of the entire MME coding region in all CH patients; no additional rare variant was detected except for a synonymous SNP (rs200455903). Clinical details of the seven CH patients heterozygous for the c.674G > C (p.G225A) variant are shown in Additional file 1: Table S6; these subjects did not show significant differences in clinical aspects distinguishing them from the rest of the CH patient sample. Other nominal suggestive results (p < 0.01) were identified, including NPY1R (Neuropeptide Y receptor type 1), BDKRB1 (B1 bradykinin receptor), SLC5A3 (Sodium/myo-inositol cotransporter), CCL26 (C-C motif chemokine 26) and GRM1 (Metabotropic glutamate receptor 1) (Table 3).
Table 3.
Results of SKAT gene-based tests in CH
| SNP | SKAT P-value |
Single SNPs P-valuea | RA | RAF case/control | RAF 1000G_all (1000G_EUR)b | RAF in ExAC_all (ExAC_NFE)c | Function | PolyPhen-2 (Prediction)d | CADD scoree |
|---|---|---|---|---|---|---|---|---|---|
|
Gene MME
(NM_000902.3) |
2.55 × 10 −5 | ||||||||
| rs61762319 | 0.42 | G | 0.030/0.021 | 0.0072(0.023) | 0.0164(0.0226) | Missense c.22A > G (p.M8V) | 0.524(P) | 15.63 | |
| rs147564881 | 2.03 × 10−5 | C | 0.035/0 | 0.001(0.002) | 0.0022(0.0033) | Missense c.674G > C (p.G225A) | 1 (D) | 21 | |
| rs61758194 | 1 | A | 0/0.0014 | 0.0002(0) | 0.0021(0.0035) | Missense c.1033G > A (p.V345I) | 0.001(B) | 11.65 | |
|
Gene NPY1R
(NM_000909.5) |
1.18 × 10 −3 | ||||||||
| rs5578 | 3.80 × 10−3 | C | 0.040/0.0084 | 0.003(0.007) | 0.0047(0.0067) | Missense c.1121A > C (p.K374T) | 0.421(B) | 12.96 | |
| rs78156188 | 1 | A | 0/0.0028 | 0.0004(0.002) | 0.0006(0.001) | Missense c.332C > T (p.A111V) | 0.023(B) | 3.073 | |
|
Gene BDKRB1
(NM_000710.3) |
3.30 × 10 −3 | ||||||||
| rs143823168 | 1 | C | 0/0.0028 | 0.0018(0.003) | 0.0024(0.0036) | Missense c.571C > G (p.L191V) | 0.999(D) | 9.214 | |
| rs45528332 | 9.23 × 10−3 | A | 0.030/0.0056 | 0.0036(0.004) | 0.0062(0.0052) | Missense c.721G > A (p.G241R) | 0.926(P) | 9.661 | |
|
Gene SLC5A3
(NM_006933.6) |
4.94 × 10 −3 | ||||||||
| rs35707420 | 0.014 | A | 0.025/0.0042 | 0.0026(0.006) | 0.0062(0.0087) | Missense c.1108G > A(p.V370M) | 1(D) | 18.46 | |
| rs199948762 | 0.39 | G | 0.0051/0.0014 | . | 0.000099 (0.0001) | Missense c.1671C > G(p.N557K) | 0.001(B) | 8.533 | |
|
Gene CCL26
(NM_006072.4) |
7.02 × 10 −3 | ||||||||
| rs41463245 | 0.022 | A | 0.020/0.0028 | 0.0036(0.016) | 0.0086(0.0131) | Nonsense c.132G > A (p.W44X) | . | 23.1 | |
|
Gene GRM1
(NM_000838.3) |
9.45 × 10 −3 | ||||||||
| rs41305288 | 0.017 | A | 0.035/0.0098 | 0.01(0.017) | 0.0172(0.0185) | Missense c.2185C > A (p.P729T) | 1(D) | 19.24 | |
| rs362936 | 1 | A | 0.0051/0.0097 | 0.004(0.017) | 0.0212(0.031) | Missense c.2651G > A (p.G884E) | 0.998(D) | 13.22 | |
| rs138759146 | 1 | C | 0/0.0014 | . | 0.0001(0.0002) | Missense c.2725A > C (p.M909L) | 0.017(B) | 1.328 | |
| rs2941 | 0.14 | A | 0.0051/0.022 | 0.0092(0.025) | 0.0132(0.0182) | Missense c.2785G > A (p.V929I) | 0.111(B) | 18.3 | |
| rs79336287 | 0.071 | A | 0.015/0.0023 | 0.005(0.004) | 0.0017(0.0013) | Missense c.3206C > T (p.P1069L) | 0(B) | 11.39 |
Abbreviations: RA rare allele, RAF rare allele frequency
aSingle SNP Fisher's exact test P-value
bRare allele frequency in 1000 Genomes all populations (1000G_all) and european populations (1000G_EUR)
cRare allele frequency in Exome Aggregation Consortium (ExAC) Non-Finnish European (NFE) populations
d HumDiv score; B benign, P possibly damaging, D probably damaging
eCADD v1.3, PHRED-like (-10*log10(rank/total)) scaled C-score
Discussion
At present, the molecular genetic background of CH remains a largely unexplored field. Molecular genetic investigations of CH have been in part hampered by the complex nature of the disorder and have been confined to single candidate gene studies. GWAS offer a hypothesis-free alternative to candidate gene studies and have recently brought novel insights into the biological and genetic underpinnings of many complex traits, including migraine [24]. However, most GWAS have focussed on common variants in the general population (>5%) with a modest effect on risk, hence requiring very large sample sizes. The diffusion of exome sequencing technology provided evidence for the role of rare coding variants in the aetiology of several complex diseases, and a cost effective alternative to exome sequencing is provided by the recent development of “exome” genotyping arrays, which were created to genotype rare coding variants identified by exome sequencing of thousands of individuals [28]. Thus, in order to comprehensively investigate genetic factors influencing risk to CH, we have used the Illumina PsychArray [15] that allows testing genome-wide common variants as well as low frequency exonic variants: the combination of these two features results in a gain in power, given the unknown genetic architecture of CH. Genotype data were exploited to carry out single marker genome-wide association tests using common SNPs, as well as gene-based association analysis focussing on rare exonic variants in candidate genes with a putative role in CH.
The most interesting result stemming from genome-wide analysis was a suggestive association of CH with a common variant of the PACAP receptor gene (ADCYAP1R1); furthermore, gene-based analysis provided evidence for a rare variant of the MME gene. Both gene products are known to have a pivotal function in pain mechanisms, thus making these associations particularly stimulating.
PACAP is a peptide involved in pain-processing in animal models [29], and recently proposed to contribute to migraine pathogenesis [30]. Notably, PACAP administration in healthy subjects induces migraine-like headache [31]. PACAP, through PACAP receptor, induces activation of neurons in the superior salivatory nucleus, sphenopalatine and Gasser ganglion [32], that is thought to be a crucial event in CH pathophysiology, and higher PACAP plasma levels were detected during ictal phase than during inter-bout periods in CH patients [33].
MME encodes for a zinc-dependent plasma membrane endopeptidase, also known as neprilysin or neutral endopeptidase (NEP), that hydrolyses several extracellular peptides. They include peptides, such as substance P, neuropeptide Y, bradykinin, enkephalins, and atrial and brain natriuretic peptides [34], involved in the central regulation of pain, vasomotion, stress and sleep-wake rhythms as well as in the modulation of trigeminal nociceptive signals, whose alterations are thought to be implied in CH and to lead to the principal clinical features of this disease [3, 35, 36]. Interestingly, constitutive NEP knockout mice showed enhanced response to noxious thermal and chemical stimuli as well as thermal hyperalgesia and allodynia, and mechanical allodynia after chronic constriction injury of sciatic nerve [37]. The MME c.674G > C (p.G225A) missense variant was absent in our unaffected controls, but it is represented at low frequency in public control datasets (MAF = 0.02%); although the prevalence of primary headache in public datasets is unknown, this could reflect incomplete penetrance and the interaction with other genetic or environmental factors. Furthermore, rare loss-of-function and missense MME variants were recently associated to late onset dominant axonal polyneuropathies (Charcot-Marie-Tooth Neuropathy Type 2, CMT2; spinocerebellar ataxia with neuropathy, SCA43) [38, 39] and to a rare autosomal recessive variant of CMT2 [40]; none of the neuropathy patients described in these studies was reported to have migraine or CH symptoms, however individuals with MME mutations were reported to experience painful sensations [38]. Given the variety of NEP target peptides and their wide expression pattern, it is plausible that MME deleterious variants may contribute to the aetiology of different neurological conditions, and further studies are needed to identify the mechanisms underlying the phenotypic differences. We can speculate that while severe, very rare deleterious mutations lead to axonal polyneuropathies, the more prevalent c.674G > C (p.G225A) missense variant might have a milder damaging effect, increasing CH susceptibility by interaction with other environmental or genetic factors.
Conclusions
In conclusion, our work provides the first evidence that genetic variants in genes involved in pain processing could confer susceptibility to CH. A major limitation in our study is the relatively small sample size, hence further investigation in larger cohorts is warranted in order to confirm our initial association results.
Acknowledgements
We gratefully acknowledge all the subjects who have participated in the study. We also thank Carlo Baraldi and Lanfranco Pellesi for editing the manuscript’s tables and bibliography.
Funding
Supported by the Italian Ministry of Health (RF2009-1549619), University of Bologna (RFO).
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contributions
EM, EB, LAP, MZ participated in the manuscript writing. LAP, MC, SG participated in patient diagnosis, recruitment, and clinical data collection. EB, CC, AM performed the experiments of DNA extraction and genotyping. EM, EB, CC participated in experimental and statistical data analysis. MZ, EM, LAP, supervised and directed the study. All authors contributed to revision and finalization of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
All study participants provided a written informed consent. This study was approved by the Modena Policlinic Ethical Committee and took place in observation of the declaration of Helsinki (protocol number 2224/2013).
Abbreviations
- ADCYAP1R1
ADCYAP receptor type I gene
- BDKRB1
B1 bradykinin receptor
- CADD
Combined Annotation–Dependent Depletion
- CCL26
C-C motif chemokine 26
- CH
Cluster Headache
- CMT2
Charcot-Marie-Tooth Neuropathy Type 2
- CPD
Cigarettes per day
- ExAC
Exome Aggregation Consortium
- FTND
Fagerström test for nicotine dependence
- GO
Gene Ontology
- GRM1
Metabotropic glutamate receptor 1
- GWAS
Genome-wide association study
- MAF
Minor allele frequency
- MME
Membrane metalloendopeptidase gene
- NEP
Neutral endopeptidase
- NPY1R
Neuropeptide Y receptor type 1
- PACAP
Pituitary adenylate cyclase-activating peptide
- PAV
Protein altering variant
- PCA
Principal component analysis
- QC
Quality control
- Q-Q
Quantile-quantile
- SCA43
Spinocerebellar ataxia with neuropathy
- SKAT
Sequence Kernel Association Test
- SLC5A3
Sodium/myo-inositol cotransporter
- SNP
Single-nucleotide polymorphism
- TNF
Tumour necrosis factor
Additional file
Supplemental Methods. Table S1. list of selected candidate genes, GO terms annotations, number of rare protein altering variants (PAV) contained in each gene, and gene-level P-values obtained from SKAT analysis. Table S2. list of rare PAV (MAF < 0.05) identified in CH cases and controls in 745 candidate genes. Table S3. P-values for single marker P-values of all tested SNPs. Table S4. logistic regression analysis. P-values for additive effect of SNPs controlling for sex as a covariate (P sex), and for sex, age and cigarettes per day (P sex, age, CPD). Table S5. Association analysis in CH cases and controls for significant migraine susceptibility SNPs emerged in meta-analysis. Figure S1. Principal component analysis (PCA) plot. Figure S2. Quantile-quantile (Q-Q) plot of Fisher’s exact test P-values for association with cluster headache (CH). Figure S3. Local association plots for chromosome 14 locus (A) and the chromosome 7 locus (B) [17, 18, 24, 41]. (ZIP 6963 kb)
Contributor Information
Elena Bacchelli, Email: elena.bacchelli@unibo.it.
Maria Michela Cainazzo, Email: cainazzo.michela@policlinico.mo.it.
Cinzia Cameli, Email: cinzia.cameli3@unibo.it.
Simona Guerzoni, Email: simona_guerzoni@libero.it.
Angela Martinelli, Email: am383@st-andrews.ac.uk.
Michele Zoli, Email: michele.zoli@unimore.it.
Elena Maestrini, Phone: +39 051 2094178, Email: elena.maestrini@unibo.it.
Luigi Alberto Pini, Phone: +39 059 4224065, Email: luigialberto.pini@unimore.it.
References
- 1.Russell MB. Epidemiology and genetics of cluster headache. Lancet Neurol. 2004;3:279–283. doi: 10.1016/S1474-4422(04)00735-5. [DOI] [PubMed] [Google Scholar]
- 2.Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition (beta version) Cephalalgia. 2013;33:629–808. doi: 10.1177/0333102413485658. [DOI] [PubMed] [Google Scholar]
- 3.Goadsby PJ. Pathophysiology of cluster headache: a trigeminal autonomic cephalgia. Lancet Neurol. 2002;1:251–257. doi: 10.1016/S1474-4422(02)00104-7. [DOI] [PubMed] [Google Scholar]
- 4.Rainero I, Rubino E, Valfre W, et al. Association between the G1246A polymorphism of the hypocretin receptor 2 gene and cluster headache: a meta-analysis. J Headache Pain. 2007;8:152–156. doi: 10.1007/s10194-007-0383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumber L, Sjostrand C, Leone M, et al. A genome-wide scan and HCRTR2 candidate gene analysis in a European cluster headache cohort. Neurology. 2006;66:1888–1893. doi: 10.1212/01.wnl.0000219765.95038.d7. [DOI] [PubMed] [Google Scholar]
- 6.Schurks M, Kurth T, Geissler I, Tessmann G, Diener HC, Rosskopf D. Cluster headache is associated with the G1246A polymorphism in the hypocretin receptor 2 gene. Neurology. 2006;66:1917–1919. doi: 10.1212/01.wnl.0000215852.35329.34. [DOI] [PubMed] [Google Scholar]
- 7.Weller CM, Wilbrink LA, Houwing-Duistermaat JJ, et al. Cluster headache and the hypocretin receptor 2 reconsidered: a genetic association study and meta-analysis. Cephalalgia. 2015;35:741–747. doi: 10.1177/0333102414557839. [DOI] [PubMed] [Google Scholar]
- 8.Sjostrand C, Giedratis V, Ekbom K, Waldenlind E, Hillert J. CACNA1A gene polymorphisms in cluster headache. Cephalalgia. 2001;21:953–958. doi: 10.1046/j.1468-2982.2001.00281.x. [DOI] [PubMed] [Google Scholar]
- 9.Sjostrand C, Modin H, Masterman T, Ekbom K, Waldenlind E, Hillert J. Analysis of nitric oxide synthase genes in cluster headache. Cephalalgia. 2002;22:758–764. doi: 10.1046/j.1468-2982.2002.00452.x. [DOI] [PubMed] [Google Scholar]
- 10.Schurks M, Neumann FA, Kessler C, et al. MTHFR 677C > T polymorphism and cluster headache. Headache. 2011;51:201–207. doi: 10.1111/j.1526-4610.2010.01780.x. [DOI] [PubMed] [Google Scholar]
- 11.Ofte HK, Tronvik E, Alstadhaug KB. Lack of association between cluster headache and PER3 clock gene polymorphism. J Headache Pain. 2015;17:18. doi: 10.1186/s10194-016-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Summ O, Gregor N, Marziniak M, Gralow I, Husstedt IW, Evers S. Cluster headache and Alpha 1-antitrypsin deficiency. Cephalalgia. 2010;30:113–117. doi: 10.1111/j.1468-2982.2009.01897.x. [DOI] [PubMed] [Google Scholar]
- 13.Zarrilli F, Tomaiuolo R, Ceglia C, et al. Molecular analysis of cluster headache. Clin J Pain. 2015;31:52–57. doi: 10.1097/AJP.0000000000000075. [DOI] [PubMed] [Google Scholar]
- 14.Fourier C, Ran C, Steinberg A, Sjostrand C, Waldenlind E, Carmine Belin A. Screening of two ADH4 variations in a Swedish cluster headache case-control material. Headache doi. 2016 doi: 10.1111/head.12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Infinium PsychArray-24 Kit | Psychiatric predisposition microarray (2016). http://www.illumina.com/products/psycharray.html. Accessed 30 Sept 2016.
- 16.John U, Meyer C, Schumann A, et al. A short form of the Fagerstrom test for nicotine dependence and the heaviness of smoking index in two adult population samples. Addict Behav. 2004;29:1207–1212. doi: 10.1016/j.addbeh.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, He J, Zhao S, et al. Illumina human exome genotyping array clustering and quality control. Nat Protoc. 2014;9:2643–2662. doi: 10.1038/nprot.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchmann R, Hazelhurst S (2016) Creating Structure Plots - Genesis Manual 0.1 | Genesis PCA and Admixture Plot Viewer. http://www.bioinf.wits.ac.za/software/genesis. Accessed 30 Sept 2016.
- 20.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit 7.20. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gormley P, Anttila V, Winsvold BS, et al. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48:856–866. doi: 10.1038/ng.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berrettini WH, Doyle GA. The CHRNA5-A3-B4 gene cluster in nicotine addiction. Mol Psychiatry. 2012;17:856–866. doi: 10.1038/mp.2011.122. [DOI] [PubMed] [Google Scholar]
- 26.Thorgeirsson TE, Gudbjartsson DF, Surakka I, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42:448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancock DB, Reginsson GW, Gaddis NC, et al. Genome-wide meta-analysis reveals common splice site acceptor variant in CHRNA4 associated with nicotine dependence. Transl Psychiatry. 2015;5 doi: 10.1038/tp.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nievergelt CM, Wineinger NE, Libiger O, et al. Chip-based direct genotyping of coding variants in genome wide association studies: utility, issues and prospects. Gene. 2014;540:104–109. doi: 10.1016/j.gene.2014.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yokai M, Kurihara T, Miyata A (2016) Spinal astrocytic activation contributes to both induction and maintenance of pituitary adenylate cyclase-activating polypeptide type 1 receptor-induced long-lasting mechanical allodynia in mice. Mol Pain. 12. doi: 10.1177/1744806916646383 [DOI] [PMC free article] [PubMed]
- 30.Tajti J, Tuka B, Botz B, Helyes Z, Vecsei L. Role of pituitary adenylate cyclase-activating polypeptide in nociception and migraine. CNS Neurol Disord Drug Targets. 2015;14:540–553. doi: 10.2174/1871527314666150429114234. [DOI] [PubMed] [Google Scholar]
- 31.Guo S, Vollesen AL, Hansen RD, et al (2016) Part I: Pituitary adenylate cyclase-activating polypeptide-38 induced migraine-like attacks in patients with and without familial aggregation of migraine. Cephalalgia. doi:10.1177/0333102416639516. [DOI] [PubMed]
- 32.Saghy E, Payrits M, Helyes Z, et al. Stimulatory effect of pituitary adenylate cyclase-activating polypeptide 6-38, M65 and vasoactive intestinal polypeptide 6-28 on trigeminal sensory neurons. Neuroscience. 2015;308:144–156. doi: 10.1016/j.neuroscience.2015.08.043. [DOI] [PubMed] [Google Scholar]
- 33.Tuka B, Szabo N, Toth E, et al. Release of PACAP-38 in episodic cluster headache patients - an exploratory study. J Headache Pain. 2016;17:69. doi: 10.1186/s10194-016-0660-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turner AJ, Isaac RE, Coates D. The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays. 2001;23:261–269. doi: 10.1002/1521-1878(200103)23:3<261::AID-BIES1036>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 35.Barloese MC. A review of cardiovascular autonomic control in cluster headache. Headache. 2016;56:225–239. doi: 10.1111/head.12730. [DOI] [PubMed] [Google Scholar]
- 36.Barloese M, Lund N, Petersen A, Rasmussen M, Jennum P, Jensen R. Sleep and chronobiology in cluster headache. Cephalalgia. 2015;35:969–978. doi: 10.1177/0333102414564892. [DOI] [PubMed] [Google Scholar]
- 37.Kramer HH, He L, Lu B, Birklein F, Sommer C. Increased pain and neurogenic inflammation in mice deficient of neutral endopeptidase. Neurobiol Dis. 2009;35:177–183. doi: 10.1016/j.nbd.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Auer-Grumbach M, Toegel S, Schabhuttl M, et al. Rare variants in MME, encoding metalloprotease neprilysin, are linked to late-onset autosomal-dominant axonal polyneuropathies. Am J Hum Genet. 2016;99:607–623. doi: 10.1016/j.ajhg.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Depondt C, Donatello S, Rai M, et al. MME mutation in dominant spinocerebellar ataxia with neuropathy (SCA43) Neurol Genet. 2016;2 doi: 10.1212/NXG.0000000000000094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higuchi Y, Hashiguchi A, Yuan J, et al. Mutations in MME cause an autosomal-recessive Charcot-Marie-Tooth disease type 2. Ann Neurol. 2016;79:659–672. doi: 10.1002/ana.24612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusions of this article are included within the article and its additional files.