

Abstract
Phosphatidic acid (PA) is a potent lipid secondary messenger whose synthesis is tightly regulated in both space and time. Established tools for detecting PA involve ex vivo analysis and do not provide information on the subcellular locations where this lipid is synthesized. Here we report a chemoenzymatic strategy for imaging sites of cellular PA synthesis by phospholipase D (PLD) enzymes. We find that PLDs can catalyze phospholipid head group exchange with alkynols to generate alkyne-labeled PA analogs within cells. Subsequent fluorophore tagging using the Cu-catalyzed azide-alkyne cycloaddition enabled both visualization by fluorescence microscopy and quantification by HPLC. Our studies revealed several intracellular sites of PLD-mediated PA synthesis. We envision applications of this approach to dissect PA-dependent signaling pathways, image PLD activity in disease, and remodel intracellular membranes with new functionality.
Keywords: Click chemistry, Hydrolases, Imaging agents, Phosphatidic acid, Phospholipids
Cells respond to environmental stimuli by transducing information from cell surface receptor binding events to activation of intracellular signaling pathways. Secondary messengers are key intermediates in these pathways. Soluble small molecules (e.g., cyclic AMP, inositol trisphosphate), lipids (e.g., diacylglycerol, phosphatidic acid), and ions (e.g., calcium) all act as secondary messengers by binding to downstream protein targets and modulating kinase cascades, metabolism, gene expression, and ultimately cellular behavior. Given the pleiotropic and context-dependent physiological effects of secondary messengers, much effort has been invested to develop molecular probes to image these species within cells[1].
Phosphatidic acid (PA) is a potent lipid secondary messenger that evokes multiple signaling outcomes in the cell. By binding to dozens of protein targets and also altering bilayer biophysical properties, PA causes diverse intracellular events such as cytoskeletal rearrangements, vesicular trafficking, and membrane biogenesis, as well as cellular behaviors including growth, migration, and survival[2,3]. Speaking to a central role of PA in cellular homeostasis, dysregulation of this lipid occurs in many diseases, including several cancers, Alzheimer's disease, thrombotic disorders, and autoimmunity[4,5].
To achieve these diverse PA-dependent physiological effects, cells must regulate the production of PA in space and time. Indeed, several enzyme isoforms responsible for PA biosynthesis – most notably members of the phospholipase D (PLD), diacylglycerol kinase, and lysophosphatidic acid acyltransferase families – have been extensively characterized, and their subcellular localizations and enzymatic activities are all carefully controlled[3,6]. Among these, the PLD family has been implicated as a major producer of PA upon cell stimulation under both physiological and pathological conditions. Given that PA causes a diversity of signaling events and is produced on several organelle membranes by different biosynthetic enzymes, it is challenging to attribute biological functions to individual subcellular pools of PA with currently available tools[7]. To dissect the multiple functions of PA, the ability to detect and resolve spatially distinct pools of PA produced by these different enzymatic pathways is critically needed.
Here we present a chemical method for imaging sites of cellular PA synthesis by PLD enzymes. Our approach builds upon the longstanding observation that PLD enzymes, which physiologically hydrolyze phosphatidylcholine (PC) to generate PA and choline, can also catalyze transphosphatidylation reactions with exogenously supplied primary alcohols (e.g., butanol) to generate unnatural phosphatidyl alcohols (Figure 1A)[8]. This transphosphatidylation reaction occurs with rapid kinetics, allowing for simple primary alcohols to effectively compete inside living cells with water in the PLD active site[2]. Classically, this method has been used with radiolabeled substrates, followed by TLC analysis. More recently, an elegant adaptation of this method, using deuterated butanol, has enabled quantification of PLD-mediated PA synthes is by mass spectrometry[9,10].
Figure 1.
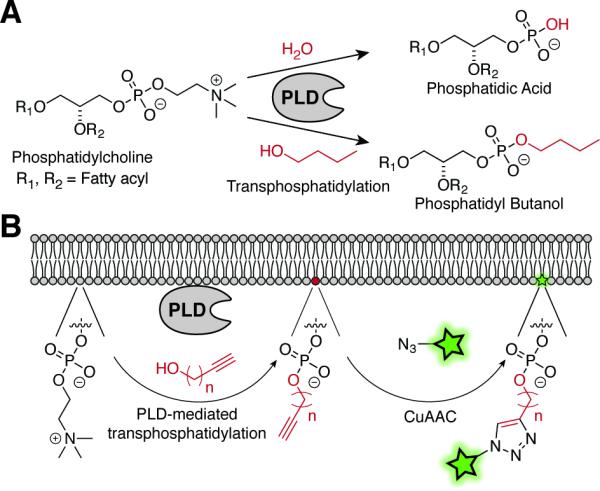
(A) PLD physiologically catalyzes hydrolysis of phosphatidylcholine to form PA (top) and can also mediate transphosphatidylation reactions with short-chain primary alcohols (e.g., butanol) to form phosphatidyl alcohols. (B) Two-step approach to image PLD-mediated PA synthesis. Cells are treated with alkynols, resulting in PLD-mediated formation of phosphatidyl alkynols, followed by CuAAC labeling with an azido fluorophore to enable visualization or quantification.
While these existing approaches allow for accurate measurement of cellular PLD activity and have seen extensive use in probing the biological effects of PA synthesis, they all require ex vivo analysis and hence do not provide spatial information on the sites of PA synthes is within the cell. Given that several PLD isoforms exist and are localized to different intracellular membrane compartments, the location of PA production is thought to be critical to the numerous physiological effects of this signaling lipid[4].
We envisioned an approach to image PLD-mediated PA synthesis within cells wherein butanol is replaced with an alkyne-functionalized alcohol (alkynol) that would be used as a transphosphatidylation substrate by PLDs and would permit subsequent detection by Cu-catalyzed azide-alkyne cycloaddition (CuAAC) with an azido fluorophore (Figure 1B). This two-step approach using the chemical reporter strategy[11] would enable both visualization and quantification of PLD-mediated PA synthesis within intact cells.
We began our studies by assessing whether PLD would accept alkynols as substrates in vitro. We evaluated a panel of five small alkynols (propynol, 3-butyn-1-ol, 4-pentyn-1-ol, 5-hexyn-1-ol, and 6-heptyn-1-ol) by incubating each with 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and a commercially available PLD. The reaction mixtures were then subjected to CuAAC with an azide-labeled rhodamine 110 derivative (Az488). HPLC separation coupled to fluorescence or electrospray ionization-mass spectrometry (ESI-MS) detection confirmed the identity of the expected fluorescent phosphatidyl alcohol products for all five alkynols (Figure 2).
Figure 2.
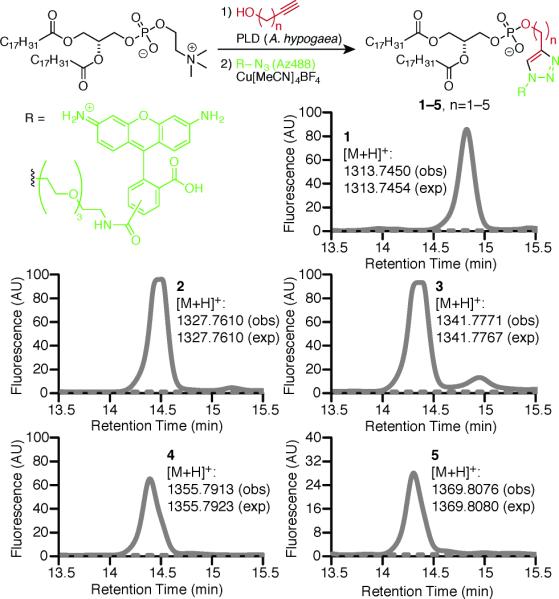
PLD accepts alkynols as substrates for transphosphatidylation. DOPC and Arachis hypogaea PLD were incubated with the indicated alkynol (solid line) or no alkynol (dotted line), followed by lipid extraction, CuAAC labeling with Az488, and HPLC analysis coupled to fluorescence or mass spectrometry detection. Shown are fluorescence traces, with masses indicated next to the curves (obs, observed; exp, expected).
We then evaluated whether mammalian PLDs would accept alkynols as substrates within live cells. We treated HeLa cells with each of the five alkynols for 20 minutes, stimulated acute PA biosynthesis pharmacologically using phorbol 12-myristate 13-acetate (PMA)[12], and then isolated total cellular lipid extracts. Following CuAAC labeling with Az488, the samples were analyzed by HPLC coupled to fluorescence detection (Figure 3A). Comparison of these spectra to those from cells labeled in the presence of a well-characterized pan-PLD inhibitor, 5-fluoro-2-indolyl deschlorohalopemide (FIPI)[13], demonstrated that fluorescent lipids were indeed produced as a result of PLD activity (Figures 3B and S1).
Figure 3.

Detection of PLD-mediated PA synthesis within mammalian cells using alkynol labeling. (A) Schematic of labeling approach. (B and C) HPLC traces of Az488-tagged lipid extracts from PMA-stimulated HeLa cells treated with (B) 12.9 mM alkynol (solid line), 750 nM FIPI + 12.9 mM alkynol (dotted line), or no alkynol (dashed line) or (C) the indicated concentration of hexynol.
While all five alkynols were accepted as transphosphatidylation substrates in HeLa cells, they were processed by cellular PLDs with different efficiencies. We found that, at the same concentration, the three longer chain alkynols (pentynol, hexynol, and heptynol) were incorporated to a greater extent into phosphatidyl alcohols compared to the shorter alkynols (Figures 3B and S2). We verified the molecular identities of the fluorescently labeled phosphatidyl alcohols (PA analogs) by analyzing labeled extracts from cells treated with each of the three longer-chain alkynols by HPLC followed by ESI-MS. These studies revealed the presence of several PA analogs differing in fatty acid chain lengths and degrees of unsaturation (Table S1).
This robust labeling with pentynol, hexynol, and heptynolled us to investigate whether we could lower the dose of alkynol. The initial concentration of 12.9 mM alkynol corresponded to the low end of the concentration range of 0.1–0.5 mol% that is typically used in the traditional TLC-based butanol transphosphatidylation assay. Such high concentrations of alcohol, however, have several negative consequences[2]. First, they are sufficient to compete with water in the PLD active site and reduce the ability of PLDs to synthesize PA, thus impacting PA-dependent signaling. Second, recent studies with PLD knockout mice and selective inhibitors such as FIPI have revealed off-target effects for short-chain alcohols when used at these high concentrations. Thus, we screened different concentrations of hexynol and we found that we could detect PA production in cells at concentrations as low as 100 μM (Figure 3C).
To image sites of PLD-mediated PA synthesis, we treated HeLa cells with each of the three longer-chain alkynols and again stimulated PLD activity acutely with PMA, in the absence or presence of FIPI. Rather than proceeding with a whole-cell lipid extraction, we fixed cells with paraformaldehyde and subjected them to CuAAC labeling with Az488 (Figure 4A). Confocal microscopy analysis revealed strong fluorescence labeling using all three alkynols, with minimal background labeling in two negative controls: (1) cells treated with alkynol and FIPI and (2) cells treated with no alkynol (Figures 4B and S3). We found that hexynol provided the highest signal-to-background ratio, and thus we used it for all subsequent imaging experiments.
Figure 4.

Imaging sites of PLD-mediated PA synthesis within intact cells. (A) Schematic of imaging strategy. (B) Confocal micrographs of cells labeled with indicated combination of FIPI, hexynol, and/or control, followed by PLD stimulation, fixation, and CuAAC labeling with Az488. (C) Colocalization analysis of hexynol-based PA tagging (PA Label) with organelle markers (PM, plasma membrane; ER, endoplasmic reticulum; Golgi, Golgi apparatus; Mito, mitochondria). Insets denote boxed area at higher magnification. Scale bars: 25 μm (B), 10 μm (C), 1 μm (C, inset).
As a further control to ensure that our protocol was principally labeling phospholipids (as opposed to cellular proteins), we found that our labeling was sensitive to treatment with the detergent Triton X-100 (Figure S4). To minimize the impact on cell metabolism and physiology, we found that hexynol concentrations as low as 100 μM would enable detection of hexynol-dependent and FIPI-sensitive fluorescence (Figure S5).
The observed labeling pattern was complex, likely the result of PA synthesis on membranes of multiple intracellular compartments. To determine the subcellular locations of the hexynol-based labeling, we performed a series of colocalization studies with markers of various organelle membranes. In these studies, we transfected HeLa cells with plasmids encoding fluorescent protein fusions to constituents of several organellar membranes and then performed the PA labeling protocol using 1 mM hexynol and, where appropriate, either Az488 or an azide-containing tetramethylrhodamine derivative.
We observed partial colocalization with markers of the plasma membrane (Lyn11-mRFP), endoplasmic reticulum (STIM1-mRFP), and Golgi apparatus (GalT-GFP) (Figure 4C). These data are consistent with the reported localizations in these compartments of PLD1 and PLD2, the two PLD isoforms responsible for the bulk of PLD-mediated PA synthesis[14]. By contrast, we did not observe substantial colocalization with markers of multiple endolysosomal compartments (Figure S6). Interestingly, though we did not observe colocalization with a mitochondrial marker (mCherry-OMP25 transmembrane domain), we did notice a close juxtaposition of the hexynol-derived fluorescence to markers of this organelle (Figure 4C).
Genetically encoded biosensors are commonly used for imaging cellular phospholipids[15]. These probes typically comprise a naturally occurring or engineered lipid-binding domain fused to a fluorescent protein. Though these sensors are widely used, they do have some drawbacks: binding to additional molecules than the desired lipid, sequestration of the target lipid (thus disrupting its function), and an inability to report on minor lipid pools because of competitive binding to major lipid pools[16].
Several PA biosensors have been reported, with the most frequently used ones containing a 40-amino acid PA-binding domain from the yeast protein Spo20p[17]. We compared our labeling to a recently reported, optimized version of this probe[18] and observed partial overlap of the fluorescence signals, particularly at the plasma membrane ( Figure S6). We did not observe complete colocalization, highlighting the differences between equilibrium binding of a biosensor to pools of PA on different membranes produced by several biosynthetic routes with our approach, which targets de novo synthesis exclusively by PLD enzymes but generates unnatural PA mimics.
In conclusion, we present here an in vivo chemoenzymatic synthetic strategy that enables direct visualization of sites of PA synthesis within intact cells by PLDs. Many different extracellular cues result in PLD-mediated PA synthesis on several intracellular membranes that lead to divergent signaling outcomes[2]. Our method will allow dissection of the spatial requirements for PA synthesis in cell signaling. Additionally, we envision applications to image elevated disease-associated PLD activities, most notably in several cancers[5]. Lastly, extension of the method with other functionalized alcohols could enable remodeling of intracellular membranes to endow them with unique physicochemical properties.
Supplementary Material
Acknowledgements
This work was supported in part by the NIH (R00 GM110121). We thank the Baird, Lammerding, and Mao laboratories and the Boyce Thompson Institute Mass Spectrometry Center for reagents and equipment. We are grateful to Jessica Daughtry and Dr. Navid Movahed for technical assistance and to Prof. Yimon Aye and Prof. Jennifer Prescher for a critical reading of the manuscript.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Newman RH, Fosbrink MD, Zhang J. Chem. Rev. 2011;111:3614–3666. doi: 10.1021/cr100002u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Selvy PE, Lavieri RR, Lindsley CW, Brown HA. Chem. Rev. 2011;111:6064–6119. doi: 10.1021/cr200296t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Su Y, Wang X. Lipid-Mediated Protein Signaling. Springer; Netherlands, Dordrecht: 2013. pp. 159–176. [Google Scholar]
- 4.Nelson RK, Frohman MA. J. Lipid Res. 2015;56:2229–2237. doi: 10.1194/jlr.R059220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Frohman MA. J. Biol. Chem. 2014;289:22567–22574. doi: 10.1074/jbc.R114.576876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wymann MP, Schneiter R. Nat. Rev. Mol. Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- 7.Neef AB, Schultz C. Angew. Chem. Int. Ed. Engl. 2009;48:1498–1500. doi: 10.1002/anie.200805507. [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009;121:1526–1528. [Google Scholar]
- 8.Walker SJ, Brown HA. Methods Mol. Biol. 2004;237:89–97. doi: 10.1385/1-59259-430-1:89. [DOI] [PubMed] [Google Scholar]
- 9.Brown HA, Henage LG, Preininger AM, Xiang Y, Exton JH. Methods Enzymol. 2007;434:49–87. doi: 10.1016/S0076-6879(07)34004-4. [DOI] [PubMed] [Google Scholar]
- 10.Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Nat. Chem. Biol. 2009;5:108–117. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grammel M, Hang HC. Nat. Chem. Biol. 2013;9:475–484. doi: 10.1038/nchembio.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDermott M, Wakelam MJO, Morris AJ. Biochem. Cell Biol. 2004;82:225–253. doi: 10.1139/o03-079. [DOI] [PubMed] [Google Scholar]
- 13.Su W, Yeku O, Olepu S, Genna A, Park J-S, Ren H, Du G, Gelb MH, Morris AJ, Frohman MA. Mol. Pharmacol. 2009;75:437–446. doi: 10.1124/mol.108.053298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng X, Frohman MA. Acta Physiologica. 2011;204:219–226. doi: 10.1111/j.1748-1716.2011.02298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemmon MA. Nat. Rev. Mol. Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 16.Maekawa M, Fairn GD. J. Cell. Sci. 2015;128:1422–1433. doi: 10.1242/jcs.164715. [DOI] [PubMed] [Google Scholar]
- 17.Kassas N, Tryoen-Tóth P, Corrotte M, Thahouly T, Bader M-F, Grant NJ, Vitale N. Methods Cell Biol. 2012;108:445–459. doi: 10.1016/B978-0-12-386487-1.00020-1. [DOI] [PubMed] [Google Scholar]
- 18.Zhang F, Wang Z, Lu M, Yonekubo Y, Liang X, Zhang Y, Wu P, Zhou Y, Grinstein S, Hancock JF, et al. Mol. Cell. Biol. 2014;34:84–95. doi: 10.1128/MCB.00987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.