

Abstract
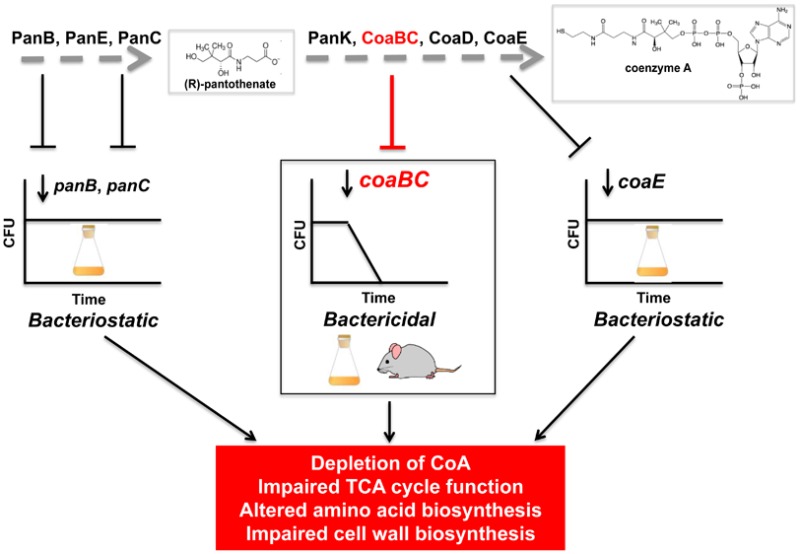
Mycobacterium tuberculosis relies on its own ability to biosynthesize coenzyme A to meet the needs of the myriad enzymatic reactions that depend on this cofactor for activity. As such, the essential pantothenate and coenzyme A biosynthesis pathways have attracted attention as targets for tuberculosis drug development. To identify the optimal step for coenzyme A pathway disruption in M. tuberculosis, we constructed and characterized a panel of conditional knockdown mutants in coenzyme A pathway genes. Here, we report that silencing of coaBC was bactericidal in vitro, whereas silencing of panB, panC, or coaE was bacteriostatic over the same time course. Silencing of coaBC was likewise bactericidal in vivo, whether initiated at infection or during either the acute or chronic stages of infection, confirming that CoaBC is required for M. tuberculosis to grow and persist in mice and arguing against significant CoaBC bypass via transport and assimilation of host-derived pantetheine in this animal model. These results provide convincing genetic validation of CoaBC as a new bactericidal drug target.
Keywords: tuberculosis, drug discovery, CoA, pantetheine, CoaBC
Tuberculosis (TB) remains a global health crisis, as evidenced by the grim statistics: 9.6 million new cases, 480,000 of which were multidrug-resistant, and 1.5 million deaths occurred from this disease in 2014.1 The ongoing emergence and spread of Mycobacterium tuberculosis (Mtb) strains resistant to available antimycobacterial drugs has underscored the urgent need to develop new drugs with novel mechanisms of action.2 However, to meet the demand for new and better drugs for the treatment of both drug-resistant and drug-susceptible forms of the disease, it is essential that the front end of the TB drug pipeline be strengthened by the identification of validated targets coupled with lead compounds.3−7
Both phenotypic3,4,6−8 and target-based approaches9−13 have been used to identify novel antimycobacterial agents with unique targets in Mtb. However, as in other areas of antimicrobial drug discovery,14 target-based approaches have met with limited success in TB drug discovery, being confounded by the issues of drug permeation, metabolism, and efflux. Moreover, basing target selection on the criterion of essentiality for growth of Mtb under in vitro conditions in the absence of knowledge of the vulnerability of that target in the host has further compromised the utility of this approach, where vulnerability, in this context, refers to the extent to which protein function must be inhibited before phenotypic consequences such as bacteriostasis or death are observed.15,16
Given these difficulties, the emphasis in recent years has shifted to using high-throughput, cell-based screening to identify hit compounds with whole-cell activity against Mtb under various conditions.2 This strategy has been more successful and has led to the development of a number of new drugs and drug candidates, including bedaquiline, delamanid, PA-824 (pretomanid), PBTZ-169, and Q-203.3,4,6,7,17 However, a limitation of this approach is that deducing the mechanisms of action of compounds with whole-cell activity is not always straightforward, which can complicate hit progression. Although whole-genome sequencing of mutants resistant to the compound provides a powerful means of target identification when the resistance-conferring mutations map to the target itself,3 this is not always the case, with a disconnect between the mechanism of resistance to a compound and its mechanism of action18 being observed when the molecule is a prodrug, when its inhibitory effects are pleiotropic,19 or when its target is not a protein.20 We and others have therefore investigated the use of target-based whole-cell screening, which integrates both phenotypic and target-led approaches to hit identification. In this approach, conditional knockdown (cKD) mutants of Mtb that are hypersensitized to target- and pathway-specific inhibitors, are used to discover novel chemical scaffolds with whole-cell activity against high-value targets or pathways and/or to assess the selectivity of inhibitors of specific targets in whole Mtb cells.15,21,22 However, it is important to note that target depletion mimics noncompetitive inhibition23 and, therefore, may not always accurately predict the chemical vulnerability of a target.22
The coenzyme A (CoA) pathway has attracted attention as a source of novel drug targets in a number of bacterial pathogens24−29 owing to its ubiquitous nature and the lack of sequence similarity between prokaryotic CoA biosynthesis enzymes and their eukaryotic counterparts.30 CoA is required both as an essential cofactor and for the regulation of key metabolic enzymes in numerous cellular pathways, with 9% of all enzymes estimated to utilize this cofactor.26,31 Of particular significance in the case of Mtb is the critical role that CoA plays in the biosynthesis of lipids, which include essential components of the cell envelope and virulence factors, as well as in the catabolism of lipids, which provide the primary source of energy for the bacillus during infection.32 Unlike mammals, prokaryotes, including Mtb, are able to synthesize pantothenate (Pan; vitamin B5), the precursor of CoA, de novo. The biosynthesis of Pan in Mtb is accomplished by PanB (Rv2225), PanD (Rv3601c), PanC (Rv3602c), and PanE (Rv2573), which constitute the first stage of the CoA pathway. In the second, universal stage, the conversion of Pan to CoA occurs in five steps, catalyzed by PanK (CoaA; Rv1092c), CoaBC (Rv1391), CoaD (Rv2965c), and CoaE (Rv1631) (Figure 1).
Figure 1.

Pan and CoA biosynthesis pathways of Mtb. CoaBC bypass occurs via PanK-mediated phosphorylation of PantSH to produce P-PantSH.
Genome-wide transposon mutagenesis screens33,34 as well as targeted gene knockout approaches29 suggested that genes in both stages of the CoA pathway are required for optimal growth of Mtb in vitro, at least in the absence of supplementation with pathway intermediates. Early studies by Jacobs and colleagues35 confirmed the essentiality of Pan biosynthesis for growth of Mtb in vitro by demonstrating a strict requirement for exogenous Pan supplement for the growth of mutants of Mtb, which lack panC and panD. This work simultaneously established the ability of Mtb to transport and assimilate Pan, although the mechanism of Pan transport remains obscure. The attenuation in mice conferred by loss of panC and panD(35) confirmed that Pan biosynthesis is required for growth of Mtb in vivo and demonstrated that the bacilli are unable to access sufficient quantities of Pan in the host to bypass the first stage of the pathway. The attenuating ΔpanCD mutation formed the backbone of double mutants of Mtb that were later developed as attenuated vaccine candidates, and have found application in TB research as Mtb strains that can be used under biosafety level (BSL) II containment.35−37 These studies, together with the availability of crystal structures of a number of CoA pathway enzymes for use in structure-guided drug design, provided further support for investigating the CoA pathway as an attractive source of new TB drug targets, encouraging the development of inhibitors against various pathway enzymes.9,13,38−41 However, despite resource-intensive efforts that led to the identification of potent inhibitors of Mtb PanC and PanK enzymes, these molecules failed to translate into lead compounds with significant whole-cell activity.13,42,43 Using cKD mutants of Mtb as tools to assess target vulnerability and the target selectivity of small-molecule inhibitors in whole Mtb cells, we21 and others43 concluded that these failures might be explained, at least in part, by the relative invulnerability of Mtb to depletion of PanC and PanK.
We reasoned that a similar genetic approach could be employed to identify one or more steps of the CoA pathway more amenable to inhibition. In this study, we generated cKD mutants in genes encoding six potential targets—PanB, PanE, PanK, CoaBC, CoaD, and CoaE—and used these to assess the impact of depleting each target on the viability of Mtb. Characterization of the cKD mutants by comparison of the impact of transcriptional silencing of individual genes on Mtb viability in vitro and in mice established CoaBC as a bactericidal target in the CoA biosynthetic pathway.
Results
Transcriptional Silencing of panB, coaBC, coaD, and coaE Inhibits the Growth of Mtb In Vitro
cKD mutants in panB, panE, panK, coaBC, coaD, and coaE were generated in a two-step process involving replacement of each native promoter with a tetracycline (Tet)-regulated promoter, Pmyc1tetO (Figure S1), by single crossover (SCO) homologous recombination.21,44 Because panB, panK, coaBC, coaD, and coaE are not predicted to be located within operons (Figure S2), promoter replacement was not expected to have any significant polar effect on the expression of their downstream genes. In contrast, the short (23 bp) intergenic spacing between the 3′-end of panE and the start codon of the downstream Rv2574 gene encoding a nonessential33,34 conserved hypothetical protein suggested the possibility of a polar effect in this case. However, this possibility was excluded by quantitative gene expression analysis of Rv2574 in H37RvMA and the panE promoter replacement mutant, which confirmed that Rv2574 transcript levels were not affected by replacement of the native panE promoter with Pmyc1tetO (data not shown). Following promoter replacement, wild-type (TetR) or reverse Tet repressors (rev-TetR) were introduced via integrative vectors to produce mutants in the Tet-ON and Tet-OFF configurations, respectively.45 For each gene, we attempted to construct two different Tet-ON configuration mutants by using promoters of different strengths to drive Tet repressor (TetR) expression, that is, strong (Tet-ONS) versus intermediate-strength (Tet-ONM). In the case of panE, panK, and coaD, mutants in all three configurations were obtained (Tet-ONS, Tet-ONM, and Tet-OFF). However, as previously observed for lysA,21 Tet-ONS mutants could not be generated for panB, coaBC, and coaE, suggesting that the expression of these genes in the presence of high levels of TetR was below the threshold required to support bacillary growth even when inducer was added.
The effect of transcriptional silencing on growth of the cKD mutants on solid medium was monitored as a function of inducer concentration. Incubation in the presence or absence of anhydrotetracycline (ATc) resulted in complete attenuation of growth of the Tet-OFF and Tet-ONM mutants in panB, coaBC, and coaE, respectively (Figure 2). In contrast, inducer-dependent phenotypes were not observed for any of the panE and panK mutants even under conditions of maximal silencing, and growth attenuation was observed only for coaD Tet-ONS at ATc concentrations ≤3.2 ng/mL (Figure 2), suggesting that strong repression of coaD expression is required to inhibit Mtb growth. The ability of exogenously supplied Pan to restore growth of the panB Tet-ONM and Tet-OFF mutants under repressed conditions (Figure 2) confirmed that growth inhibition was due to interference with Pan biosynthesis. Similarly, restoration of growth of the coaBC Tet-ONM and Tet-OFF mutants by exogenous pantethine (PantS) (Figure 2) confirmed the functionality of CoaBC bypass in Mtb via phosphorylation of pantetheine (PantSH)—the reduced form of PantS—by the type I PanK (CoaA) to produce 4′-phosphopantetheine (P-PantSH)46,47 (Figure 1). In contrast, P-PantSH could not rescue growth of these mutants on agar when supplied at concentrations up to 2.5 mg/mL (Figure S3), confirming that this CoA pathway intermediate, which enters eukaryotic cells,48 cannot be assimilated by Mtb in vitro. Together, the growth phenotyping data suggested that panB, coaBC, and coaE grouped with panC(21) at one extreme, being amenable to inducer-dependent growth inhibition using this transcriptional silencing system; panE and panK grouped at the other extreme, being refractory to inducer-dependent growth attenuation; and coaD fell between the two.
Figure 2.

ATc dose dependence of growth of cKD mutants of Mtb. Strains were grown to early log phase and equivalent numbers of cells were inoculated onto 7H10 agar containing the indicated concentrations of ATc (ng/mL) and/or Pan (25 μg/mL) and PantS (2.5 mg/mL) and incubated for 9 days. Tet-ONM, TetR expressed from intermediate-strength promoter; Tet-OFF, reverse TetR expressed from intermediate-strength promoter; Tet-ONS, TetR expressed from strong promoter.
We postulated that the amenability of Mtb to inducer-dependent modulation of growth inhibition by transcriptional silencing of a given gene would depend upon the basal transcript level of that gene in the wild-type strain. To test this, we used droplet digital PCR (ddPCR) to measure the concentration of CoA pathway gene transcripts in exponentially growing H37Rv as compared to the cKD mutants in the presence and absence of a range of inducer concentrations (Figures S4 and S5; Table S1). Dose-dependent repression of panB, panC, coaBC, and coaE was shown to correlate with progressively reduced growth of the corresponding Tet-ONM and Tet-OFF mutants, and growth inhibition was observed only when the transcript levels were reduced to below the corresponding levels in H37Rv (Figure S4; Table S1). Under the conditions tested, panE, panK, and coaD transcript levels were substantially lower than those of panB, panC, coaBC, and coaE in wild-type Mtb (Figure S5). This would explain the lack of responsiveness of the panE mutants, where basal expression is very low and could not be reduced further by any of the regulated systems, although this gene has also been reported to be nonessential.33,34 Interestingly, replacement of the coaD native promoter with Pmyc1tetO resulted in a >60-fold induction of expression of coaD, and transcript levels could be reduced only to below those observed in H37Rv upon maximal silencing of the Tet-ONS cKD, which is in accordance with the observed growth phenotype. Although panK transcript levels were reduced by ∼50% relative to wild-type in the fully repressed panK Tet-ONS conditional mutant, this was not sufficient to confer a growth phenotype. These findings are consistent with those of Reddy et al.,43 who, during the course of our study, reported that Mtb was able to grow unimpeded in the absence of detectable levels of PanK, suggesting invulnerability of this target.
Although the regulated gene expression system used to generate the cKD mutants characterized in this study was unable to reduce panE, panK, and coaD transcripts to levels sufficiently low to confer a growth phenotype, the recent development of newer tools with improved silencing capabilities may facilitate more efficient silencing of genes such as these, which are expressed at low basal levels in Mtb. These include systems for regulated target protein degradation,15,49 dual control (DUC) systems that enable the simultaneous down-regulation of target genes as well as degradation of their encoded proteins,50,51 and clustered regularly interspaced short palindromic repeat interference (CRISPRi)-regulated systems.52,53
Silencing of coaBC Is Bactericidal in Mtb In Vitro
Having confirmed the association between transcriptional silencing of panB, panC, coaBC, or coaE and inhibition of Mtb growth on agar (Figure 2), we asked what impact silencing of these genes would have on the viability of Mtb. Mutants in the Tet-ONM and Tet-OFF configurations were cultured in standard Middlebrook 7H9 medium in the absence or presence of ATc, respectively, and aliquots sampled daily over a 9-day time course were assessed for viability by scoring colony-forming units (CFUs) on Middlebrook 7H10 agar with or without ATc. All controls behaved as expected: equivalent, ATc-independent growth was observed for the wild-type and promoter-replacement strains, and the absence of colonies from cultures plated under conditions of silencing confirmed maintenance of ATc-dependent growth for all eight cKD mutants throughout the course of the experiment (Figure 3). Following silencing of panB, panC, and coaE by incubation of the Tet-ONM mutants in liquid culture in the absence of ATc for 9 days, the number of CFUs recovered on plates containing 200 ng/mL ATc (i.e., permissive for growth) was equivalent to that present in the starting inoculum, suggesting that depletion of PanB, PanC, and CoaE is bacteriostatic in Mtb (Figure 3). Similarly, equivalent numbers of CFUs were recovered after plating on agar containing no ATc following incubation of the panB, panC, and coaE Tet-OFF mutants in the presence of ATc for 9 days (Figure 3). In contrast, however, the coaBC cKD mutants showed a rapid decline in CFU counts between days 2 and 3 (Tet-ONM) or days 4 and 5 (Tet-OFF), which remained below the limit of detection for the remainder of the experiment (Figure 3). The difference in timing of the CFU decline between the coaBC Tet-ONM and Tet-OFF mutants can be attributed to the fact that the coaBC Tet-ONM inoculum was grown in the presence of PantS supplement but without ATc for 3 days, thereby allowing coaBC silencing to occur prior to commencing the experiment. Together, these results suggest that following transcriptional silencing of coaBC, residual CoaBC already present in the cell can sustain the growth of Mtb for approximately 6 days, whereafter depletion of CoaBC becomes bactericidal.
Figure 3.

Effect of transcriptional silencing of panB, panC, coaBC, and coaE on the viability of Mtb in vitro. Tet-ONM and Tet-OFF cultures of each conditional mutant were grown in the presence and absence of ATc prior to plating serial dilutions of each on 7H10 agar in the presence (200 ng/mL) and absence of ATc. CFUs were scored at the indicated time points, and the results are representative of three independent experiments. The limit of detection was 1 CFU. Error bars represent standard deviation. SCO, promoter replacement mutant generated by single crossover (SCO) homologous recombination; (ATc–/−), incubated in the absence of ATc and plated in the absence of ATc; (AT−/+), incubated in the absence of ATc and plated in the presence of ATc; (ATc+/−), incubated in the presence of ATc and plated in the absence of ATc; (ATc+/+), incubated in the presence of ATc and plated in the presence of ATc.
To investigate whether the distinct effect of coaBC silencing on Mtb viability could be due to a relatively high rate of CoaBC protein depletion, CoaBC-specific peptides in the coaBC Tet-OFF cKD were quantified by mass spectrometry as a function of time under conditions of silencing analogous to those used previously to analyze the impact of panC silencing on PanC levels in the panC Tet-OFF mutant.21 In the gene silencing protocol employed here, PantS was included in the culture medium to enable CoaBC bypass and, thus, ensure equivalent growth of the coaBC Tet-OFF mutant cultured under repressed and derepressed conditions. CoaBC peptide abundance in the coaBC Tet-OFF mutant was found to be decreased by 61 and 93% after 3 and 6 days of silencing, respectively (Figure 4). This result confirmed the association of CoaBC depletion with loss of viability of Mtb. By comparison, panC silencing of the panC Tet-OFF cKD under similar conditions was found to result in ≥95% depletion of PanC after 4 days of exposure to ATc, as determined by Western analysis.21 These findings suggest that differential kinetics of protein depletion are unlikely to explain why silencing of coaBC is bactericidal in Mtb, whereas silencing of panC is bacteriostatic over the same time course (Figure 3).
Figure 4.
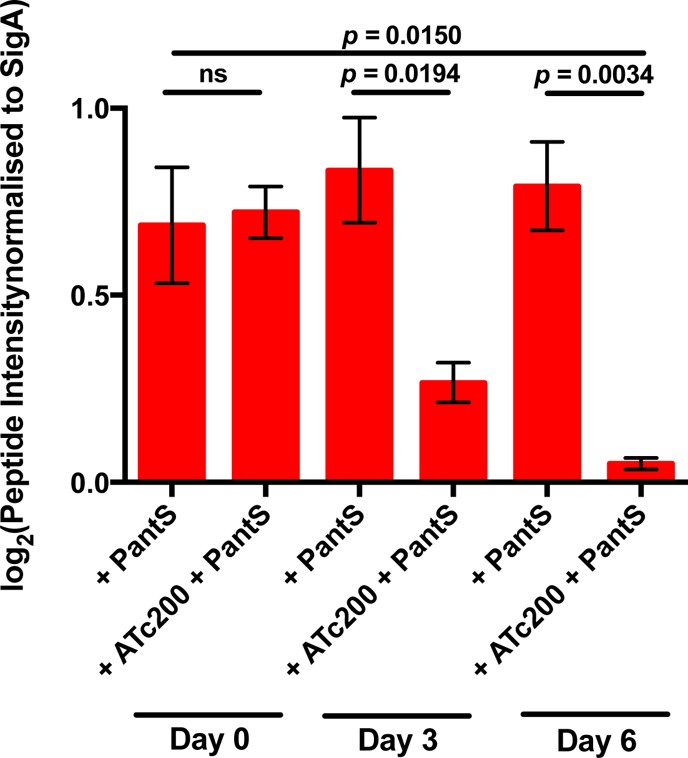
Mass spectrometric quantitation of CoaBC-specific peptides. Label-free quantitative analysis of six CoaBC-specific peptides in the coaBC Tet-OFF mutant with and without silencing for 3 and 6 days. Strains were grown in the presence of PantS to prevent the emergence of unresponsive repressor mutants and to ensure equivalent growth rates of all strains. All data are representative of three biological replicates and were normalized to SigA peptide abundance. ATc200, anhydrotetracycline (200 ng/mL); PantS, pantethine (2.5 mg/mL).
The CFU decline accompanying coaBC silencing could be due to either cell death or impaired culturability. To distinguish these possibilities, we reasoned that nongrowing but metabolically active (NGMA)54 cells with impaired culturability might be rescuable through CoaBC bypass with exogenously supplied PantS. Indeed, inclusion of PantS (2.5 mg/mL) in the media used to score the viability of silenced coaBC Tet-OFF cells over an extended (32-day) time course had an impact on culturability of this strain, as evidenced by the ability to culture CFUs on plates containing PantS supplement versus PantS-free controls over a longer period of time (Figure 5). The rescue of culturable organisms by PantS was, however, time-limited, being sustained for a further 20 days before the effect was lost (Figure 5). Moreover, the rescue was PantS dose-dependent, being less pronounced and sustainable at lower concentrations of supplement (Figure 5).
Figure 5.

PantS supplementation temporarily rescues the bactericidal effect of coaBC silencing in Mtb in vitro in a dose-dependent manner. The coaBC Tet-OFF cKD mutant was grown in the presence and absence of ATc prior to plating serial dilutions on 7H10 agar in the presence (200 ng/mL) and absence of ATc, as well as on 7H10 agar containing various concentrations of PantS (mg/mL). CFUs were scored at the indicated time points, and the results are representative of three independent experiments. The limit of detection was 1 CFU. Error bars represent standard deviation. (ATc–/−), incubated in the absence of ATc and plated in the absence of ATc; (ATc−/+), incubated in the absence of ATc and plated in the presence of ATc; (ATc+/−), incubated in the presence of ATc and plated in the absence of ATc; (ATc+/+), incubated in the presence of ATc and plated in the presence of ATc.
Metabolic Consequences of panB, panC, coaBC, and coaE Silencing
Postulating that the distinct impact of coaBC silencing on the culturability and viability of Mtb might be attributable to the accumulation of one or more toxic metabolites, we analyzed the intracellular pool size changes of a set of 69 metabolites associated with Pan and CoA metabolism by assessing the impact of ATc-mediated silencing of the coaBC Tet-OFF cKD for 1.5 or 3 days relative to the ATc-free control. For comparison, the panB, panC, and coaE Tet-OFF cKD mutants, treated under identical conditions, were analyzed in parallel (Figures 6, S6, and S7).
Figure 6.

Metabolic impact of genetic silencing of pantothenate and CoA biosynthesis enzymes in ATc-regulated cKD mutant strains. Steps in the biosynthetic pathway that were subject to transcriptional silencing are indicated in orange diamonds with black lettering; other steps are shown as pink diamonds with gray lettering. Relative metabolite abundances (based on ion intensities) are indicated in heatmap format below each pathway intermediate, with columns indicating the duration of silencing (1.5 or 3 days) and rows denoting the specific gene silenced. Asterisks indicate metabolites for which levels varied in direction between two independent experiments after 3 days of silencing. Primary data are included in Figures S6 and S7.
The metabolite profiles of the four cKD mutants sampled after 1.5 and 3 days of silencing were qualitatively and quantitatively similar. In particular, there was no clear evidence of accelerated metabolic derangement resulting from silencing of coaBC compared to panB, panC, or coaE, as would be expected if CoaBC was degraded more rapidly than PanB, PanC, or CoaE. The metabolite profiles of all four mutants were broadly consistent with the metabolic adaptation that is expected to occur as a consequence of CoA depletion.55 Importantly, the levels of CoA itself were depleted in all four cKD mutants, showing more pronounced depletion after 3 days of transcriptional silencing than after 1.5 days (Figures 6 and S6). ATc-dependent depletion of acetyl-CoA, phosphoenolpyruvate, α-ketoglutarate, fumarate, and malate was also observed in all four strains following 1.5 and 3 days of silencing (Figures S6 and S7), consistent with functional impairment of the tricarboxylic acid (TCA) cycle. Various levels of accumulation or depletion of amino acids and amino acid precursors were observed upon silencing of the four genes, reflecting an impact of CoA depletion on amino acid biosynthesis as a result of TCA cycle dysfunction.55 Of the CoA thioesters included in this analysis, pools of malonyl-CoA, a key metabolite utilized in fatty acid biosynthesis, were most markedly depleted in all strains, more so than its biosynthetic precursor, acetyl-CoA. In contrast to malonyl-CoA and acetyl-CoA, and unlike in Escherichia coli where CoA starvation resulted in profound depletion of succinyl-CoA,55 changes in succinyl-CoA levels were minor and variable in the Mtb strains. Other metabolites depleted in all strains included UDP-glucose and UDP N-acetyl-d-glucosamine, which are involved in the synthesis of trehalose and peptidoglycan, respectively—both critical components of the Mtb cell envelope.
A focused analysis of the subset of metabolites that comprise the substrates and intermediates of the CoA pathway revealed that the levels of the pathway intermediates did not exhibit accumulations and depletions readily predicted by the specific step blocked by silencing (Figure 6). Moreover, strain classification according to profiles of CoA biosynthetic intermediates showed that coaBC clustered more closely with panC than with panB and coaE (data not shown). This analysis thus indicates that the distinct phenotypic impact of coaBC silencing on viability and culturability is likely mediated by a mechanism more complex than alteration of its specific substrate and product levels.
CoaBC Is Required for Growth and Persistence of Mtb in Mice
The ability of PantS to restore growth of CoaBC-deficient Mtb in vitro suggested that CoaBC would be rendered nonessential should CoaBC bypass be operational in vivo. To investigate this possibility, we analyzed the impact of coaBC silencing on Mtb growth and survival in mice. Both coaBC Tet-ONM and Tet-OFF mutants failed to establish an infection when coaBC transcription was silenced at the point of infection (Figure S8). Furthermore, neither mutant was detectable in mouse lungs by ∼10 days post-infection. The effect of depleting CoaBC during acute or chronic infection was then investigated by measuring the bacillary burdens in mouse lungs (Figure 7a) and spleens (Figure 7b) after silencing of coaBC Tet-OFF at 8 or 35 days post-infection, respectively. Initiation of silencing at both of these later time point resulted in a rapid decline in CFUs recovered from the lungs and spleens of infected mice compared to animals that did not receive doxycycline (doxy). Mice fed doxy from day 8 showed decreased lung pathology by day 56 post-infection as compared to untreated controls and, similarly, less inflammation was observed by day 140 in mice fed doxy from day 35 (Figure S9). These results confirm that CoaBC is required for growth and persistence of Mtb in mice and argue against CoaBC bypass by salvage of PantS(H) in this infection model. Critically, plating of lung homogenate from mice administered doxy from day 8 and sacrificed on day 35 on agar containing PantS (2.5 mg/mL) yielded a significantly higher CFU count than on plates without PantS supplement (Figure 7a), although this difference was resolved at later time points (Figure 7a). Thus, as observed in vitro, high concentrations of PantS can transiently rescue nonculturable, CoaBC-deficient Mtb from mouse lungs before CoaBC depletion becomes bactericidal.
Figure 7.

CoaBC is required for growth and persistence of Mtb in mouse lungs (a) and spleen (b). Mice were infected with the coaBC Tet-OFF mutant and received food containing doxycycline starting from the day of infection (day 0), at day 8 (during acute infection), at day 35 (during chronic infection), or not at all, as indicated. The limit of detection was 4 CFU in lungs and spleens. The data are representative of four mice per time point; error bars represent standard deviation. Doxy day 8 + PantS, lung homogenate from mice fed with doxy from day 8 post-infection inoculated onto agar containing PantS (2.5 mg/mL); Doxy day 35 + PantS, lung homogenate from mice fed with doxy from day 35 post-infection inoculated onto agar containing PantS (2.5 mg/mL).
Discussion
The set of cKD mutants reported here and previously21 provided a powerful resource for investigating the impact of blocking the CoA biosynthesis pathway at specific steps in both stages on the physiology and metabolism of Mtb. Clear growth attenuation was observed upon transcriptional silencing of panB, coaBC, and coaE, providing further confirmation of the essentiality of these enzymes for the growth of Mtb in vitro.33,34 Although comparable inhibition of bacillary growth was observed as a result of transcriptional silencing of panB, panC,21coaBC, and coaE, silencing of coaBC was distinguished by the fact that it resulted in a bactericidal phenotype, whereas panB, panC, or coaE silencing was bacteriostatic under identical conditions. Two lines of evidence argued against this difference being attributable to a comparatively high rate of protein degradation of CoaBC: First, quantification by mass spectrometry of CoaBC-specific peptides during the course of silencing of the coaBC Tet-OFF cKD under conditions analogous to those used to analyze the impact of panC silencing on PanC levels in the panC Tet-OFF cKD by Western analysis21 suggested that CoaBC is not degraded more rapidly than PanC. Second, there was no evidence of a more rapid change in metabolite levels in Mtb following silencing of coaBC compared to panB, panC, or coaE. The finding that PantS temporarily restored culturability of CoaBC-depleted Mtb in a dose-dependent manner suggests that although the bacilli were unable to grow to form visible colonies on agar, they were able to detect, transport, and assimilate PantS to produce CoA, which enabled regrowth. At a practical level, these findings highlight the limitations of using colony-forming ability as a proxy for viability of Mtb, particularly when central metabolism may be functionally impaired. Importantly, however, the state of impaired culturability inferred from PantS rescue was maintained only transiently with sustained coaBC silencing leading to Mtb cell death, both in vitro and in mice. One possible reason for the time-limited rescue is that PantS became exhausted during the course of these experiments. If this were the case, it would imply that coaBC transcript levels in the coaBC Tet-OFF mutant were not restored during the period of PantS rescue and that the expression system was not reversible following removal of ATc. However, this explanation is unlikely for the following reasons. First, there is no obvious reason why the system should be irreversible only in the case of coaBC. Second, because the coaBC cKD mutants in the Tet-OFF and Tet-ON configurations showed similar phenotypes in response to silencing (Figures 2, 3, and S4), the system would need to be irreversible in both configurations. Finally, Tet-OFF regulated expression systems controlled by transcription (as used in this study), proteolysis, or both have been analyzed in Mycobacterium smegmatis using the phenotypically neutral reporter, luciferase, and were found to be reversible, regaining luciferase activity with similar kinetics.50 As an alternative hypothesis, we propose that the time-dependent lack of CoaBC activity might result instead from the misincorporation of less than fully elaborated pantethine precursors into CoA analogues that are used to post-translationally modify CoA-dependent proteins. This would lead to an irreversible accumulation of inappropriately modified CoA-dependent enzymes and thus result in metabolic poisoning of the organism. Experiments to test this hypothesis are currently in progress.
Comparative metabolomic analyses did not reveal any specific alterations in the levels of metabolite pools that could account for the distinct physiology associated with coaBC silencing. Moreover, levels of the pathway intermediates did not exhibit accumulations and depletions readily predicted by the specific step that was blocked by silencing, suggesting more complex regulatory mechanisms in Mtb and/or differences/variations in the pathway structure. These possibilities are the subject of ongoing investigation. These questions notwithstanding, the metabolomic analysis provided a signature that reflects the pleiotropic effects of CoA starvation on Mtb metabolism while raising questions regarding the hierarchy/ordering of metabolic changes that accompany a decline in the intracellular levels of CoA. For example, does the early depletion of malonyl-CoA signal preferential use of declining levels of acetyl-CoA to drive the TCA cycle at the expense of lipid biosynthesis, and, if so, how might this be affected—if at all—by the step at which the CoA pathway is blocked? In this regard, a class of CoA thioesters predicted to be affected by CoA depletion are the holo-form acyl carrier proteins (ACPs) as well as ACP domains in the two fatty acid synthase systems and the multiple type-I polyketide synthases of Mtb. These proteins/domains are characteristically rich in acidic residues and carry a P-PantSH prosthetic group attached to an invariant serine. Because the in vitro silencing experiments reported here were performed in media replete in glutamic acid, CoA depletion might lead to apo-form ACP accumulation,56 which could be toxic in Mtb, as reported in Escherichia coli.57 The bactericidal effect of transcriptional silencing of pptT, which encodes the 4′-phosphopantetheinyl transferase that transfers P-PantSH to the ACP domains of type I PKSs58 and AcpM,59 is consistent with this notion. Thus, comparative lipidomic and proteomic analyses of the pathway mutants might be particularly informative.
A critical question in the context of metabolic drug targets in a pathogen such as Mtb is whether, and to what extent, bypass/salvage mechanisms enabled by metabolite scavenge from the host might subvert a target’s essentiality in vivo. We directly addressed this in the case of CoaBC by analyzing the effect of its depletion on the growth and persistence of Mtb in mice. Our results confirmed that Mtb does not have access to sufficient PantSH to sustain CoaBC bypass in this animal model. PantSH levels in the host are limited by pantetheinases, which convert PantSH into Pan and cysteamine for reuse in CoA biosynthesis.60 Pantetheinase activity has been demonstrated in a variety of tissues in mammals, including mice and humans. The high degree of amino acid sequence similarity shared by mouse and human pantetheinases (79.1%)61 suggests that these enzymes likely play a similar role in limiting PantSH levels in both.
There have been reports of a possible association between CoA biosynthesis and resistance of Mtb to pyrazinamide (PZA) through mutations in the aspartate decarboxylase, PanD,62,63 although whether there is a direct link between the two remains unclear.64 However, Gopal and colleagues65 recently demonstrated that exposure of Mycobacterium bovis BCG to pyrazinoic acid, the active form of the prodrug PZA, resulted in depletion of intracellular CoA levels. These results suggest that treatment with a combination of a CoA biosynthesis inhibitor and PZA—one of only two TB drugs known to contribute to treatment shortening—could potentially act synergistically to reduce the duration of treatment even further. In this context, the data reported here have validated CoaBC as an attractive new target for TB drug development. Potent and selective inhibitors of E. coli, Enterococcus faecalis, and Streptococcus pneumoniae CoaBC have been synthesized, but none of these showed whole-cell activity.66 Although this is likely attributable to their physicochemical properties,66 it may also be due to the fact that they are noncompetitive inhibitors and, as such, are predicted to be less effective at inhibiting CoA pathway flux than mixed inhibitors would be.43 There are currently no known inhibitors of Mtb CoaBC. However, the druggability of the CoaBC homologue from Staphylococcus aureus was confirmed by Strauss and colleagues, who showed that the natural product, CJ-15,801 is converted into a tight-binding inhibitor of the 4′-phosphopantothenoyl-l-cysteine synthetase activity of CoaBC through activation by the S. aureus type II PanK.27 Although CJ-15,801 was not active against Mtb (data not shown), the most likely explanation for its lack of activity is the absence of a type II PanK in Mtb rather than intrinsic undruggability and/or invulnerability of the Mtb enzyme.
Finally, the results presented here underscore the need to move beyond simple “essentiality” of a specific gene/protein to a more nuanced pathway-focused estimation of the consequences of engaging specific targets in their innate cellular context. Particularly as we endeavor to identify agents that might shorten the course of TB treatment, targets whose inhibition leads to lethal consequences in vivo are much more valuable than targets that simply inhibit growth, reinforcing the importance of understanding the mechanisms that distinguish cidal from static phenotypes. This study has provided important insights in this regard while raising important questions whose answers await further investigation.
Methods
Bacterial Strains and Growth Conditions
Bacterial strains and growth conditions are described in the Supporting Information.
Construction and Genotypic Characterization of Promoter Replacement Mutants
The integrative plasmids and primers used for generation of the promoter replacement mutants are listed in Tables S2 and S3. The procedures used to construct and genotypically characterize the promoter replacement mutants are described in the Supporting Information.
Gene Expression Analysis of Conditional Knockdown Mutants Using ddPCR
Total cellular RNA was extracted, DNase-treated, reverse transcribed, and used in ddPCR assays as described in the Supporting Information.
Effect of Transcriptional Silencing on Viability of Mtb
The effect of transcriptional silencing of panB, panC, coaBC, and coaE on the viability of Mtb was determined as described in the Supporting Information.
Proteomic Analysis of coaBC Conditional Knockdown Mutants
The coaBC Tet-OFF mutant was grown in a volume of 60 mL of 7H9 medium supplemented with pantethine (2.5 mg/mL) and antibiotic selection. Cells were grown to OD600 = 0.4, following which each culture was divided into two volumes of 30 mL, and ATc (200 ng/mL) was added to one of each. Total protein was extracted from 10 mL of culture and analyzed by MS-MS as previously described,67 following 0, 3, and 6 days of incubation in the presence and absence of ATc. Raw MS files were processed for peptide identification as previously described,67 and the total peptide intensities of six CoaBC-specific peptides were normalized to three reliably quantifiable SigA peptides defined in the Mtb proteome library.68 The data are representative of three independent biological replicates, and P values were calculated by unpaired t test.
Mtb Metabolite Extraction and Metabolomic Profiling
Mtb H37Rv and the panB Tet-OFF, panC Tet-OFF, coaBC Tet-OFF, and coaE Tet-OFF mutants were cultured at 37 °C in Middlebrook 7H9 broth or on 7H10 (Difco) supplemented with 0.2% acetate, 0.04% Tyloxapol (broth only), 0.5 g/L bovine serum albumin, and 0.085% NaCl. Mtb-laden filters used for metabolomic studies were generated as previously described69 and incubated at 37 °C for 4 days to expand biomass. Mtb-laden filters were then transferred onto a fresh 7H10 agar plate with or without ATc (500 ng/mL) for a further 1.5 or 3 days. Mtb-laden filters were metabolically quenched by plunging filters into a mixture of acetonitrile/methanol/H2O (40:40:20) precooled to −40 °C; metabolites were extracted by mechanical lysis with 0.1 mm zirconia beads in a Precellys tissue homogenizer for 3 min (6500 rpm) twice under continuous cooling at or below 2 °C. Lysates were clarified by centrifugation and then filter sterilized across a 0.22 μm filter. All data obtained by metabolomics were the average of independent triplicates.
LC-MS-based metabolomic profiling was utilized to describe the metabolic landscapes of the wild-type and conditional knockdown mutants. As described previously, extracted metabolites were separated on a Cogent Diamond Hydride Type C column. The mass spectrometer used was an Agilent Accurate Mass 6220 TOF coupled to an Agilent 1200 LC system. Metabolites were identified on the basis of accurate-mass matching (mass tolerance < 0.005 Da) and, when available, retention time matching against chemical standards. Metabolite ion intensities were extracted using Profinder 8.0 and Qualitative Analysis 6.0 (Agilent). The CoA biosynthesis pathway was visualized using OMIX. A metabolite heatmap and hierarchical clustering trees were generated using Cluster 3.0 and Java TreeView 1.0.
Genetic Validation of CoaBC in Vivo
Female C57BL/6 mice (Jackson Laboratory) were infected with early log-phase Mtb cultures as described in the Supporting Information. Procedures involving mice were performed following the National Institutes of Health guidelines for the housing and care of laboratory animals and were reviewed and approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College.
Acknowledgments
This work was supported by grants from the TB Drug Accelerator program of the Bill and Melinda Gates Foundation (to C.E.B. III, K.Y.R, D.S., and V.M.); the South African Medical Research Council; the National Research Foundation of South Africa, and an International Research Scholar’s grant from the HHMI (to V.M.); and, in part, by the Intramural Research Program of NIAID (H.I.M.B. and C.E.B. III). We thank Erick Strauss for valuable discussions and for kindly providing P-PantSH, Jonathan Blackburn and Nelson Soares for advice and assistance with mass spectrometry, and Digby Warner for helpful discussions and for critically reviewing the manuscript.
Glossary
Abbreviations
- ATc
anhydrotetracycline
- CFU
colony-forming unit
- CoaB
4′-phosphopantotheinoyl-l-cysteine synthetase (PPCS)
- CoaC
4′-phosphopantotheinoyl-cysteine decarboxylase (PPCDC)
- CoaD
phosphopantetheine adenylyltransferase
- CoaE
dephospho-CoA kinase
- ddPCR
droplet digital PCR
- Mtb
Mycobacterium tuberculosis
- Pan
pantothenate (vitamin B5)
- P-Pan
phosphopantothenate
- PanB
ketopantoate hydroxymethyltransferase
- PanC
pantothenate synthetase
- PanD
aspartate decarboxylase
- PanE
ketopantoate reductase
- PanK
type I pantothenate kinase
- PantS
pantethine
- PantSH
pantetheine
- P-PanSH
phosphopantetheine
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.6b00150.
Supplemental methods, three tables, and nine figures (PDF)
Author Present Address
∥ Department of Molecular Microbiology and Immunology, Keck School of Medicine, University of Southern California, Zilkha Neurogenetic Institute, 1501 San Pablo Street, Los Angeles, California 90033, USA.
Author Contributions
J.C.E., H.I.M.B., C.E.B. III, K.Y.R.. and V.M. designed research; J.C.E., C.T., Z.W.. and H.E. performed research; J.C.E., C.T., Z.W., H.E., S.E., D.S., K.Y.R.. and V.M. analyzed data; J.C.E. and V.M. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- (2015) Global Tuberculosis Report, WHO. [Google Scholar]
- Zumla A.; Nahid P.; Cole S. T. (2013) Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discovery 12 (5), 388–404. 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Gohlmann H. W.; Neefs J. M.; Winkler H.; Van Gestel J.; Timmerman P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307 (5707), 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Matsumoto M.; Hashizume H.; Tomishige T.; Kawasaki M.; Tsubouchi H.; Sasaki H.; Shimokawa Y.; Komatsu M. (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 3 (11), e466. 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar M.; Nathan C.; Rhee K. Y. (2014) Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 5, 4306. 10.1038/ncomms5306. [DOI] [PubMed] [Google Scholar]
- Pethe K.; Bifani P.; Jang J.; Kang S.; Park S.; Ahn S.; Jiricek J.; Jung J.; Jeon H. K.; Cechetto J.; Christophe T.; Lee H.; Kempf M.; Jackson M.; Lenaerts A. J.; Pham H.; Jones V.; Seo M. J.; Kim Y. M.; Seo M.; Seo J. J.; Park D.; Ko Y.; Choi I.; Kim R.; Kim S. Y.; Lim S.; Yim S. A.; Nam J.; Kang H.; Kwon H.; Oh C. T.; Cho Y.; Jang Y.; Kim J.; Chua A.; Tan B. H.; Nanjundappa M. B.; Rao S. P.; Barnes W. S.; Wintjens R.; Walker J. R.; Alonso S.; Lee S.; Kim J.; Oh S.; Oh T.; Nehrbass U.; Han S. J.; No Z.; Lee J.; Brodin P.; Cho S. N.; Nam K.; Kim J. (2013) Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 19 (9), 1157–1160. 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Stover C. K.; Warrener P.; VanDevanter D. R.; Sherman D. R.; Arain T. M.; Langhorne M. H.; Anderson S. W.; Towell J. A.; Yuan Y.; McMurray D. N.; Kreiswirth B. N.; Barry C. E.; Baker W. R. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405 (6789), 962–966. 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- Protopopova M.; Hanrahan C.; Nikonenko B.; Samala R.; Chen P.; Gearhart J.; Einck L.; Nacy C. A. (2005) Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 56 (5), 968–974. 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- Ciulli A.; Scott D. E.; Ando M.; Reyes F.; Saldanha S. A.; Tuck K. L.; Chirgadze D. Y.; Blundell T. L.; Abell C. (2008) Inhibition of Mycobacterium tuberculosis pantothenate synthetase by analogues of the reaction intermediate. ChemBioChem 9 (16), 2606–2611. 10.1002/cbic.200800437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger I. V.; Freundlich J. S.; Gawandi V. B.; Roberts J. P.; Gawandi V. B.; Sun Q.; Owen J. L.; Fraile M. T.; Huss S. I.; Lavandera J. L.; Ioerger T. R.; Sacchettini J. C. (2012) Structure-guided discovery of phenyl-diketo acids as potent inhibitors of M. tuberculosis malate synthase. Chem. Biol. 19 (12), 1556–1567. 10.1016/j.chembiol.2012.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Kawate T.; Iwase N.; Shimizu M.; Clatworthy A. E.; Kazyanskaya E.; Sacchettini J. C.; Ioerger T. R.; Siddiqi N. A.; Minami S.; Aquadro J. A.; Grant S. S.; Rubin E. J.; Hung D. T. (2013) Diarylcoumarins inhibit mycolic acid biosynthesis and kill Mycobacterium tuberculosis by targeting FadD32. Proc. Natl. Acad. Sci. U. S. A. 110 (28), 11565–11570. 10.1073/pnas.1302114110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willand N.; Dirie B.; Carette X.; Bifani P.; Singhal A.; Desroses M.; Leroux F.; Willery E.; Mathys V.; Deprez-Poulain R.; Delcroix G.; Frenois F.; Aumercier M.; Locht C.; Villeret V.; Deprez B.; Baulard A. R. (2009) Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 15 (5), 537–544. 10.1038/nm.1950. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Yin W.; Martinelli L. K.; Evans J.; Chen J.; Yu Y.; Wilson D. J.; Mizrahi V.; Qiao C.; Aldrich C. C. (2014) Reaction intermediate analogues as bisubstrate inhibitors of pantothenate synthetase. Bioorg. Med. Chem. 22 (5), 1726–1735. 10.1016/j.bmc.2014.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Wei J. R.; Krishnamoorthy V.; Murphy K.; Kim J. H.; Schnappinger D.; Alber T.; Sassetti C. M.; Rhee K. Y.; Rubin E. J. (2011) Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U. S. A. 108 (10), 4176–4181. 10.1073/pnas.1018301108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barve A.; Gupta A.; Solapure S. M.; Kumar A.; Ramachandran V.; Seshadri K.; Vali S.; Datta S. (2010) A kinetic platform for in silico modeling of the metabolic dynamics in Escherichia coli. Adv. Appl. Bioinform Chem. 3, 97–110. 10.2147/AABC.S14368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Mollmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. (2009) Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324 (5928), 801–804. 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Mizrahi V. (2016) Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discovery Today 10.1016/j.drudis.2016.09.010. [DOI] [PubMed] [Google Scholar]
- Kohanski M. A.; Dwyer D. J.; Wierzbowski J.; Cottarel G.; Collins J. J. (2008) Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135 (4), 679–690. 10.1016/j.cell.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L. L.; Schneider T.; Peoples A. J.; Spoering A. L.; Engels I.; Conlon B. P.; Mueller A.; Schaberle T. F.; Hughes D. E.; Epstein S.; Jones M.; Lazarides L.; Steadman V. A.; Cohen D. R.; Felix C. R.; Fetterman K. A.; Millett W. P.; Nitti A. G.; Zullo A. M.; Chen C.; Lewis K. (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517 (7535), 455–459. 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahams G. L.; Kumar A.; Savvi S.; Hung A. W.; Wen S.; Abell C.; Barry C. E. 3rd; Sherman D. R.; Boshoff H. I.; Mizrahi V. (2012) Pathway-selective sensitization of Mycobacterium tuberculosis for target-based whole-cell screening. Chem. Biol. 19 (7), 844–854. 10.1016/j.chembiol.2012.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans J. C.; Mizrahi V. (2015) The application of tetracyclineregulated gene expression systems in the validation of novel drug targets in Mycobacterium tuberculosis. Front. Microbiol. 6, 812. 10.3389/fmicb.2015.00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran V.; Singh R.; Yang X.; Tunduguru R.; Mohapatra S.; Khandelwal S.; Patel S.; Datta S. (2013) Genetic and chemical knockdown: a complementary strategy for evaluating an anti-infective target. Adv. Appl. Bioinform. Chem. 6, 1–13. 10.2147/AABC.S39198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. S.; Jia K. F.; Chen T.; Chang S. Y.; Lin M. S.; Yin H. S. (2013) Experimentally validated novel inhibitors of Helicobacter pylori phosphopantetheine adenylyltransferase discovered by virtual high-throughput screening. PLoS One 8 (9), e74271. 10.1371/journal.pone.0074271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Tempel W.; Smil D.; Bolshan Y.; Schapira M.; Park H. W. (2013) Crystal structures of Klebsiella pneumoniae pantothenate kinase in complex with N-substituted pantothenamides. Proteins: Struct., Funct., Genet. 81 (8), 1466–1472. 10.1002/prot.24290. [DOI] [PubMed] [Google Scholar]
- Spry C.; Kirk K.; Saliba K. J. (2008) Coenzyme A biosynthesis: an antimicrobial drug target. FEMS Microbiol Rev. 32 (1), 56–106. 10.1111/j.1574-6976.2007.00093.x. [DOI] [PubMed] [Google Scholar]
- van der Westhuyzen R.; Hammons J. C.; Meier J. L.; Dahesh S.; Moolman W. J.; Pelly S. C.; Nizet V.; Burkart M. D.; Strauss E. (2012) The antibiotic CJ-15,801 is an antimetabolite that hijacks and then inhibits CoA biosynthesis. Chem. Biol. 19 (5), 559–571. 10.1016/j.chembiol.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L.; Allanson N. M.; Thomson S. P.; Maclean J. K. F.; Barker J. J.; Primrose W. U.; Tyler P. D.; Lewendon A. (2003) Inhibitors of phosphopantetheine adenylyltransferase. Eur. J. Med. Chem. 38 (4), 345–349. 10.1016/s0223-5234(03)00047-3. [DOI] [PubMed] [Google Scholar]
- Ambady A.; Awasthy D.; Yadav R.; Basuthkar S.; Seshadri K.; Sharma U. (2012) Evaluation of CoA biosynthesis proteins of Mycobacterium tuberculosis as potential drug targets. Tuberculosis (Oxford, U. K.) 92 (6), 521–528. 10.1016/j.tube.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Genschel U. (2004) Coenzyme A biosynthesis: reconstruction of the pathway in archaea and an evolutionary scenario based on comparative genomics. Mol. Biol. Evol. 21 (7), 1242–1251. 10.1093/molbev/msh119. [DOI] [PubMed] [Google Scholar]
- Begley T. P.; Kinsland C.; Strauss E. (2001) The biosynthesis of coenzyme A in bacteria. Vitam. Horm. 61, 157–71. 10.1016/S0083-6729(01)61005-7. [DOI] [PubMed] [Google Scholar]
- Lee W.; VanderVen B. C.; Fahey R. J.; Russell D. G. (2013) Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288 (10), 6788–6800. 10.1074/jbc.M112.445056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin J. E.; Gawronski J. D.; Dejesus M. A.; Ioerger T. R.; Akerley B. J.; Sassetti C. M. (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7 (9), e1002251. 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti C. M.; Boyd D. H.; Rubin E. J. (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48 (1), 77–84. 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy V. K.; Wang X.; Chen B.; Russell R. G.; Derrick S.; Collins F. M.; Morris S. L.; Jacobs W. R. Jr. (2002) A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 8 (10), 1171–1174. 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- Sambandamurthy V. K.; Derrick S. C.; Hsu T.; Chen B.; Larsen M. H.; Jalapathy K. V.; Chen M.; Kim J.; Porcelli S. A.; Chan J.; Morris S. L.; Jacobs W. R. Jr. (2006) Mycobacterium tuberculosis DeltaRD1 DeltapanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24 (37–39), 6309–6320. 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- Sampson S. L.; Dascher C. C.; Sambandamurthy V. K.; Russell R. G.; Jacobs W. R. Jr.; Bloom B. R.; Hondalus M. K. (2004) Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect. Immun. 72 (5), 3031–3037. 10.1128/IAI.72.5.3031-3037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timofeev V.; Smirnova E.; Chupova L.; Esipov R.; Kuranova I. (2012) X-ray study of the conformational changes in the molecule of phosphopantetheine adenylyltransferase from Mycobacterium tuberculosis during the catalyzed reaction. Acta Crystallogr. D Biol. Crystallogr. 68 (12), 1660–1670. 10.1107/S0907444912040206. [DOI] [PubMed] [Google Scholar]
- Venkatraman J.; Bhat J.; Solapure S. M.; Sandesh J.; Sarkar D.; Aishwarya S.; Mukherjee K.; Datta S.; Malolanarasimhan K.; Bandodkar B.; Das K. S. (2012) Screening, identification, and characterization of mechanistically diverse inhibitors of the Mycobacterium tuberculosis enzyme, pantothenate kinase (CoaA). J. Biomol. Screening 17 (3), 293–302. 10.1177/1087057111423069. [DOI] [PubMed] [Google Scholar]
- Walia G.; Gajendar K.; Surolia A. (2011) Identification of critical residues of the mycobacterial dephosphocoenzyme a kinase by site-directed mutagenesis. PLoS One 6 (1), e15228. 10.1371/journal.pone.0015228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia G.; Surolia A. (2011) Insights into the regulatory characteristics of the mycobacterial dephosphocoenzyme A kinase: implications for the universal CoA biosynthesis pathway. PLoS One 6 (6), e21390. 10.1371/journal.pone.0021390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkelid C.; Bergfors T.; Raichurkar A. K.; Mukherjee K.; Malolanarasimhan K.; Bandodkar B.; Jones T. A. (2013) Structural and biochemical characterization of compounds inhibiting Mycobacterium tuberculosis pantothenate kinase. J. Biol. Chem. 288 (25), 18260–18270. 10.1074/jbc.M113.476473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. K.; Landge S.; Ravishankar S.; Patil V.; Shinde V.; Tantry S.; Kale M.; Raichurkar A.; Menasinakai S.; Mudugal N. V.; Ambady A.; Ghosh A.; Tunduguru R.; Kaur P.; Singh R.; Kumar N.; Bharath S.; Sundaram A.; Bhat J.; Sambandamurthy V. K.; Bjorkelid C.; Jones T. A.; Das K.; Bandodkar B.; Malolanarasimhan K.; Mukherjee K.; Ramachandran V. (2014) Assessment of Mycobacterium tuberculosis pantothenate kinase vulnerability through target knockdown and mechanistically diverse inhibitors. Antimicrob. Agents Chemother. 58 (6), 3312–3326. 10.1128/AAC.00140-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt S.; Guo X. V.; Hickey C. M.; Ryou M.; Monteleone M.; Riley L. W.; Schnappinger D. (2005) Controlling gene expression in mycobacteria with anhydrotetracycline and Tet repressor. Nucleic Acids Res. 33 (2), e21. 10.1093/nar/gni013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotzsche M.; Ehrt S.; Schnappinger D. (2009) Improved tetracycline repressors for gene silencing in mycobacteria. Nucleic Acids Res. 37 (6), 1778–1788. 10.1093/nar/gkp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balibar C. J.; Hollis-Symynkywicz M. F.; Tao J. (2011) Pantethine rescues phosphopantothenoylcysteine synthetase and phosphopantothenoylcysteine decarboxylase deficiency in Escherichia coli but not in Pseudomonas aeruginosa. J. Bacteriol. 193 (13), 3304–3312. 10.1128/JB.00334-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolman W. J.; de Villiers M.; Strauss E. (2014) Recent advances in targeting coenzyme A biosynthesis and utilization for antimicrobial drug development. Biochem. Soc. Trans. 42 (4), 1080–1086. 10.1042/BST20140131. [DOI] [PubMed] [Google Scholar]
- Srinivasan B.; Baratashvili M.; van der Zwaag M.; Kanon B.; Colombelli C.; Lambrechts R. A.; Schaap O.; Nollen E. A.; Podgorsek A.; Kosec G.; Petkovic H.; Hayflick S.; Tiranti V.; Reijngoud D. J.; Grzeschik N. A.; Sibon O. C. (2015) Extracellular 4′-phosphopantetheine is a source for intracellular coenzyme A synthesis. Nat. Chem. Biol. 11 (10), 784–792. 10.1038/nchembio.1906. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Wei J. R.; Wallach J. B.; Robbins R. S.; Rubin E. J.; Schnappinger D. (2011) Protein inactivation in mycobacteria by controlled proteolysis and its application to deplete the beta subunit of RNA polymerase. Nucleic Acids Res. 39 (6), 2210–2220. 10.1093/nar/gkq1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H.; O’Brien K. M.; Sharma R.; Boshoff H. I.; Rehren G.; Chakraborty S.; Wallach J. B.; Monteleone M.; Wilson D. J.; Aldrich C. C.; Barry C. E. 3rd; Rhee K. Y.; Ehrt S.; Schnappinger D. (2013) A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc. Natl. Acad. Sci. U. S. A. 110 (47), 19095–19100. 10.1073/pnas.1315860110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolly G. S.; Sala C.; Vocat A.; Cole S. T. (2014) Assessing essentiality of transketolase in Mycobacterium tuberculosis using an inducible protein degradation system. FEMS Microbiol. Lett. 358 (1), 30–35. 10.1111/1574-6968.12536. [DOI] [PubMed] [Google Scholar]
- Choudhary E.; Thakur P.; Pareek M.; Agarwal N. (2015) Gene silencing by CRISPR interference in mycobacteria. Nat. Commun. 6, 6267. 10.1038/ncomms7267. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Carette X.; Potluri L. P.; Sharp J. D.; Xu R.; Prisic S.; Husson R. N. (2016) Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res. gkw625. 10.1093/nar/gkw625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manina G.; McKinney J. D. (2013) A single-cell perspective on non-growing but metabolically active (NGMA) bacteria. Curr. Top. Microbiol. Immunol. 374, 135–161. 10.1007/82_2013_333. [DOI] [PubMed] [Google Scholar]
- Jackowski S.; Rock C. O. (1986) Consequences of reduced intracellular coenzyme A content in Escherichia coli. J. Bacteriol. 166 (3), 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D. H.; Zhang Y.; Cronan J. E. Jr. (1996) The apparent coupling between synthesis and posttranslational modification of Escherichia coli acyl carrier protein is due to inhibition of amino acid biosynthesis. J. Bacteriol. 178 (9), 2662–2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating D. H.; Carey M. R.; Cronan J. E. (1995) The unmodified (apo) form of Escherichia coli acyl carrier protein is a potent inhibitor of cell growth. J. Biol. Chem. 270, 22229–22235. 10.1074/jbc.270.38.22229. [DOI] [PubMed] [Google Scholar]
- Leblanc C.; Prudhomme T.; Tabouret G.; Ray A.; Burbaud S.; Cabantous S.; Mourey L.; Guilhot C.; Chalut C. (2012) 4′-Phosphopantetheinyl transferase PptT, a new drug target required for Mycobacterium tuberculosis growth and persistence in vivo. PLoS Pathog. 8 (12), e1003097. 10.1371/journal.ppat.1003097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimhony O.; Schwarz A.; Raitses-Gurevich M.; Peleg Y.; Dym O.; Albeck S.; Burstein Y.; Shakked Z. (2015) AcpM, the meromycolate extension acyl carrier protein of Mycobacterium tuberculosis, is activated by the 4′-phosphopantetheinyl transferase PptT, a potential target of the multistep mycolic acid biosynthesis. Biochemistry 54 (14), 2360–2371. 10.1021/bi501444e. [DOI] [PubMed] [Google Scholar]
- Kaskow B. J.; Proffitt J. M.; Blangero J.; Moses E. K.; Abraham L. J. (2012) Diverse biological activities of the vascular non-inflammatory molecules – the Vanin pantetheinases. Biochem. Biophys. Res. Commun. 417 (2), 653–658. 10.1016/j.bbrc.2011.11.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras B.; Barra D.; Dupre S.; Pitari G. (1999) Is pantetheinase the actual identity of mouse and human vanin-1 proteins?. FEBS Lett. 461 (3), 149–152. 10.1016/S0014-5793(99)01439-8. [DOI] [PubMed] [Google Scholar]
- Shi W.; Chen J.; Feng J.; Cui P.; Zhang S.; Weng X.; Zhang W.; Zhang Y. (2014) Aspartate decarboxylase (PanD) as a new target of pyrazinamide in Mycobacterium tuberculosis. Emerging Microbes Infect. 3 (8), e58. 10.1038/emi.2014.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Chen J.; Shi W.; Liu W.; Zhang W.; Zhang Y. (2013) Mutations in panD encoding aspartate decarboxylase are associated with pyrazinamide resistance in Mycobacterium tuberculosis. Emerging Microbes Infect. 2 (6), e34. 10.1038/emi.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon N. A.; Peterson N. D.; Rosen B. C.; Baughn A. D. (2014) Pantothenate and pantetheine antagonize the antitubercular activity of pyrazinamide. Antimicrob. Agents Chemother. 58 (12), 7258–7263. 10.1128/AAC.04028-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal P.; Yee M.; Sarathy J.; Low J. L.; Sarathy J. P.; Kaya F.; Dartois V.; Gengenbacher M.; Dick T. (2016) Pyrazinamide resistance is caused by two distinct mechanisms: prevention of coenzyme a depletion and loss of virulence factor synthesis. ACS Infect. Dis. 2, 616–626. 10.1021/acsinfecdis.6b00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone J. D.; Yao J.; Scott N. E.; Dotson G. D. (2009) Selective inhibitors of bacterial phosphopantothenoylcysteine synthetase. J. Am. Chem. Soc. 131 (45), 16340–16341. 10.1021/ja906537f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V.; Brecik M.; Mukherjee R.; Evans J. C.; Svetlikova Z.; Blasko J.; Surade S.; Blackburn J.; Warner D. F.; Mikusova K.; Mizrahi V. (2015) The complex mechanism of antimycobacterial action of 5-fluorouracil. Chem. Biol. 22 (1), 63–75. 10.1016/j.chembiol.2014.11.006. [DOI] [PubMed] [Google Scholar]
- Schubert O. T.; Mouritsen J.; Ludwig C.; Rost H. L.; Rosenberger G.; Arthur P. K.; Claassen M.; Campbell D. S.; Sun Z.; Farrah T.; Gengenbacher M.; Maiolica A.; Kaufmann S. H.; Moritz R. L.; Aebersold R. (2013) The Mtb proteome library: a resource of assays to quantify the complete proteome of Mycobacterium tuberculosis. Cell Host Microbe 13 (5), 602–612. 10.1016/j.chom.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar M.; Prosser G. A.; de Carvalho L. P.; Rhee K. (2015) Metabolomics of Mycobacterium tuberculosis. Methods Mol. Biol. 1285, 105–115. 10.1007/978-1-4939-2450-9_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.