

Abstract
Oligoflexus tunisiensis Shr3T is the first strain described in the newest (eighth) class Oligoflexia of the phylum Proteobacteria. This strain was isolated from the 0.2-μm filtrate of a suspension of sand gravels collected in the Sahara Desert in the Republic of Tunisia. The genome of O. tunisiensis Shr3T is 7,569,109 bp long and consists of one scaffold with a 54.3% G + C content. A total of 6,463 genes were predicted, comprising 6,406 protein-coding and 57 RNA genes. Genome sequence analysis suggested that strain Shr3T had multiple terminal oxidases for aerobic respiration and various transporters, including the resistance-nodulation-cell division-type efflux pumps. Additionally, gene sequences related to the incomplete denitrification pathway lacking the final step to reduce nitrous oxide (N2O) to nitrogen gas (N2) were found in the O. tunisiensis Shr3T genome. The results presented herein provide insight into the metabolic versatility and N2O-producing activity of Oligoflexus species.
Keywords: Oligoflexia, Proteobacteria, RND-type efflux pump, Denitrification, Nitrous oxide (N2O)
Introductions
The phylum Proteobacteria traditionally comprises five classes of Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Deltaproteobacteria and Epsilonproteobacteria [1, 2], with two additional classes ‘Zetaproteobacteria’ and Acidithiobacillia proposed by Emerson et al. [3] and Williams and Kelly [4], respectively. Proteobacteria hosts the greatest number of isolates and sequenced genomes among the prokaryotic phyla [5] and contains members exhibiting extremely diversified metabolisms relevant to global carbon, nitrogen, and sulfur cycles [2]. This phylum recently gained the eighth (or seventh if yet-to-be-validated ‘Zetaproteobacteria’ is excluded) class Oligoflexia with the cultured species Oligoflexus tunisiensis type strain Shr3T [6]. The class Oligoflexia includes environmentally-derived 16S rRNA gene sequences, otherwise known as environmental clones or phylotypes, recovered from a variety of habitats including soils, the Taklamakan Desert, glacial ice, lake water, seawater, human skin, and the guts of earthworms [6]. In contrast to their wide distribution, Oligoflexia-affiliated clones have rarely been found in clone libraries [7]; accordingly, it has been suggested that the Oligoflexia members show a small population size, belonging to the so-called rare biosphere [8].
At the time of writing, O. tunisiensis Shr3T was the only cultured species within the class Oligoflexia. Physiological and biochemical features of strain Shr3T could not be fully characterized because of restrictive culture conditions owing to the slow-growing nature of this strain [6]. The phenotypic information is essential for understanding its ecological role and biotechnological potentials. Here, we compensated for the limited knowledge regarding Oligoflexia members by conducting genomic analysis of strain Shr3T.
Organism information
Classification and features
During a study of ultramicro-sized bacteria that could pass through 0.2-μm pore-size filters, which are generally used for sterile filtration to remove microorganisms, we isolated the bacterium designated isolate Shr3 [9]. The isolation source of this bacterium was a 0.2-μm filtrate of the suspension of sand gravels collected in December 2008 in Matmata (33° 31’ N 9° 57’ E) on the eastern margin of the Sahara Desert in the Republic of Tunisia. Isolate Shr3 was thereafter described as the type strain of Oligoflexus tunisiensis, the first cultured representative of the novel class Oligoflexia [6].
Figure 1 shows the phylogenetic position of O. tunisiensis and related environmental clones in a 16S rRNA-based evolutionary tree. The sequence of the three 16S rRNA gene copies in the genome was 100% identical to the previously published 16S rRNA gene sequence (DDBJ/EMBL/GenBank accession no. AB540021 [6]). The database search showed that seven environmental clones had a >97% high similarity with the O. tunisiensis 16S rRNA gene sequence [7]. The seven clones were from rice paddy soil, cyanobacterial blooms in a hypereutrophic lake, a microalgal photobioreactor, a bio-filter, and human skin [7]. Strain Shr3T has been deposited in the Japan Collection of Microorganisms and the National Collection of Industrial, food and Marine Bacteria under accession numbers JCM 16864T and NCIMB 14846T, respectively. The general features of strain Shr3T are reported in Table 1.
Fig. 1.
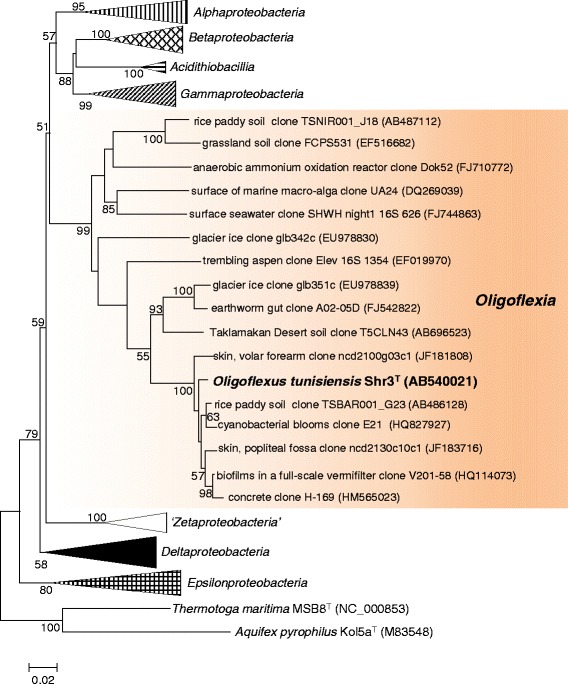
Phylogenetic relationships between O. tunisiensis Shr3T and related environmental clones in the phylum Proteobacteria based on 16S rRNA gene sequences. At the time of writing, strain Shr3T was the only cultured species within the class Oligoflexia. The tree, generated with MEGA 6.0 [34] using the neighbor-joining method [35], is based on a comparison of approximately 1130 nucleotides. Bootstrap values >50%, expressed as percentages of 1000 replicates, are shown above and below branches. Bar: 0.02 substitutions per nucleotide position
Table 1.
Classification and general features of Oligoflexus tunisiensis type strain Shr3T according to MIGS standards [30]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [31] | |
| Phylum Proteobacteria | TAS [32] | ||
| Class Oligoflexia | TAS [6] | ||
| Order Oligoflexales | TAS [6] | ||
| Family Oligoflexaceae | TAS [6] | ||
| Genus Oligoflexus | TAS [6] | ||
| Species Oligoflexus tunisiensis | TAS [6] | ||
| Type strain: Shr3T | TAS [6] | ||
| Gram stain | negative | TAS [6] | |
| Cell shape | filamentous-shaped | TAS [6, 7] | |
| Motility | non-motile | TAS [6] | |
| Sporulation | none | TAS [6] | |
| Temperature range | 20–37 °C | TAS [6] | |
| Optimum temperature | 25–30 °C | TAS [6] | |
| pH range; Optimum | 7.0–9.5; 7.0–8.0 | TAS [6] | |
| Carbon source | heterotrophic | TAS [6] | |
| MIGS-6 | Habitat | desert | TAS [6] |
| MIGS-6.3 | Salinity | 0–0.5% (w/v) NaCl | TAS [6] |
| MIGS-22 | Oxygen requirement | aerobic | TAS [6] |
| MIGS-15 | Biotic relationship | free-living | TAS [6] |
| MIGS-14 | Pathogenicity | not reported | |
| MIGS-4 | Geographic location | Matmata, Republic of Tunisia | TAS [6] |
| MIGS-5 | Sample collection | December 2008 | TAS [6] |
| MIGS-4.1 | Latitude | 33.53 | TAS [6] |
| MIGS-4.2 | Longitude | 9.96 | TAS [6] |
| MIGS-4.4 | Altitude | not determined |
aEvidence codes – IDA: Inferred from Direct Assay; TAS: Traceable Author Statement (i.e., a direct report exists in the literature); NAS: Non-traceable Author Statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [33]
O. tunisiensis Shr3T is a Gram-negative, aerobic, non-motile, filamentous bacterium of 0.4–0.8 μm in width when cultivated under the experimental culture conditions [6]. Some cells exhibited a spiral, spherical (or curled), or curved rod morphology [7]. Although the factors controlling the cell shapes are still unclear, the morphological flexibility is likely associated with their ability to pass through 0.2-μm filters. Strain Shr3T grows in the R2A medium [6]. The cells showed slow growth, with 3–5 days required before colonies could be seen by the naked eye [6]. The growth occurs at NaCl concentrations <1.0% (w/v), 20–37 °C (optimum 25–30 °C), and pH 7.0–9.5 (optimum pH 7.0–8.0) [6]. Enzyme activities of esterase lipase, leucine arylamidase, trypsin, naphthol-AS-BI-phosphohydrolase and α-mannosidase are positive [6]. Transmission electron microscopy revealed that cells contained many low electron-dense particles (Fig. 2). Some, but not all, particles were stained by Sudan black B upon staining PHB or lipophilic particles. Because cells swollen by accumulated PHB were not observed when grown on PHB-containing medium [6], the particles stained with Sudan black B are likely lipophilic granules.
Fig. 2.
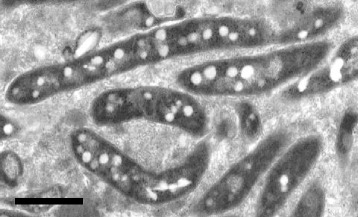
Transmission electron micrograph of O. tunisiensis Shr3T. Many low electron-density particles were observed. Cells were grown on R2A medium for 7 days at 25 °C. Scale: 1 μm
Chemotaxonomy
The major respiratory quinone was menaquinone-7 (MK-7) [6]. The dominant cellular fatty acids were C16 : 1 ω5c (65.7%) and C16 : 0 (27.5%), the major hydroxy fatty acid was C12 : 0 3-OH (1.3%), and the minor fatty acids included C10:0, C12:0, C15:0, C17:0, C18:0 and C18:1 ω5c [6]. The fatty acid, C16 : 1 ω5c, was also detected in myxobacteria of Cystobacterineae in the class Deltaproteobacteria, but at only 15–39% [10].
Genome sequencing information
Genome project history
Phenotypic features of strain Shr3T are described above, but could not be fully tested because of restrictive culture conditions [6]. Therefore, this organism was selected for genome sequencing to investigate the basis of its ecological role and biotechnological potentials. The genome project is deposited in the Genomes OnLine Database [11] under the accession number Gp0139475. The information genome sequence is available from the DDBJ/EMBL/GenBank database. A summary of this genome project is shown in Table 2.
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | High-quality draft |
| MIGS-28 | Libraries used | Pair-end library and mate-pair library |
| MIGS 29 | Sequencing platforms | Illumina HiSeq 2000 |
| MIGS 31.2 | Fold coverage | 149 × |
| MIGS 30 | Assemblers | SOAPdenovo version 2.04 |
| MIGS 32 | Gene calling method | Prodigal |
| Locus Tag | Ga0118670 (IMG-ER) | |
| GenBank ID | BDFO01000001 | |
| GenBank Date of Release | 30 June 2016 | |
| GOLD ID | Gp0139475 | |
| BIOPROJECT | PRJDB4872 | |
| MIGS 13 | Source Material Identifier | JCM 16864, NCIMB 14846 |
| Project relevance | ecology, biotechnology |
Growth conditions and genomic DNA preparation
A culture of O. tunisiensis Shr3T grown aerobically in R2A broth (DAIGO; Nihon Pharmaceutical Co., Ltd., Tokyo, Japan) at 30 °C was used to prepare genomic DNA. The genomic DNA was extracted using Qiagen Genomic-Tip 500/G columns according to the manufacturer’s instructions. The quantity and purity of the extracted DNA was checked by spectrophotometric measurement at 260 nm and agarose gel electrophoresis.
Genome sequencing and assembly
The genome sequence was generated using paired-end sequencing (2 × 90 bp) on an Illumina HiSeq 2000 platform at the BGI with the pair-end library and mate-pair library of two different insert sizes, 456 to 496 bp and 6310 to 6350 bp. After trimming of low quality reads, 1130 Mb was obtained and assembled into 19 contigs in one scaffold using SOAPdenovo version 2.04 [12]. The assembly result was locally optimized according to the paired-end and overlap relationship via mapping reads to obtained contigs. A summary of this genome sequence is shown in Table 3.
Table 3.
Genome statistics
| Attribute | Value | % of Total |
|---|---|---|
| Genome size (bp) | 7,569,109 | 100.00 |
| DNA coding (bp) | 6,849,121 | 90.49 |
| DNA G + C (bp) | 4,113,347 | 54.34 |
| DNA scaffolds | 1 | 100.00 |
| Total genes | 6,463 | 100.00 |
| Protein coding genes | 6,406 | 99.12 |
| RNA genes | 57 | 0.88 |
| Pseudogenes | not determined | not determined |
| Genes in internal clusters | 1,494 | 23.12 |
| Genes with function prediction | 4,051 | 62.68 |
| Genes assigned to COGs | 2,938 | 45.46 |
| Genes with Pfam domains | 4,268 | 66.04 |
| Genes with signal peptides | 1,084 | 16.77 |
| Genes with transmembrane helices | 1,393 | 21.55 |
| CRISPR repeats | 8 |
Genome annotation
Gene sequences were identified via the Prodigal V2.6.3 [13] as part of the DOE-JGI genome annotation pipeline in the Integrated Microbial Genomes–Expert Review (IMG-ER) system [14]. Gene functional annotation as well as data visualization was conducted within the IMG-ER [15]. The predicted coding sequences were translated and used to search the National Center for Biotechnology Information non-redundant, UniProt, TIGR-Fam, Pfam, KEGG, COG, and InterPro databases. Identification of RNA gene sequences and miscellaneous features were carried out using HMMER 3.1b2 [16] and INFERNAL 1.0.2 and 1.1.1 [17]. Additional functional prediction was performed with the RAST server [18] under accession number 708132.3. Candidate CRISPR regions were detected using the CRISPRFinder program [19].
Genome Properties
The genome of O. tunisiensis Shr3T consists of a 7,569,109 bp long chromosome with a 54.3% G + C content (Table 3). Of the 6463 predicted genes, 6406 were protein-coding genes and 57 were RNA genes (three rRNA operons, 46 tRNAs, and two miscRNAs). The majority of the protein-coding genes (62.7%) were assigned to a putative function. The remaining ones were annotated as hypothetical proteins. The distribution of genes classified into COGs functional categories is shown in Table 4 and Fig. 3.
Table 4.
Number of genes associated with general COG functional categories
| Code | Value | %age | Description |
|---|---|---|---|
| J | 228 | 6.91 | Translation, ribosomal structure and biogenesis |
| A | 1 | 0.03 | RNA processing and modification |
| K | 154 | 4.67 | Transcription |
| L | 103 | 3.12 | Replication, recombination and repair |
| B | 1 | 0.03 | Chromatin structure and dynamics |
| D | 31 | 0.94 | Cell cycle control, Cell division, chromosome partitioning |
| V | 87 | 2.64 | Defense mechanisms |
| T | 314 | 9.52 | Signal transduction mechanisms |
| M | 229 | 6.94 | Cell wall/membrane biogenesis |
| N | 125 | 3.79 | Cell motility |
| U | 44 | 1.33 | Intracellular trafficking and secretion |
| O | 159 | 4.82 | Posttranslational modification, protein turnover, chaperones |
| C | 172 | 5.21 | Energy production and conversion |
| G | 142 | 4.30 | Carbohydrate transport and metabolism |
| E | 264 | 8.00 | Amino acid transport and metabolism |
| F | 73 | 2.21 | Nucleotide transport and metabolism |
| H | 170 | 5.15 | Coenzyme transport and metabolism |
| I | 193 | 5.85 | Lipid transport and metabolism |
| P | 156 | 4.73 | Inorganic ion transport and metabolism |
| Q | 100 | 3.03 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 337 | 10.22 | General function prediction only |
| S | 153 | 4.64 | Function unknown |
| - | 3,525 | 54.54 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Fig. 3.
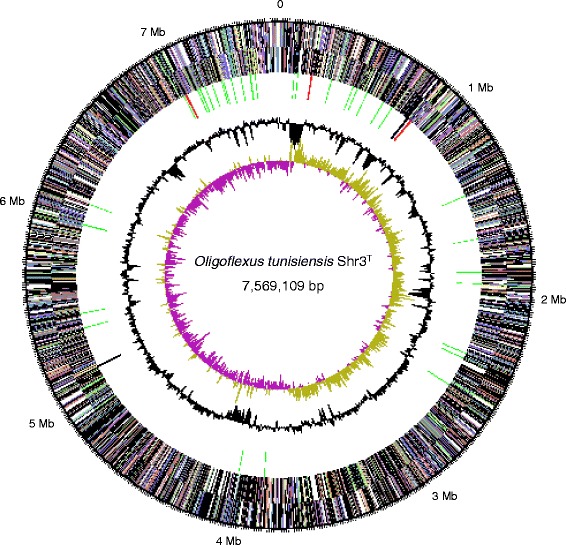
Graphical circular map of the chromosome of O. tunisiensis Shr3T. From outside to the center: genes on forward strand (color by COG categories), genes on reverse strand (color by COG categories), RNA genes (tRNAs green, rRNAs red, other RNAs black), GC content, GC skew
Insights from the genome sequence
The genome of O. tunisiensis Shr3T encoded genes for ABC transporters of amino acid, oligopeptide/dipeptide, and phosphonate, ammonium and nitrate/nitrite transporters, as well as RND-type efflux pumps. One of the amino acid sequences (Ga0118670_114686) classified as an RND pump showed a high similarity (67% identity and 99% coverage) to sequences of the pathogenic bacteria Achromobacter xylosoxidans and Pseudomonas aeruginosa. The RND-type efflux system is widely distributed in Gram-negative bacteria and known to promote resistance to various kinds of antimicrobial substances, termed as multidrug resistance [20].
In support of its aerobic growth, gene sequences assigned to different terminal oxidases including aa 3- and cbb 3-type cytochrome c oxidases (COG0843 and COG3278) and cytochrome bd-type quinol oxidase (COG1271 and COG1294) were found in the Shr3T genome.
The Shr3T genome contained a nirK gene coding for a copper-dependent nitrite reductase (Nir) (Ga0118670_114712) involved in denitrification, a major component of the nitrogen cycle [21]. Denitrification is the dissimilatory reduction of nitrate or nitrite to nitrogen gas (NO3 − → NO2 − → NO → N2O → N2) [22] that usually occurs under oxygen-limiting conditions [21]. The key steps releasing gaseous products NO, N2O, and N2 are catalyzed by Nir, nitric-oxide reductase (Nor) and nitrous oxide reductase (Nos), respectively [23, 24]. There are two structurally different nitrite reductases among denitrifiers: a copper-containing type (Cu-Nir) encoded by the nirK gene and a cytochrome cd 1-containg one (cd 1-Nir) encoded by the nirS gene [24]. The nirS gene was absent from the O. tunisiensis Shr3T genome.
The NirK deduced amino acid sequence of O. tunisiensis Shr3T was most closely related to that of Bdellovibrio bacteriovorus of the class Deltaproteobacteria, with 70% identity and 96% coverage. B. bacteriovorus has an incomplete denitrifying pathway with a Cu-Nir, a cytochrome c–dependent Nor (cNor), and no Nos [25, 26]. O. tunisiensis Shr3T also had a partial pathway containing the Cu-Nir described above, a quinol-dependent Nor (qNor), and no Nos inferred from the genome data. Strain Shr3T has two copies of the gene encoding qNor (Ga0118670_112818 and Ga0118670_114769). NorR protein is known to regulate Nor expression in response to NO [27, 28]. The transcription regulator norR gene (Ga0118670_114771) was nearly adjacent to one of two copies of the qNor-encoding gene in the genome.
Our results suggest that the Oligoflexus species has the capability to produce N2O as a final product of the incomplete denitrification lacking the last step (reduction of N2O to N2). N2O is known as a strong greenhouse gas, as well as an ozone-depleting substance [29]. Accordingly, future studies should examine the N2O-producing phenotype of strain Shr3T.
Conclusions
In this study, we characterized the genome of O. tunisiensis Shr3T, the first cultured representative of the novel proteobacterial class Oligoflexia. The genome sequence gives us insight into the metabolic versatility and incomplete denitrification pathway of Oligoflexus species. The genome information will facilitate future systematics and comparative genomics studies within the phylum Proteobacteria.
Acknowledgements
The computations for this work were partially performed on the NIG supercomputer at the ROIS National Institute of Genetics.
Funding
This work was supported by a Grant for Basic Science Research Projects from the Sumitomo Foundation (no. 130894 to RN). RN was supported by a Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Young Scientists (no. JP13J03441). This work was partially funded by a JSPS Grant-in-Aid for Young Scientists (A) (no. JP15H05620 to RN).
Authors’ contributions
RN coordinated the study, annotated the genome and drafted the manuscript. RN, FK, SS, HI, and TN maintained and cultured the strain. RN conducted the wet-lab work, MN performed the electron microscopy. RN, TF, YN, TB, and HN discussed the bioinformatics analysis. RN, TB, MN, TN, and HN discussed the data. All authors read and approved the final manuscript.
Competing interests
None of the authors has any competing interests.
Abbreviations
- PHB
Polyhydroxybutyrate
- RND
Resistance-nodulation-cell division
References
- 1.Gupta RS. The phylogeny of proteobacteria: relationships to other eubacterial phyla and eukaryotes. FEMS Microbiol Rev. 2000;24:367–402. doi: 10.1111/j.1574-6976.2000.tb00547.x. [DOI] [PubMed] [Google Scholar]
- 2.Kerters K, De Vos P, Gillis M, Swings J, Vandamme P, Stackebrandt E. Introduction to the Proteobacteria. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The Prokaryotes, A Handbook on the Biology of Bacteria, Volume 5: Proteobacteria: Alpha and Beta Subclasses. 3. New York: Springer; 2006. pp. 3–37. [Google Scholar]
- 3.Emerson D, Rentz JA, Lilburn TG, Davis RE, Aldrich H, Chan C, Moyer CL. A novel lineage of proteobacteria involved in formation of marine Fe-oxidizing microbial mat communities. PLoS One. 2007;2:e667. doi: 10.1371/journal.pone.0000667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Williams KP, Kelly DP. Proposal for a new class within the phylum Proteobacteria, Acidithiobacillia classis nov., with the type order Acidithiobacillales, and emended description of the class Gammaproteobacteria. Int J Syst Evol Microbiol. 2013;63:2901–6. doi: 10.1099/ijs.0.049270-0. [DOI] [PubMed] [Google Scholar]
- 5.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature. 2013;499:431–7. doi: 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 6.Nakai R, Nishijima M, Tazato N, Handa Y, Karray F, Sayadi S, et al. Oligoflexus tunisiensis gen. nov., sp. nov., a Gram-negative, aerobic, filamentous bacterium of a novel proteobacterial lineage, and description of Oligoflexaceae fam. nov., Oligoflexales ord. nov. and Oligoflexia classis nov. Int J Syst Evol Microbiol. 2014;64:3353–9. doi: 10.1099/ijs.0.060798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakai R, Nagamura T. Oligoflexia, the newest class of the phylum Proteobacteria, consisting of only one cultured species and uncultured bacterial phylotypes in diverse habitats. J Phylogen Evol Biol. 2015;3:141. [Google Scholar]
- 8.Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc Natl Acad Sci U S A. 2006;103:12115–20. doi: 10.1073/pnas.0605127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakai R, Shibuya E, Justel A, Rico E, Quesada A, Kobayashi F, et al. Phylogeographic analysis of filterable bacteria with special reference to Rhizobiales strains that occur at cryospheric habitats. Antarct Sci. 2013;25:219–28. doi: 10.1017/S0954102012000831. [DOI] [Google Scholar]
- 10.Shimkets LJ, Dworkin M, Reichenbach H. The Myxobacteria. In: Dworkin M, Falkow S, Rosenberg E, Schleifer K-H, Stackebrandt E, editors. The Prokaryotes, A Handbook on the Biology of Bacteria, Volume 7: Proteobacteria: Delta and Epsilon Subclasses. Deeply Rooting Bacteria. 3. New York: Springer; 2006. pp. 3–37. [Google Scholar]
- 11.Liolios K, Chen IM, Mavromatis K, Tavernarakis N, Hugenholtz P, Markowitz VM, Kyrpides NC. The Genomes On Line Database (GOLD) in 2009: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res. 2010;38:D346–54. doi: 10.1093/nar/gkp848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;1:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mavromatis K, Ivanova NN, Chen IM, Szeto E, Markowitz VM, Kyrpides NC. The DOE-JGI standard operating procedure for the annotations of microbial genomes. Stand Genomic Sci. 2009;1:63–7. doi: 10.4056/sigs.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–8. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- 16.Finn DR, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25:1335–7. doi: 10.1093/bioinformatics/btp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genomics. 2008;9:75–89. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–7. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poole K. Multidrug resistance in Gram-negative bacteria. Curr Opin Microbiol. 2001;4:500–508. doi: 10.1016/S1369-5274(00)00242-3. [DOI] [PubMed] [Google Scholar]
- 21.Canfield DE, Glazer AN, Falkowski PG. The evolution and future of Earth’s nitrogen cycle. Science. 2010;330:192–6. doi: 10.1126/science.1186120. [DOI] [PubMed] [Google Scholar]
- 22.Knowles R. Denitrification. Microbiol Rev. 1982;46:43–70. doi: 10.1128/mr.46.1.43-70.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraft B, Strous M, Tegetmeyer HE. Microbial nitrate respiration-genes, enzymes and environmental distribution. J Biotechnol. 2011;155:104–17. doi: 10.1016/j.jbiotec.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 24.Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev. 1997;61:533–616. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rendulic S, Jagtap P, Rosinus A, Eppinger M, Baar C, Lanz C, et al. A predator unmasked: life cycle of Bdellovibrio bacteriovorus from a genomic perspective. Science. 2004;303:689–92. doi: 10.1126/science.1093027. [DOI] [PubMed] [Google Scholar]
- 26.Shapleigh JP. Denitrifying Prokaryotes. In: Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F, editors. The Prokaryotes–Prokaryotic Physiology and Biochemistry. 4. Berlin Heidelberg: Springer; 2013. pp. 405–25. [Google Scholar]
- 27.Pohlmann A, Cramm R, Schmelz K, Friedrich B. A novel NO-responding regulator controls the reduction of nitric oxide in Ralstonia eutropha. Mol Microbiol. 2000;38:626–38. doi: 10.1046/j.1365-2958.2000.02157.x. [DOI] [PubMed] [Google Scholar]
- 28.Spiro S. Regulators of bacterial responses to nitric oxide. FEMS Microbiol Rev. 2007;31:193–211. doi: 10.1111/j.1574-6976.2006.00061.x. [DOI] [PubMed] [Google Scholar]
- 29.Ravishankara AR, Daniel JS, Portmann RW. Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century. Science. 2009;326:123–5. doi: 10.1126/science.1176985. [DOI] [PubMed] [Google Scholar]
- 30.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87:4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrity GM, Bell JA, Phylum XIV LT. Proteobacteria phyl. nov. In: Brenner DJ, Krieg NR, Staley JT, Garrity GM, editors. Bergey’s Manual of Systematic Bacteriology, second edition, vol. 2 (The Proteobacteria), part B (The Gammaproteobacteria) New York: Springer; 2005. p. 1. [Google Scholar]
- 33.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–25. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]