

Abstract
Objective
Although hypertension is the most common risk factor for thoracic aortic diseases, it is not understood how increased pressures on the ascending aorta lead to aortic aneurysms. We investigated the role of Ang II type 1 (AT1) receptor activation in ascending aortic remodeling in response to increased biomechanical forces using a transverse aortic constriction (TAC) mouse model.
Approach and Results
Two weeks after TAC, the increased biomechanical pressures led to ascending aortic dilatation, aortic wall thickening and medial hypertrophy. Significant adventitial hyperplasia and inflammatory responses in TAC ascending aortas were accompanied by increased adventitial collagen, elevated inflammatory and proliferative markers, and increased cell density due to accumulation of myofibroblasts and macrophages. Treatment with losartan significantly blocked TAC induced vascular inflammation and macrophage accumulation. However, losartan only partially prevented TAC induced adventitial hyperplasia, collagen accumulation and ascending aortic dilatation. Increased Tgfb2 expression and phosphorylated-Smad2 staining in the medial layer of TAC ascending aortas was effectively blocked with losartan. In contrast, the increased Tgfb1 expression and adventitial phospho-Smad2 staining were only partially attenuated by losartan. In addition, losartan significantly blocked Erk activation and ROS production in the TAC ascending aorta.
Conclusions
Inhibition of the AT1 receptor using losartan significantly attenuated the vascular remodeling associated with TAC but did not completely block the increased TGF- β1 expression, adventitial Smad2 signaling and collagen accumulation. These results help to delineate the aortic TGF-β signaling that is dependent and independent of the AT1 receptor after TAC.
Keywords: Transverse aortic constriction, thoracic aortic aneurysms and dissections, AT1 receptor, Transforming growth factor-β, mouse model
Thoracic aortic aneurysms leading to acute aortic dissections (TAAD) are a common cause of premature deaths in United States.1 Hypertension is the most common risk factor for thoracic aortic disease. More than 75% of patients with thoracic aortic aneurysms or acute aortic dissections have hypertension.2 Despite the common association of hypertension and thoracic aortic disease, it is not understood how increased pressures on the ascending aorta lead to TAAD.
An established model system to study aortic disease is acute angiotension II (Ang II) infusion in mice, which leads to aortic disease characterized by aortic aneurysm formation and subsequent dissection/rupture primarily in the suprarenal aorta but also in the ascending thoracic aorta.3, 4 With Ang II infusion, the medial layer of the ascending thoracic aorta thickens with expansion between the elastic lamellae.4 Additionally, the adventitial fibroblasts secrete proinflammatory cytokines, including IL-6 and MCP-1, which recruit monocytes to the aortic wall, which further activate fibroblast proliferation and myofibroblast activation, adventitial thickening, and additional cytokine production.5
Another established mouse model of thoracic aortic aneurysms is the Marfan syndrome (MFS) mouse, which has been engineered to harbor an Fbn1 missense mutation. Studies in this mouse model have suggested that promiscuous activation of transforming growth factor-β (TGF-β) from stores in the microfibrils in the extracellular matrix (ECM) drives aortic aneurysm formation6. Blocking TGF-β signaling using a TGF-β neutralizing antibody or an Ang II type 1 receptor (AT1) blocking agent, losartan, prevented aneurysms in the MFS mouse model.6
Transverse aortic constriction (TAC), created by contracting the aortic lumen between the innominate artery and the left carotid artery, is a common experimental method to induce pressure overload cardiac hypertrophy and heart failure. While TAC initially leads to compensatory hypertrophy of the heart, over time the response to the chronic hemodynamic overload becomes maladaptive, resulting in cardiac dilatation and heart failure.7, 8 Although studies have shown increased aortic diameter and thickening of the aortic wall, the molecular signaling pathways responsible for this remodeling have not been identified. Evidence of increased TGF-β signaling has been suggested by increased phosphorylated (p) Smad2 levels in medial SMCs. Parallel activation of extracellular signal–regulated kinase (ERK1/2) signaling is also present in this model. Inhibition of both TGF-β and ERK1/2 signaling decreased the extent of aortic dilatation with TAC.8
Since Ang II can activate ERK1/2 signaling and increase expression of TGF-β in vascular SMCs, we sought to determine if signaling through the AT1 receptor was responsible for both the increased TGF-β and ERK1/2 signaling in the ascending aorta of the TAC mouse model8. Our results indicate that inhibition of the AT1 receptor using losartan significantly attenuated the vascular remodeling associated with TAC but did not completely block the increased TGF- β1 expression, adventitial Smad2 signaling and collagen accumulation. Thus, these results help to delineate aortic TGF-β signaling that is dependent and independent of the AT1 receptor after TAC.
Results
TAC induces ascending aortic dilatation and remodeling of the aortic wall
Twelve weeks old C57BL ⁄ 6 male mice were subjected to an established TAC protocol (Supplemental Figure IA) that reproducibly causes left ventricular hypertrophy.9 Doppler analysis performed 1 week after TAC demonstrated that the right carotid artery flow velocity ratio was 5–10-fold higher than the left carotid artery9. Echocardiography identified an increase in the ascending aortic diameter proximal to the aortic band of 23% (± 2.5%) in TAC mice compared with sham operated mice (referred to as controls), similar to the dilatation observed in previous studies (Figure 1A).8
Figure 1. Transverse aortic constriction (TAC) induces ascending aortic dilatation and structural remodeling in mice.
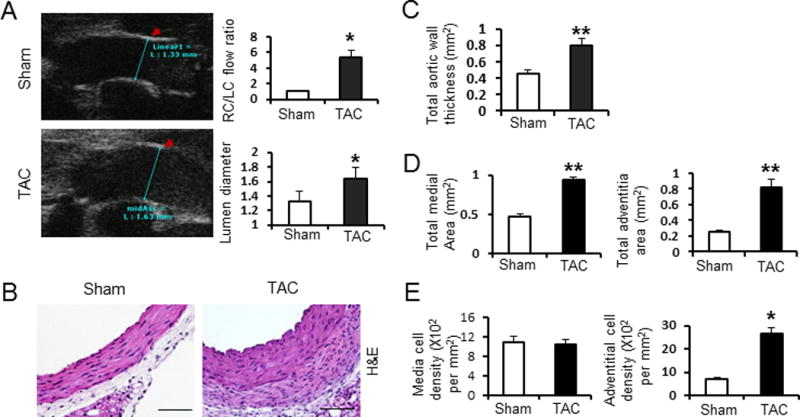
A. Representative echo-image of the ascending aorta (measurement indicated by blue lines). Right top bar graph shows the flow velocity ratio of the right carotid artery (RC) versus the left carotid artery (LC) 1 week after TAC. The mean diameter of the ascending aortic lumen at 2 weeks after TAC is also shown. Sham: Sham-operated; TAC: transverse aortic constriction for 2 weeks. These recordings demonstrate a successful TAC was achieved. B. Haematoxylin and eosin (H&E) staining of representative cross sections of mouse ascending aorta from mice 2 weeks after sham-operation or TAC at 200x magnification. C. Bar graphs show total aortic wall thickness. D. Bar graphs show total medial area (left) and total adventitial area (right). E. Bar graphs show total medial cell density (left) and total adventitial cell density (right). Ascending aortic sections were from mice 2 weeks after sham-operated (white bar) or TAC (black bar). n = 5 per group. *, P<0.05. **, P<0.01.
Aortas from the TAC and control mice were harvested with all three layers of the aorta preserved (intima, media, and adventitia). Microscopic examination of these aortas revealed no evidence of dissecting aneurysms or intramural thrombus. At two weeks after TAC, we measured a significant increase in the thickness of both the adventitia and media of the ascending aorta compared with control aortas (0.8±0.1 versus 0.42± 0.1, P<0.01, Figure 1B and 1C). Histomorphometric analysis confirmed increased medial and adventitial area (P < 0.05; Figure 1D). The cell density (total cell number divided by total area) did not significantly change in the medial layer, whereas the adventitial layer had a significant increase in cell density (Figure 1E). To further analyze the aortic remodeling with TAC, various components of the ascending aorta were further characterized. Verhoeff-van Gieson (VVG) staining of elastin showed no elastin fiber fragmentation with TAC (Figure 2A). The percent of elastin content in the medial layer was decreased with TAC, possibly due to the increased medial area or thinning of the elastin fibers (P<0.05; Figure 2B). Sirius red staining identified increased collagen content in the ascending aorta at 2 weeks after TAC, which was most pronounced in the adventitial layer (P< 0.05; Figure 2C). Medial and adventitial thickness, as well as collagen and elastin content of the descending aorta, did not change after TAC (data not shown).
Figure 2. Transverse aortic constriction (TAC) induces ascending aortic dilatation and structural remodeling in mice.
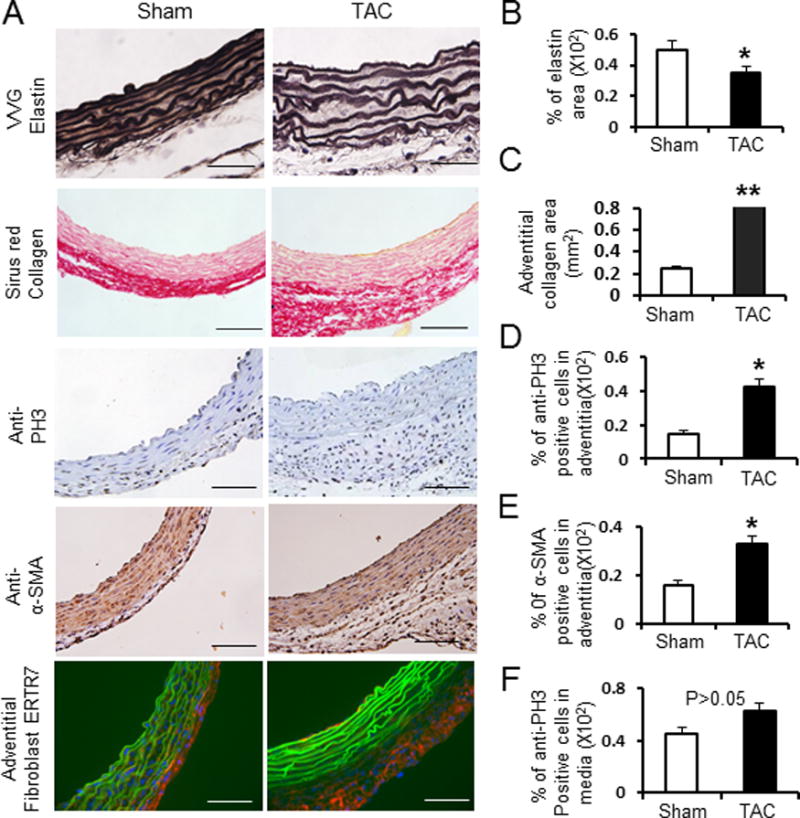
A. Representative cross sections of mouse ascending aorta from sham-operated and TAC mice with Verhoeff-van Gieson staining (VVG, for elastin), Sirius red staining (for collagen), anti-smooth muscle α-actin staining (α –SMA, for smooth muscle cells), anti-ERTR7 staining (for adventitial fibroblasts) and anti-Phospho-histone H3 staining (PH3), (for proliferative cells). B–F. bar-graphs show the quantification of percent elastin area, adventitial collagen area, percent anti-α-SMA positive cells in the adventitia, percent anti-PH3 positive cells in the adventitia and percentage of anti- anti-PH3 positive cells in the media. Ascending aortic sections were from sham-operated mice (white bar) and TAC mice for 2 weeks (black bar). n = 5 per group. *, P<0.05. **, P<0.01.
The cell density of adventitial layer in the ascending aorta increased after TAC when compared to controls. Immunostaining for the nuclear cell mitosis marker, phospho-histone H3 (Ser10) (PH3) confirmed increased number of proliferating cells in the adventitial layer after TAC (P< 0.05; Figure 2D). The number of cells that express α-smooth muscle actin (α-SMA), a marker for myofibroblasts, was also significantly increased in the adventitial layer after TAC (P < 0.05; Figure 2E). Immunofluorescence using an antibody to ERTR7, a marker for fibroblast, was significantly increased in the adventitia after TAC (Figure 2A bottom). However, the percentage of cells that stained for pH3 and α-SMA in the medial layer of the ascending aorta did not significantly change.(P > 0.05, Figure 2F and data not shown), suggesting that medial thickening was due to hypertrophy of the SMCs and/or accumulation of matrix proteins between the elastin layers. These results suggest that adventitial thickening after TAC is driven in part by myofibroblast proliferation and activation, along with collagen accumulation. It is notable that these histological changes with TAC are similar to the changes observed in the ascending aortic with Ang II infusion3, 4.
We have previously shown that Ang II infusion in mice leads to aortic production of IL-6 and MCP-1, which are cytokines that promote vascular inflammation and macrophage recruitment in cooperation with adventitial fibroblasts.3 Q-PCR analysis of RNA harvested from the ascending aortas of TAC and control mice showed a 10-fold increase in IL-6 expression and a 15-fold increase in MCP-1 expression in TAC aortas (P<0.05, Figure 3A and Supplemental Figure II). Immunostaining of aortic cross sections from the ascending aorta demonstrated that the levels of IL-6 or MCP-1 were significantly increased in the adventitial and medial layers of TAC aortas compared with controls (P<0.05, Figure 3B). Macrophage recruitment to the aortas was also confirmed through immunostaining with monoclonal antibodies to CD68 and MOMA2, and positively stained cells were detected predominantly in the adventitial layer (Figure 3C). Inflammatory cells produce proteases,5 and Mmp2 and Mmp9 expression was increased by 10- and 9-fold, respectively, in TAC mice as compared with controls (P<0.05, Figure 3D). Therefore, a local inflammatory response occurs in the ascending aorta after TAC that is similar to that observed with Ang II infusion.
Figure 3. TAC induced ascending aortic inflammatory changes without and with losartan treatment.
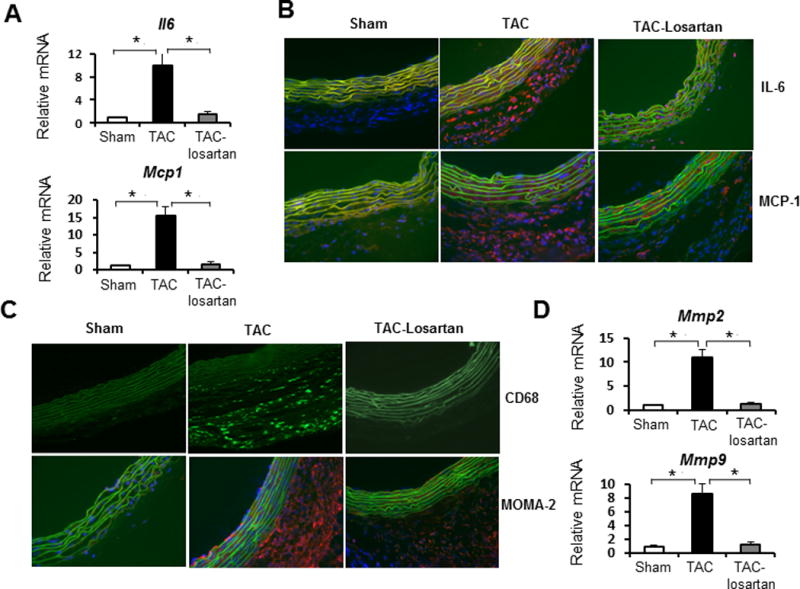
A. Q-PCR analysis of IL-6 and Mcp-1 expression in ascending aortic tissues from sham-operated (white bars), TAC for 2 weeks (black bars) and TAC mice treated with losartan for 2 weeks (grey bar). Gene expression levels were normalized to Gapdh. n = 5 per group, *, P<0.05. B and C. Immunofluorescence staining for IL-6, MCP-1(B); macrophage marker CD68 and MOMA-2 (C). Transverse cryosections (7 μm) of ascending aorta were prepared from sham-operated mice, TAC mice for 2 weeks and TAC mice treated with losartan for 2 weeks. Nuclei were stained with DAPI (blue). Elastic lamellae of the media are green (autofluorescence). Positive staining is red (Texas red-conjugated secondary antibody). Original magnification: 400X. D. Q-PCR analysis of expression levels of Mmp2 and Mmp9 in ascending aortic tissues from sham-operated (white bars), TAC mice (black bars), and TAC mice treated with losartan for 2 weeks (grey bar). Gene expression levels were normalized to Gapdh. n = 5 per group. *, P<0.05.
Inhibition of AT1 activation attenuates remodeling of the aorta with TAC but does not completely block TGF-β1 expression and adventitial Smad2 signaling
Since pressure-induced remodeling of the ascending aorta leads to histologic changes similar to those observed with Ang II infusion, we sought to test the hypothesis that this remodeling is driven by the activation of AT1 receptor by treating the TAC mice with the AT1 blocker losartan.6 The losartan was administered by oral at a dose of 0.6g/l in drinking water for 3 days prior to TAC and mice were continued on oral therapy for 2 weeks after TAC.6 Doppler analysis demonstrated that there was no statistical difference in arterial flow velocity ratios between TAC animals in the treatment or placebo groups (Figure 1A right top and data not shown). Echocardiograms also showed that systolic flow velocities in the ascending aortas of TAC mice were 4.9 to 5.3-fold higher than those of shan-operated mice. Systolic flow velocities in the ascending aortas of mice in the losartan treated and placebo groups were not significantly different (Supplemental Figure IB). Echocardiography revealed that losartan treatment attenuated ascending aortic dilatation associated with TAC (Figure 4, upper right side panel). Morphological and histological analysis revealed that losartan treatment also significantly attenuated ascending aortic wall thickness, medial hypertrophy, adventitial cellular hyperplasia and collagen accumulation but did not block these histologic changes completely (Figure 4). Losartan also significantly reduced the induction of Mcp1, IL6, Mmp2 and Mmp9 expression in TAC ascending aortas (Figure 3A, D; P<0.05). Inflammatory responses and macrophage recruitment, as determined by anti-IL6, MCP1, CD68 and MOMA2 staining, was also dramatically decreased in the adventitial layer of the ascending aorta after losartan treatment (Figure 3B and 3C). These data indicate that the inflammatory response after TAC was effectively inhibited when AT1 receptor activation was blocked with losartan. However, losartan did not completely inhibit aortic enlargement or medial and adventitial thickening.
Figure 4. Losartan attenuates TAC induced ascending aortic medial and adventitial remodeling.
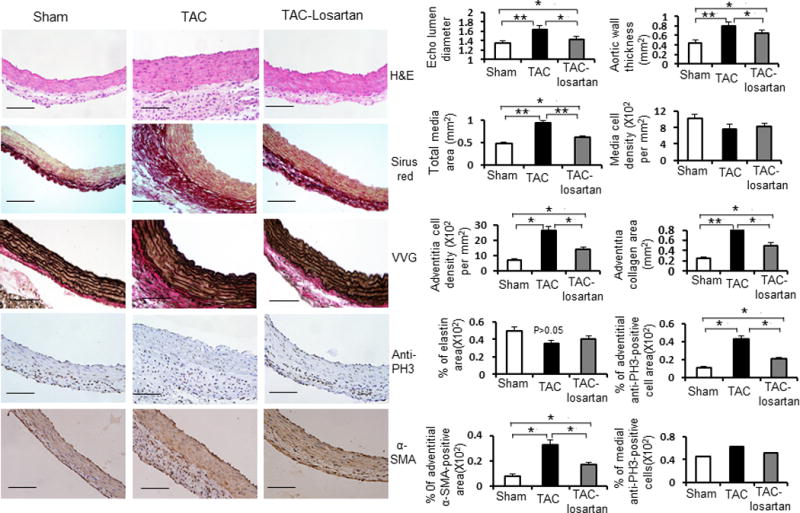
Left side: Representative cross sections of ascending aortas from sham-operated, TAC mice for 2 weeks, and TAC mice treated with losartan for 2 weeks by H&E staining, Sirius red staining for collagens, Verhoeff-van Gieson (VVG) staining for elastin, anti-phospho-histone H3 (PH3) staining for proliferative cells and anti-smooth muscle α-actin (α–SMA) staining for smooth muscle cells separately. Right side: Bar graph show mean ascending aortic lumen diameters of sham-operated mice (white bars), TAC mice (black bars) and TAC mice treated with losartan for 2 weeks (gray bars) (n = 5 per group). Bar graphs show quantification of aortic wall thickness, total media area, medial cell density, adventitial cell density, adventitial collagen area, percent elastin area, percent adventitial anti-PH3 positive area, percent medial anti-PH3 positive area and percent adventitial anti-α-SMA positive area from sham-operated mice (white bars), TAC mice (black bars) and TAC mice treated with losartan for 2 weeks (gray bars). n = 5 per group. *, P<0.05. **, P<0.01.
Since losartan treatment with TAC attenuated but did not completely prevent adventitial hyperplasia and collagen accumulation, we sought to determine if TGF-β signaling independent of AT1 activation could be responsible for these changes. Tgfb2 (TGF-β2) expression increased with TAC, and this increase was effectively blocked by losartan treatment. However, Tgfb1 (TGF-β1) expression was also induced by TAC, and its expression was attenuated but not completely blocked by losartan (Figure 5A). Induction of Col1a1 and Col1a3 expression was also incompletely blockedin the TAC aortas with losartan treatment (Figure 5B). Immunoblot analysis showed increased levels of phosphorated (p) Smad2 in the ascending aortas of TAC mice. Immunostaining of ascending aortic tissues detected increased number of nuclear pSmad2-positive cells in both the medial and adventitial layer after TAC (as quantitated by percent of pSmad2 positive cells in media divided by total media area; percent of pSmad2 positive cells in adventitia divided by total adventitia area). While losartan treatment dramatically decreased nuclear pSmad2 staining in the media, positive pSmad2-staining cells remained significantly more abundant in the adventitia in comparison with sham-operated controls (Figure 5D, P<0.05). These observations suggest that residual Tgfb1 expression and adventitial Smad2 signaling independent of AT1 receptor activation may account for some of the aortic histopathologic changes induced by TAC.
Figure 5. TAC without and with losartan treatment alters angiotensin II type I receptor (AT1) and TGF-β signaling.
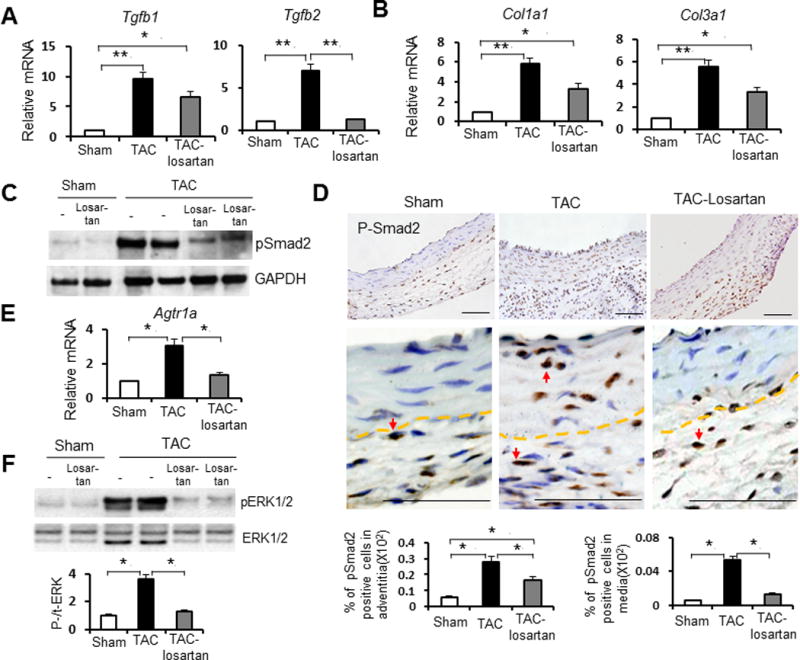
A and B. Q-PCR analysis of Tgfb1 and Tgfb2 (A) and Col1a1 and Col3a1 (B) expression levels in ascending aortic tissues from sham-operated mice (white bars), TAC mice (black bars) and TAC mice treated withlosartan for 2 weeks (gray bars) mice. Gene expression levels were normalized to Gapdh. n = 5 per group. *, P<0.05. **, P<0.01.
C. Immunoblots using antibodies against phosphorylated (p) Smad2 and GAPDH proteins. Lysates were prepared from ascending aortas of sham-operated mice, TAC mice and TAC mice treated with losartan. n = 5 per group. D. Immunostaining for representative cross sections of ascending aorta stained with anti-pSmad2 antibody from sham-operated mice, TAC mice and TAC mice treated with losartan (low and high magnification separately for each). Arrow indicates nuclear anti-p-Smad2 staining in cells. Dashed line indicates the boundary between media and adventitia. Below bar graphs represent the quantification of the adventitial and medial nuclear p-Smad2 positive area from sham-operated mice (white bar), TAC mice (black bar) and TAC mice treated with losartan (gray bar) (n = 5 per group) separately. *, P<0.05. E. Agtr1a expression by Q-PCR analysis in ascending aortic tissues from sham-operated mice (white bar), TAC mice (black bar) and TAC mice treated with losartan (gray bar). Gene expression levels were normalized to Gapdh (n = 5 per group). *, P<0.05. F. Effect of TAC and losartan treatment on pERK1/2 levels. Left, Immunoblots using antibodies that recognize pERK1/2 and total ERK1/2 proteins. Immunoblots were prepared using antibodies that recognize pERK1/2 and total ERK1/2 proteins and lysates from ascending aortas of sham-operated mice, TAC mice and TAC mice treated with losartan. Western blot signals were quantified by densitometry, and the ratio of p-ERK/ERK were calculated (mean+SEM; n=5 per group) and shown by bar graphs. *, P<0.05.
Activation of AT1 receptor with TAC
Previous studies have demonstrated that losartan attenuated both canonical (Samd dependent) and noncanonical (predominantly extracellular signal-regulated kinase (ERK) 1/2) TGF-β signaling cascades in a TGF-β- and AT1 receptor–dependent manner.10 To further characterize signaling through the AT1 receptor with pressure-induced aortic remodeling in mice, ascending aortic tissue was harvested 2 weeks after TAC, and Agtr1a (encoding AT1 receptor) expression was measured by Q-PCR. Agtr1a message levels were elevated 3-fold two weeks after TAC in comparison with controls (Figure 5E, P<0.05). Inhibition of AT1 signaling by losartan treatment significantly decreased Agtr1a message levels in the TAC ascending aortas to levels similar to controls (Figure 5E, P<0.05). To determine the effect of mechanical stress induced by TAC on the activation of extracellular signal–regulated kinase (ERK) 1/2, a kinase that can be activated by AT1, immunoblotting was performed with anti-phospho (p) ERK1/2 antibody.7 TAC significantly increased pERK1/2 levels in ascending aortic tissue, and this activation was significantly attenuated by losartan (Figure 5F, P<0.05).
Increased production of reactive oxygen species (ROS) via NAD(P)H oxidase has been shown to occur with ascending aortic aneurysm formation after Ang II infusion3. To determine whether ROS are increased in aortic SMCs in response to TAC, we performed in situ dihydroethidium (DHE) staining using frozen ascending aortic sections from TAC and control animals. DHE is a ROS-sensitive nuclear dye.3. Widespread and enhanced DHE staining was observed in the TAC ascending aortic regions compared with controls (Supplemental Figure IIIA). The intensity of DHE staining was significantly attenuated in TAC mice after losartan treatment, and reduced to levels similar to control. Cyba (encoding p22-PHOX) encodes the alpha subunit of cytochrome b(−245), which is a component of the NAD(P)H oxidase that is markedly upregulated after Ang II infusion.11 Cyba expression was increased in TAC aortas compared with controls (Supplemental Figure IIIB, P <0.05), and losartan effectively reduced Cyba expression to that of controls (Supplemental Figure IIIB, P<0.05). Thus, pERK1/2 levels, ROS production, and Cyba expression are increased in the ascending aorta after TAC and are dramatically reduced by losartan, supporting the hypothesis that AT1 receptor activation is central to the pathological changes that occur after TAC.
Discussion
Aortic remodeling after TAC is characterized by medial thickening, adventitial hyperplasia and collagen deposition.12 In this study, we demonstrate that TAC leads to inflammatory changes in the ascending aorta that are similar to those observed with Ang II infusion, including increased expression of Il6 and Mcp1 and macrophage accumulation.3, 5 Furthermore, our data also indicate that an AT1 receptor antagonist, losartan, effectively blocked these TAC-induced inflammatory changes. Since the observed inflammatory changes are similar to those associated with Ang II infusion and were effectively blocked by losartan treatment, these data support the conclusion that losartan effectively blocked AT1 signaling associated with TAC. Despite this evidence of effective AT1 blockade on TAC-induced inflammatory responses, other features of aortic remodeling associated with TAC were attenuated but not completely blocked with losartan treatment, including increased lumen diameter and thickening of the medial and adventitial layer. Based on previous studies, the dose of losartan we used in this study was comparable to other studies in investigating the role of AT1 receptor in mice.6, 13–15 Therefore, this model provides insight into AT1-dependent and independent aortic remodeling with TAC.
Our results indicate that Tgfb1 expression was attenuated but not completely blocked by losartan treatment. Similarly, molecular changes downstream of TGF-β signaling, such as myofibroblast proliferation and activation, Col1a1 and Col3a1 expression, adventitial collagen accumulation, and nuclear pSmad2 accumulation, were attenuated but not completely blocked by losartan. Induction of Tgfb2 expression and pSmad2 accumulation in the media were both effectively blocked by losartan, suggesting that these components of TGF-β signaling are dependent on AT1 activation. Activation of α-SMA expression is an important marker of myofibroblast differentiation, which contributes to vascular remodeling through cell proliferation, cell migration and synthesis of ECM proteins.8, 16 Our data suggest that TAC-induced TGF-β1 expression and activation in adventitial fibroblasts leads to myofibroblast differentiation, activation and fibrosis in the adventitia that is independent of AT1 activation. It is important to note that losartan did attenuate adventitial myofibroblast activation and collagen deposition. Previous data showed that losartan may attenuate Ang II-driven monocyte recruitment, which helps to drives fibroblast proliferation, myofibroblast activation, adventitial thickening, and cytokine production.3, 5 Since losartan effectively blocked monocyte recruitment with TAC, the monocyte-driven component of myofibroblast activation was most likely blocked.8
The observed AT1-independent, pressure-induced fibroblast to myofibroblast transition after TAC is similar to a well-characterized transition in response to mechanical stress. TGF-β1 is synthesized as a propeptide that binds to the latent TGF-β1-binding protein-1, which is part of the ECM. Transmission of forces on cell via integrins receptors leads to release and activation of latent TGF-β1 in the ECM,17 and the released TGF-β1promotes the differentiation of fibroblasts to myofibroblasts. Therefore, AT1-independent myofibroblast transition and collagen deposition in the adventitial layer with TAC could result from locally increased TGF-β1 due to mechanical stress on the aortic wall. Increased expression of Tgfb1 was also identified in the aorta after TAC. The autocrine production of TGF-β by myofibroblasts has been shown to be important for proper wound healing, and the biomechanical forces could similar increase autocrine production of TGF-β1.18 Similar to our results, remodeling of vein grafts due to increased hemodynamic stress is also characterized by increased Tgfb1 expression and myofibroblast activation in the adventitial layer.19 Previous studies have shown that TGF-β-mediated ERK1/2 activation is the predominant driver of aneurysm progression in MFS.10 ERK1/2 activation occurs after TAC, but we did not determine whether this activation is driven by AT1 versus TGF-β signaling. We did show that losartan effectively blocks TAC-induced ERK1/2 activation and nuclear pSMAD2 accumulation in medial SMCs. Further studies are needed to determine if ERK1/2 signaling is due to AT1 activation or downstream of AT1-driven TGF-β signaling. It is notable that exposure of smooth muscle cells (SMCs) to Ang II leads to rapid EKR1/2 activation.20 At the same time, Ang II increases TGF-β expression in SMCs, and Ang II can also lead to Smad signaling that is not dependent on TGF-β.21, 22
Our data indicate that activation of the AT1 receptor is responsible for many aspects of aortic remodeling induced by TAC. However, the molecular mechanism by which the AT1 receptor is activated by increased intraluminal pressure is unknown. AT1 is a G-protein coupled receptor (GPCR). The β-arrestins (β-arrestin-1 and -2) function to regulate agonist-mediated GPCR signaling by mediating both receptor desensitization and resensitization processes. Recent work suggests that load-induced membrane stretch on cardiomyocytes activates AT1 receptor signaling in a ligand-independent manner.23 This ligand-independent activation of the AT1 receptor occurs via transient receptor potential channels that depend on G protein coupling and the recruitment of β-arrestin to act as a biased agonist for the AT1 receptor. Mechanical stretch of cardiomyocytes also increases inward potassium currents, by ligand-independent mechanical activation of AT1 receptors.24 Therefore, the ligand-independent activation of the AT1 receptor after TAC could result from a similar mechanical activation due to the increased mechanical forces induced by the increased pulsatile stretch on aortic SMCs. Alternatively, the increased mechanical stress on SMCs could induce ROS formation and increase the sensitivity of SMCs to Ang II activation.3 AT1 activation would further increase ROS production and AT1 activation in a feedback loop. Finally, local production of Ang II due to mechanical stretch may be augmented by increased SMC expression of rennin-angiotensin pathways genes.
In summary, this study indicates that AT1 receptor activation plays a critical role in the ascending thoracic aortic remodeling that occurs due to increased biomechanical stress associated with TAC (Supplemental Figure IV). This study also illustrates that pressure-induced aortic remodeling involves increased expression of Tgfb1 and Tgfb2, as well as activation of pSmad2. Activation of these TGF-β signaling components is dependent on AT1 receptor activation in the media but occurs partially independently of AT1 receptor activation in the adventitia. Furthermore, we have identified histopathological and molecular changes in the medial versus the adventitial layer of the aorta that are differentially dependent on AT1 activation. These findings have important implications for the understanding the role of AT1 and TGF-β signaling in the aorta with increased pressures.
Supplementary Material
Significance.
Although hypertension is the most common risk factor for thoracic aortic diseases, it is not understood how increased pressures on the ascending aorta may lead to TAAD. This is the first study to investigate the role of AT1 receptor activation in ascending aortic remodeling in response to increased biomechanical forces, using the TAC mouse model. Although previous studies indicate that angiotensin II can activate Erk signaling and increase TGFβ expression in vascular smooth muscle cells, our results implicate the AT1 receptor in both increased TGFβ downstream signaling and Erk activation. Since many hypertensive patients with thoracic aortic aneurysms are already on losartan treatment, our finding may provide molecular basis for treatment of TAAD.
Acknowledgments
Sources of Funding
The following sources provided funding to D.M.M. for these studies: P50HL083794-01, Whitaker Foundation, Richard T. Pasani Foundation, and Vivian Smith Foundation. ARB was supported by P50HL083794 and HL70925.
Non-standard abbreviations and acronyms
- Ang II
angiotensin II
- AT1
angiotensin II type 1 receptor
- α-SMA
α smooth muscle actin
- DHE
dihydroethidium
- ERK
extracellular signal–regulated kinase
- ECM
extracellular matrix
- MFS
Marfan syndrome
- pERK
phosphorylated extracellular signal–regulated kinase (ERK)
- pSmad2
phosphorylated Smad2
- pH3
phosphorylated histone H3 (Ser10)
- ROS
reactive oxygen species
- SMC
smooth muscle cell
- TAC
transverse aortic constriction
- TAAD
thoracic aortic aneurysms and/or aortic dissections
- TGF-β
transforming growth factor-β
- VVG
Verhoeff-van Gieson
Footnotes
Disclosures
None.
References
- 1.Hiratzka LF, Bakris GL, Beckman JA, et al. ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the Diagnosis and Management of Patients With Thoracic Aortic Disease: Executive Summary. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121:e266–e369. doi: 10.1161/CIR.0b013e3181d4739e. [DOI] [PubMed] [Google Scholar]
- 2.LeMaire SA, McDonald ML, Guo DC, et al. Genome-wide association study identifies a susceptibility locus for thoracic aortic aneurysms and aortic dissections spanning FBN1 at 15q21.1. Nat Genet. 2011;43:996–1000. doi: 10.1038/ng.934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tieu BC, Lee C, Sun H, Lejeune W, Recinos A, III, Ju X, Spratt H, Guo DC, Milewicz D, Tilton RG, Brasier AR. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J Clin Invest. 2009;119:3637–51. doi: 10.1172/JCI38308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010;118:681–9. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tieu BC, Ju X, Lee C, Sun H, Lejeune W, Recinos A, III, Brasier AR, Tilton RG. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J Vasc Res. 2011;48:261–72. doi: 10.1159/000320358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Habashi JP, Judge DP, Holm TM,DB, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–21. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA. Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest. 2011;121:2301–12. doi: 10.1172/JCI44824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, Dietz HC, Gabrielson KL, Kass DA, Semenza GL. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-beta signaling. Proc Natl Acad Sci U S A. 2012;109:E841–E850. doi: 10.1073/pnas.1202081109. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.deAlmeida AC, van Oort RJ, Wehrens XH. Transverse aortic constriction in mice. J Vis Exp. 2010;38:1729. doi: 10.3791/1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, Chen Y, Modiri AN, Judge DP, Dietz HC. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. 2011;332:361–365. doi: 10.1126/science.1192152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Q, 4th, Taylor WR, Harrison DG, de Leon H, Wilcox JN, Griendling KK. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Wu J, Li L, Zou YZ, Zhu DL, Gao PJ. Effect of an acute mechanical stimulus on aortic structure in the transverse aortic constriction mouse model. Clin Exp Pharmacol Physiol. 2011;38:570–576. doi: 10.1111/j.1440-1681.2011.05544.x. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Zhou N, Gong H, Wu J, Lin L, Komuro I, Ge J, Zou Y. Comparison of angiotensin II type 1-receptor blockers to regress pressure overload-induced cardiac hypertrophy in mice. Hypertens Res. 2010;33:1289–1297. doi: 10.1038/hr.2010.182. [DOI] [PubMed] [Google Scholar]
- 14.Meems LM, Cannon MV, Mahmud H, Voors AA, van Gilst WH, Silljé HH, Ruifrok WP, de Boer RA. The vitamin D receptor activator paricalcitol prevents fibrosis and diastolic dysfunction in a murine model of pressure overload. Journal of Steroid Biochemistry & Molecular Biology. 2012;132:282–289. doi: 10.1016/j.jsbmb.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Knowles JW, Esposito G, Mao L, Hagaman JR, Fox JE, Smithies O, Rockman HA, Maeda N. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor A–deficient mice. J Clin Invest. 2001;107:975–984. doi: 10.1172/JCI11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YH, Hsieh CY, Wang DL, Chung HC, Liu SL, Chao TH, Shi GY, Wu HL. Remodeling of carotid arteries is associated with increased expression of thrombomodulin in a mouse transverse aortic constriction model. Thromb Haemost. 2007;97:658–664. [PubMed] [Google Scholar]
- 17.Hinz B. Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission. Eur J Cell Biol. 2006;85:175–181. doi: 10.1016/j.ejcb.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 18.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 19.Jiang Z, Yu P, Tao M, Fernandez C, Ifantides C, Moloye O, Schultz GS, Ozaki CK, Berceli SA. TGF-beta- and CTGF-mediated fibroblast recruitment influences early outward vein graft remodeling. Am J Physiol Heart Circ Physiol. 2007;293:H482–H488. doi: 10.1152/ajpheart.01372.2006. [DOI] [PubMed] [Google Scholar]
- 20.Kusuhara M, Takahashi E, Peterson TE, Abe J, Ishida M, Han J, Ulevitch R, Berk BC. p38 Kinase is a negative regulator of angiotensin II signal transduction in vascular smooth muscle cells: effects on Na+/H+ exchange and ERK1/2. Circ Res. 1998;83:824–831. doi: 10.1161/01.res.83.8.824. [DOI] [PubMed] [Google Scholar]
- 21.Gibbons GH, Pratt RE, Dzau VJ. Vascular smooth muscle cell hypertrophy vs. hyperplasia. Autocrine transforming growth factor-beta 1 expression determines growth response to angiotensin II. J Clin Invest. 1992;90:456–461. doi: 10.1172/JCI115881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation. 2005;111:2509–2517. 11. doi: 10.1161/01.CIR.0000165133.84978.E2. [DOI] [PubMed] [Google Scholar]
- 23.Rakesh K, Yoo B, Kim IM, Salazar N, Kim KS, Rockman HA. beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal. 2010;3(125):ra46. doi: 10.1126/scisignal.2000769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Mello WC. Mechanical stretch reduces the effect of angiotensin II on potassium current in cardiac ventricular cells of adult Sprague Dawley rats. On the role of AT1 receptors as mechanosensors. J Am Soc Hypertens. 2012;6:369–374. doi: 10.1016/j.jash.2012.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.