

SUMMARY
The master regulatory transcription factor GATA-2 triggers hematopoietic stem and progenitor cell generation. GATA2 haploinsufficiency is implicated in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), and GATA2 overexpression portends a poor prognosis for AML. However, the constituents of the GATA-2-dependent genetic network mediating pathogenesis are unknown. We described a p38-dependent mechanism that phosphorylates GATA-2 and increases GATA-2 target gene activation. We demonstrate that this mechanism establishes a growth-promoting chemokine/cytokine circuit in AML cells. p38/ERK-dependent GATA-2 phosphorylation facilitated positive autoregulation of GATA2 transcription and expression of target genes, including IL1B and CXCL2. IL-1β and CXCL2 enhanced GATA-2 phosphorylation, which increased GATA-2-mediated transcriptional activation. p38/ERK-GATA-2 stimulated AML cell proliferation via CXCL2 induction. As GATA2 mRNA correlated with IL1B and CXCL2 mRNAs in AML-M5 and high expression of these genes predicted poor prognosis of cyto-genetically normal AML, we propose that the circuit is functionally important in specific AML contexts.
In Brief
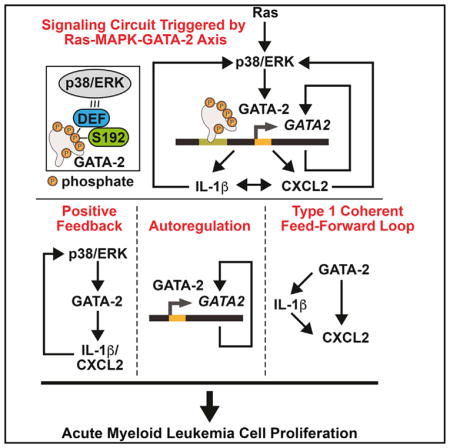
Katsumura et al. uncover a signaling mechanism that amplifies GATA-2 activity at select target genes in acute myeloid leukemia cells. Expression of GATA-2 target genes encoding the chemokine CXCL2 and cytokine IL-1β correlates with GATA-2 expression in a subtype of human AML, and high GATA-2/CXCL2 expression predicts poor prognosis.
INTRODUCTION
The heterogeneous malignancy acute myeloid leukemia (AML) is characterized by aberrant myeloid cell proliferation and differentiation (Coombs et al., 2016). AML prognosis in geriatric patients has a 5-year survival of 5%–10% (Klepin et al., 2014), and 30%–40% of pediatric patients do not experience long-term survival (Zwaan et al., 2015). Whereas defects in signaling and gene expression mechanisms controlling hematopoiesis can cause AML, many questions remain regarding the signals, factors, and circuits.
RAS and KIT mutations, which can be mutually exclusive or co-occur in AML patients, yield aberrant signaling molecules that stimulate AML cell proliferation (Boissel et al., 2006; Goemans et al., 2005). Recently, GATA-2, a master regulator of hematopoietic stem and progenitor cell (HSPC) genesis/function (Tsai et al., 1994), was implicated in AML. Heterozygous GATA2 mutations cause a primary immunodeficiency (Mono-MAC) associated with myelodysplastic syndrome (MDS) that progresses to AML (Dickinson et al., 2011; Hahn et al., 2011; Hsu et al., 2011; Ostergaard et al., 2011). GATA2 mutations were detected in 7% of pediatric MDS patients (Wlodarski et al., 2016). These mutations attenuate GATA-2 chromatin binding, thus disrupting the GATA-2-dependent genetic network (Katsumura et al., 2014). Heterozygous mutations of a Gata2 intronic enhancer (+9.5 kb), which normally increases Gata2 expression in hemogenic endothelium, hematopoietic stem cells (HSCs), and myeloid progenitors (Gao et al., 2013; Grass et al., 2006; Johnson et al., 2012; Sanalkumar et al., 2014), cause MonoMAC with a phenotype resembling patients with coding region mutations (Hsu et al., 2013; Johnson et al., 2012).
A distinct mechanism deregulates GATA2 in poor prognosis 3q21-q26 AML, which constitutes −2% of AML. An inversion repositions a GATA-2-binding GATA2 cis element (−77 kb) (Grass et al., 2006) to a region upstream of the distant oncogene EVI1, increasing EVI1 and decreasing GATA2 expression (Gröschel et al., 2014; Yamazaki et al., 2014). Deletion of the −77 kb site reduces Gata2 expression in myeloid progenitors, confers a differentiation blockade, and is embryonic lethal (Johnson et al., 2015). These results suggest that reduced GATA-2 expression in progenitors and ectopic EVI1 expression underlie leukemogenesis. Epigenetic alterations can decrease GATA2 expression in normal karyotype AML (Celton et al., 2014). While decreased GATA2 expression is linked to MDS/AML, increased GATA2 expression correlates with poor prognosis adult and pediatric AML (Luesink et al., 2012; Vicente et al., 2012). Gain-of-function mutations in chronic myeloid leukemia (Zhang et al., 2008) and GATA-2 overexpression in murine bone marrow suppress hematopoiesis (Persons et al., 1999). GATA-2 activity must be maintained within a physiological window, as decreases or increases disrupt the GATA-2-dependent genetic network, initiating or promoting leukemogenesis. The vital constituents of the network and their circuits are largely unknown.
Ras-p38 signaling stimulates GATA-2 S192 phosphorylation, which promotes multi-site GATA-2 phosphorylation and enhances GATA-2-mediated transcriptional activation in pro-erythroblast and endothelial cells (Katsumura et al., 2014). GATA-2 and oncogenic Ras cooperatively promote non-small-cell lung cancer and colon cancer (Kumar et al., 2012; Shen et al., 2014; Steckel et al., 2012). NRAS, KRAS, and HRAS mutations occur in 10%, 5%, and 5% of AML patients (Ward et al., 2012). Considering that Ras-p38 signaling stimulates GATA-2 activity, we asked whether the Ras-GATA-2 axis functions in AML cells. p38/ERK functions through a GATA-2 docking site for ERK FXF (DEF) motif (Jacobs et al., 1999) to phosphorylate GATA-2 in AML cells, and DEF motifs have not been implicated in GATA factor mechanisms. This mechanism enhances GATA-2-mediated activation of select target genes, including genes implicated in leukemogenesis (GATA2, TAL1, IL1B, and CXCL2). These results revealed a positive-feedback circuit in which Ras-p38/ERK-GATA-2 upregulate CXCL2 expression, CXCL2 stimulates AML (Kasumi-1) cell proliferation, and CXCL2 acts on GATA-2-expressing cells to stimulate the signal-dependent GATA-2 mechanism. Coupled with insights from AML patient data and the poor prognosis of AML highly expressing the CXCL2 receptor CXCR2 (Schinke et al., 2015), the p38/ERK-GATA-2 axis may inform AML therapeutics development.
RESULTS
Ras-p38/ERK- and GATA-2 DEF Motif-Mediated GATA-2 Phosphorylation and Transcriptional Activation in AML Cells
Given that GATA-2 levels/activity must be tightly controlled to ensure normal hematopoiesis, we tested whether the p38-GATA-2 pathway functions in AML cells. We analyzed GATA-2 phosphorylation in Kasumi-1 cells harboring KIT and RUNX1 mutations, which were derived from a pediatric M2 stage AML patient (Asou et al., 1991). Previously, we described GATA-2 phosphorylation sites that create a slow mobility GATA-2 isoform detected by SDS-PAGE. We demonstrated that λ-phosphatase converts phosphorylated GATA-2 to a dephosphorylated, fast-migrating isoform (Katsumura et al., 2014). In Kasumi-1 cells, λ-phosphatase decreased the slow mobility phosphorylated isoform of endogenous GATA-2 (Figure 1A). Identical results were obtained with Kasumi-3 cells (Figure S1A), an AML cell line derived from adult M0 AML with t (3; 7) (q27; q22) and high EVI1 expression (Asou et al., 1996). To determine the influence of MAPK signaling on GATA-2 phosphorylation in AML cells, we expressed constitutively active oncogenic Ras, Ras(G12V). Ras(G12V) induced the slow mobility isoform of endogenous (Figure 1B) and expressed GATA-2 (Figures 1C and S1B). In our prior G1E proerythroblast and HEK293 analyses (Katsumura et al., 2014), Ras(G12V) stimulated GATA-2 S192 phosphorylation, inducing multi-site phosphorylation, which yields the slow-migrating isoform. S192A mutation abrogated Ras(G12V)-induced GATA-2 hyperphosphorylation in Kasumi-1 and Kasumi-3 cells (Figures 1C and S1B).
Figure 1. Ras-p38/ERK- and GATA-2 DEF Motif-Mediated GATA-2 Phosphorylation in AML Cells.

(A) Total Kasumi-1 cell protein was incubated with or without λ-phosphatase and analyzed by western blotting with anti-GATA-2 antibody.
(B) Left: western blot analysis of endogenous GATA-2 in Kasumi-1 cells with or without expression of H-Ras(G12V). Right: densitometric analysis of relative protein levels is shown. The ratio of intensities of the upper to lower bands from control Kasumi-1 cells was designated as 1 (n = 3, mean ± SE, *p < 0.05).
(C) Left: western blot analysis of MAP kinases and wild-type and mutant proteins transiently expressed in Kasumi-1 cells with or without H-Ras(G12V). Expressed GATA-2 was detected with anti-HA antibody. Right: densitometric analysis of relative protein levels is shown. The ratio of intensities of the upper to lower bands from HA-GATA-2-expressing Kasumi-1 cells was designated as 1 (n = 3, mean ± SE, **p < 0.01).
(D) Top: western blot analysis of substrates of MAP kinases, GATA-2, and Ras proteins transiently expressed in Kasumi-1 cells with or without H-Ras(G12V) and MAP kinase inhibitors. Expressed GATA-2 was detected with anti-HA antibody. ERK, HSP27, and c-Jun phosphorylation was assessed to test inhibitor specificities. Bottom: densitometric analysis of relative protein levels is shown. The ratio of intensities of the upper to lower bands from Ras(G12V)-expressing Kasumi-1 cells was designated as 1 (n = 3, mean ± SE, *p < 0.05 and **p < 0.01).
(E) Left: western blot analysis of MAP kinases, substrates of MAP kinases, and GATA-2 proteins transiently expressed in Kasumi-1 cells with or without constitutively active MEK1 or p38. Expressed GATA-2 protein was detected with anti-HA antibody. Right: densitometric analysis of relative protein levels is shown. The ratio of intensities of the upper to lower bands from GATA-2-expressing Kasumi-1 cells was designated as 1 (n = 3, mean ± SE, **p < 0.01).
(F) DEF motif in GATA-2 is shown.
(G) Left: western blot analysis of MAP kinases and wild-type and mutant proteins transiently expressed in Kasumi-1 cells with or without H-Ras(G12V). Expressed GATA-2 was detected with anti-HA antibody. Right: densitometric analysis of relative protein levels is shown. The ratio of intensities of upper to lower bands from HA-GATA-2-expressing Kasumi-1 cells was designated as 1 (n = 3, mean ± SE, **p < 0.01).
(H) Total protein from Kasumi-1 cells expressing GATA-2 proteins was incubated with or without λ-phosphatase. Proteins were analyzed by western blotting with anti-HA antibody.
(I) Left: western blot analysis of HA-GATA-2 and HA-GATA-2ΔDEF transiently expressed in MAE cells with or without Ras(G12V). Right: qRT-PCR analysis of Hdc mRNA levels in MAE cells transiently expressing HA-GATA-2 and HA-GATA-2ΔDEF with or without Ras(G12V) is shown (n = 3, mean ± SE, *p < 0.05).
See also Figure S1.
Distinct MAPKs phosphorylate similar target sequences, and sequences surrounding the phosphorylated residues (S73, S119, and S192) are predicted to generate phosphorylation sites for p38, ERK, and JNK. To determine which MAPK mediates GATA-2 hyperphosphorylation in AML cells, GATA-2 hyperphosphorylation was assessed in SB203580- (p38 MAPK inhibitor), U0126- (MEK inhibitor), and SP600125- (JNK inhibitor) treated Kasumi-1 cells. Whereas SB203580 and U0126 suppressed GATA-2 hyperphosphorylation, SP600125 had no effect (Figure 1D). Similar results were obtained in Kasumi-3 cells (Figure S1C). Constitutively active MEK1 and constitutively active p38α induced GATA-2 hyperphosphorylation (Figure 1E).
GATA-2 contains an FXFP sequence, and these DEF motifs are recognized by ERK2 and p38α, but not JNK3 (Jacobs et al., 1999). ERK2 and p38α have a DEF pocket that binds DEF motif-containing substrates (Tzarum et al., 2013). As DEF motifs have not been characterized in GATA factors and the conserved GATA-2 DEF motif (lacking in GATA-1) is located 20 amino acids N-terminal to S192 (Figure 1F), we tested whether it mediates signal-dependent GATA-2 phosphorylation. Mutating the DEF motif (F171A and F173A) abrogated Ras(G12V)-induced GATA-2 hyperphosphorylation (Figure 1G). The DEF mutant exhibited steady-state phosphorylation, as λ-phosphatase reduced its mobility (Figure 1H); presumably, other kinases can phosphorylate the mutant. To analyze DEF motif function, we quantitated the capacity of wild-type and DEF motif mutant GATA-2 to induce endogenous Hdc mRNA expression in mouse aortic endothelial (MAE) cells. The MAE assay allows one to uniquely analyze GATA-2-mediated endogenous gene regulation, and Hdc is a direct GATA-2 target gene (Katsumura et al., 2014). The DEF motif mediated Ras(G12V)-induced GATA-2 hyperphosphorylation in MAE cells (Figure 1I), resembling the Kasumi-1 cells. In the presence of Ras(G12V), the DEF motif mutant was less active than wild-type GATA-2 (Figure 1I). Thus, p38 and ERK mediate GATA-2 phosphorylation in AML cells, and a DEF motif is required for signal-dependent GATA-2 function.
To dissect how the p38/ERK-GATA-2 axis functions in AML cells, we tested whether the signaling mechanism impacts GATA-2 target gene expression. Previously, we described GATA-2-regulated stem cell and inflammatory genes in the mouse aorta, gonad, mesonephros (AGM) region (Gao et al., 2013) and in human umbilical vein endothelial cells (HUVECs) (e.g., CXCL2 and IL1B) (Linnemann et al., 2011). To investigate whether GATA-2 regulates all or a cohort of these genes, we used Kasumi-1 cells stably infected with retroviruses expressing control small hairpin RNA (shRNA) targeting luciferase mRNA (sh-luc) (Kasumi-1/sh-luc cells) or sh-GATA2 (Kasumi-1/sh-G2 cells) (Figure 2A). GATA2 knockdown decreased TAL1 expression by 50% (p < 0.001) in Kasumi-1 cells, while expression of KIT, GFI1, and SFPI1, which are also GATA-2 target genes, was unaffected (Figure 2B).
Figure 2. Ras-p38/ERK Enhance GATA-2-Mediated Target Gene Transcription in AML Cells.

(A) Top: western blot analysis of endogenous GATA-2 in Kasumi-1 cells stably infected with sh-luc virus or sh-GATA2 virus. Bottom: densitometric analysis of relative protein levels is shown. The protein expression in Kasumi-1 cells infected with sh-luc virus was designated as 1.
(B) Real-time RT-PCR analysis of GATA2 mRNA and transcripts of GATA-2 target genes in Kasumi-1 cells infected with sh-luc virus or sh-GATA2 virus is shown (n = 5; mean ± SE; *p < 0.05, **p < 0.01, and ***p < 0.001).
(C) ChIP-seq analysis of endogenous GATA-2 occupancy at GATA2, IL1B, and CXCL2 loci in Kasumi-3 cells, TF-1 cells (Mazumdar et al., 2015), and human CD34-positive hematopoietic cells (Beck et al., 2013) is shown.
(D) Real-time RT-PCR analysis of transcripts of GATA-2 target genes in Kasumi-1 cells treated with 40 μM SB203580 (n = 4, mean ± SE). Samples were harvested at the designated times.
(E) Western blot analysis of endogenous GATA-2 in Kasumi-1 cells treated with 40 μM SB203580 is shown (n = 5, mean ± SE, *p < 0.05).
(F) Real-time RT-PCR analysis of GATA-2 target genes in Kasumi-1 cells treated with 20 μM U0126 (n = 4, mean ± SE). Samples were harvested at the designated times.
(G) Western blot analysis of endogenous GATA-2 in Kasumi-1 cells treated with 20 μM U0126 is shown (n = 5, mean ± SE, *p < 0.05).
(H) Left: western blot analysis of endogenous GATA-2 and MAP kinase proteins in Kasumi-1 cells expressing Ras(G12V). Right: real-time RT-PCR analysis of IL1B and CXCL2 expression in Kasumi-1 cells expressing Ras(G12V) is shown (n = 3, mean ± SE, *p < 0.05 and **p < 0.01).
(I) Real-time RT-PCR analysis of IL1B and CXCL2 expression in Kasumi-1/sh-luc cells or Kasumi-1/sh-G2 cells expressing Ras(G12V) (n = 6, mean ± SE, *p < 0.05 and **p < 0.01).
See also Figure S2.
CXCL2 functions through the CXCR2 receptor, CXCR2 antagonism impairs AML cell growth in vivo, and high CXCR2 expression predicts poor prognosis of human AML (Schinke et al., 2015). IL-1β supports AML cell survival and proliferation (Dubois et al., 1994; Estrov et al., 1999; Turzanski et al., 2004), and antagonizing IL-1β signaling inhibits AML cell proliferation (Ågerstam et al., 2015; Rambaldi et al., 1991). IL-1β-mediated GM-CSF induction contributes to this growth effect (Delwel et al., 1989; Bradbury et al., 1990). Thus, we tested whether GATA-2 regulates CXCL2 and IL1B expression. IL1B and CXCL2 mRNA expression decreased by 82% and 73%, respectively, in Kasumi-1/sh-G2 cells (Figure 2B). Using a distinct shRNA (sh-G2-2), TAL1, IL1B, and CXCL2 expression decreased similarly (Figure 2B). While TAL1 is an established GATA-2 target gene (Lugus et al., 2007), IL1B and CXCL2 were not known to be direct GATA-2 targets. We tested whether endogenous GATA-2 occupies IL1B and CXCL2 using chromatin immunoprecipitation sequencing (ChIP-seq) data from Kasumi-3 cells. GATA-2 occupied chromatin at or near IL1B and CXCL2, as well as the intronic +9.5 kb GATA2 enhancer (Grass et al., 2006; Johnson et al., 2012) (Figure 2C). GATA-2 occupied the same sites in human CD34-positive cells and TF-1 myeloerythroid cells (Beck et al., 2013; Mazumdar et al., 2015), and the sites were DNase hypersensitive in CD34-positive cells (Figure 2C). The GATA2 knockdown did not affect genes flanking IL1B (Figure S2A).
We tested whether p38 and ERK regulate GATA-2 target genes. SB203580 or U0126 reduced TAL1, IL1B, and CXCL2 expression in Kasumi-1 cells (Figures 2D, 2F, S2B, and S2C), without affecting KIT, GFI1, SFPI1, and GAPDH expression (Figures S2B and S2C). SB203580 and U0126 also decreased GATA2 expression (Figures 2D, 2F, S2B, and S2C), consistent with the model in which GATA-2 positively autoregulates GATA2 transcription through binding −77 kb and +9.5 kb enhancers (Bresnick et al., 2010; Grass et al., 2006). GATA2 primary transcripts were regulated with kinetics resembling GATA2 mRNA (Figures 2D, right and 2F, right). Whereas SB203580 and U0126 reduced GATA-2 target gene expression within 6 hr, total GATA-2 protein was unaffected at this time (Figures 2E and 2G). This analysis suggested that decreased GATA-2 target gene mRNAs did not result from a rapid decline in total GATA-2 protein. As SB203580 and U0126 increased the hypophosphorylated isoform (Figures 2E and 2G), reduced GATA-2 target gene expression was linked to lower GATA-2 phosphorylation.
To complement the pharmacological analysis, we tested whether oncogenic Ras-mediated MAPK activation enhanced GATA-2-dependent transcription. Ras(G12V) expression in Kasumi-1 cells increased IL1B and CXCL2 mRNA levels in a GATA-2-dependent manner (Figures 2H and 2I). Whereas Ras(G12V) induced IL1B expression 12-fold and CXCL2 expression 1.7-fold in control cells, GATA-2 downregulation decreased the induction to 3.5- and 1.3-fold (Figure 2I). ERK and p38 activation by the phosphatase inhibitor okadaic acid induced S192-dependent GATA-2 hyperphosphorylation (Figure S2D). Okadaic acid increased GATA2, IL1B, TAL1, and CXCL2 mRNA levels 10-, 40-, 1.5-, and 3-fold, respectively (Figure S2E). The okadaic acid activity to regulate GATA-2 phosphorylation and induce GATA-2 target genes was attenuated by SB203580, and treatment of cells with both SB203580 and U0126 yielded more inhibition (Figures S2F and S2G). p38 and ERK also regulated GATA-2 target genes in Kasumi-3 cells (Figures S2H and S2I). These data indicate that p38 and ERK increase GATA-2-mediated target gene activation in AML cells.
We tested whether the Ras-p38/ERK-GATA-2 axis regulates GATA-2 chromatin occupancy at CXCL2 and IL1B using functionally important GATA2 cis elements as controls. GATA-2 occupied GATA2 −77 kb and +9.5 kb enhancers (Figure 3A), the disruption of which is linked to leukemogenesis (Gröschel et al., 2014; Johnson et al., 2012; Yamazaki et al., 2014). SB203580 and U0126 decreased GATA-2 occupancy at −77 kb and +9.5 kb sites 75% and 25%, respectively (Figure 3A). SB203580 and U0126 reduced GATA-2 occupancy at IL1B and CXCL2 sites identified by ChIP-seq (Figure 3B). Formaldehyde-assisted isolation of regulatory elements (FAIRE) analysis (Sanalkumar et al., 2014) revealed p38 and ERK inhibition reduced chromatin accessibility at the GATA-2-occupied −77 kb, IL1B, and CXCL2 sites (Figure 3C). In Kasumi-1/sh-G2 cells, chromatin accessibility was decreased and MAPK inhibitors did not alter chromatin accessibility (Figure S3A). GATA-2 dephosphorylation preceded (SB203580) or occurred concomitantly (U0126) with decreased GATA-2 occupancy at the −77 kb site (Figures 3D–3G). GATA-2 overexpression attenuated the SB203580-induced decrease of IL1B and CXCL2 mRNA (Figure 3H). Thus, the signaling mechanism increased GATA-2 chromatin occupancy and GATA-2-mediated activation of select GATA2 target genes in AML cells. AP-1 and necrosis factor κB (NF-κB) occupy chromatin near the GATA-2 site at IL1B (Figures S3B and S3C). Although GATA-2 directly regulates IL-1B and CXCL2 expression, one cannot rule out the possibility that GATA-2 functionally interfaces with these factors that can mediate the induction of inflammatory cytokines.
Figure 3. p38/ERK Signaling Promotes GATA-2 Chromatin Occupancy and Chromatin Remodeling.

(A) Gata2 locus map. Numbers represent distance to mouse 1S transcription start site. 1G is another GATA2 transcription start site. Quantitative ChIP analysis of GATA-2 occupancy in Kasumi-1 cells treated with 40 μM SB203580 or 20 μM U0126 is shown (n = 4, mean ± SE, *p < 0.05 and **p < 0.01). The western blot (anti-GATA-2 antibody) (inset) illustrates GATA-2 expression in representative samples used for ChIP.
(B) Quantitative ChIP analysis of GATA-2 occupancy at GATA-2 target genes in Kasumi-1 cells treated with 40 μM SB203580 or 20 μM U0126 is shown (n = 4, mean ± SE, *p < 0.05).
(C) Quantitative FAIRE analysis of chromatin accessibility in Kasumi-1 cells treated with 40 μM SB203580 or 20 μM U0126 is shown (n = 4, mean ± SE, *p < 0.05 and **p < 0.01).
(D) Kinetics of GATA-2 expression and phosphorylation are shown (n = 3, mean ± SE, *p < 0.05).
(E) GATA-2 occupancy at the −77 kb site during 40 μM SB203580 treatment is shown (n = 3, mean ± SE, *p < 0.05).
(F) Kinetics of GATA-2 protein expression and phosphorylation are shown (n = 3, mean ± SE, *p < 0.05).
(G) GATA-2 occupancy at the −77 kb site during 20 μM U0126 treatment is shown (n = 3, mean ± SE, *p < 0.05).
(H) Left: rescue assay. Right: qRT-PCR analysis of IL1B and CXCL2 in Kasumi-1 cells expressing GATA-2 or control vector with or without 10 μM SB203580 is shown (n = 4; mean ± SE; *p < 0.05; PI, preimmune).
See also Figure S3.
p38/ERK-GATA-2 Positive-Feedback Circuit Promotes Kasumi-1 Cell Proliferation
Chemokine/cytokine signaling through cognate receptors can activate the MAPK pathway. The CXCL2 receptor CXCR2 activates Ras (Knall et al., 1996) and IL-1β activates p38 (Suzuki et al., 2001). AML cells express CXCR2 and IL1R1 at levels comparable to control bone marrow cells, and IL1RAP, which associates with IL1R1, is overexpressed in AML cells as described (Figure S4; Barreyro et al., 2012). We tested whether the GATA-2-dependent increase in IL1B and CXCL2 expression promotes IL-1β and CXCL2 signaling in GATA-2-expressing cells to further increase GATA-2 activity through a p38/ERK-dependent mechanism. Recombinant IL-1β or CXCL2 increased endogenous GATA-2 phosphorylation in Kasumi-1 cells (Figures 4A and 4B). While IL-1β activated p38, CXCL2 activated p38 and ERK (Figures 4A and 4B). IL-1β and CXCL2 increased GATA-2 occupancy at GATA2 −77 kb and +9.5 kb sites in Kasumi-1 cells (Figure 4C), and they induced accumulation of the slow mobility GATA-2 isoform (Figure 4D) and increased GATA2 mRNA (Figure 4E) in patient-derived primary AML cells. p38/ERK-GATA-2-mediated induction of IL1β and CXCL2, therefore, constitutes a positive-feedback loop in AML cells.
Figure 4. p38/ERK-GATA-2 Axis Establishes a Chemokine/Cytokine-Dependent Positive-Feedback Circuit.

(A) Left: western blot analysis of endogenous GATA-2 in Kasumi-1 cells treated with 10 ng/ml recombinant human IL-1β. The cells were serum-starved overnight before IL-1β treatment. Right: densitometric analysis is shown. The ratio of intensities of the upper to lower bands from at 0 min was designated as 1 (n = 3, mean ± SE, *p < 0.05).
(B) Western blot analysis of endogenous GATA-2 in Kasumi-1 cells treated with 100 ng/ml recombinant human CXCL2. Cells were serum-starved overnight before CXCL2 treatment. Right: densitometric analysis is shown. The ratio of intensities of the upper to lower bands at 0 min was designated as 1 (n = 3, mean ± SE, *p < 0.05).
(C) Quantitative ChIP analysis of GATA-2 occupancy at GATA-2 target genes in Kasumi-1 cells treated with 10 ng/ml IL-1β or 100 ng/ml CXCL2 (n = 4, mean ± SE). The cells were serum-starved overnight before treatment with IL-1β or CXCL2 (*p < 0.05).
(D) Left: western blot analysis of endogenous GATA-2 in primary AML cells treated with 10 ng/ml recombinant human IL-1β or 100 ng/ml recombinant human CXCL2 for 5 min. The cells were serum-starved for 90 min before treatment. Right: densitometric analysis is shown. The ratio of intensities of the upper to lower bands from the control sample was designated as 1 (n = 4, mean ± SE, *p < 0.05).
(E) Real-time RT-PCR analysis of GATA2 mRNA in primary AML cells treated with 10 ng/ml IL-1β or 100 ng/ml CXCL2 (n = 4, mean ± SE). Primary AML cells were serum-starved for 90 min and treated with IL-1β or CXCL2 for 30 min (*p < 0.05).
See also Figure S4.
Target genes of the p38/ERK-GATA-2 axis (GATA2, IL1B, and CXCL2) are implicated in AML. We tested whether the GATA-2-dependent chemokine/cytokine circuit impacts AML cell proliferation. Kasumi-1/sh-G2 cells proliferated slower than control Kasumi-1/sh-luc cells (Figure 5A). Fluorescence-activated cell sorting (FACS) analysis of Kasumi-1/sh-G2 and Kasumi-1/sh-luc cell proliferation revealed reduced proliferation of Kasumi-1/sh-G2 cells (Figures 5B and 5C). Since downregulating GATA-2 in Kasumi-1 cells reduced IL1B and CXCL2 expression and the cells proliferated slower, we tested whether IL-1β and CXCL2 stimulate Kasumi-1 cell proliferation. Human CXCL2, but not IL-1β, restored Kasumi-1/sh-G2 cell proliferation (Figures 5D and S3A). FACS analysis also revealed CXCL2-mediated restoration of Kasumi-1/sh-G2 cell proliferation (Figures 5E, 5F, and S3B). These results support a model in which the p38/ERK-GATA-2-instigated chemokine/cytokine circuit controls AML cell proliferation.
Figure 5. p38/ERK-GATA2 Axis Stimulates Kasumi-1 Cell Proliferation.

(A) Comparison of growth rates of Kasumi-1 cells stably infected with sh-luc virus or sh-GATA2 virus. The Kasumi-1/sh-luc cells and Kasumi-1/sh-G2 cells (1 × 105) were plated, and cells were counted every second day. Cells were passaged at a density of 1 × 105 cells at day 4 (n = 9, mean ± SE, *p < 0.05 and **p < 0.01).
(B) Top: proliferation analysis. Bottom: representative plots from proliferation analysis of Kasumi-1/sh-luc cells and Kasumi-1/sh-G2 cells using Cell-Trace Violet dye are shown. A greater number of generations indicates the cells underwent more rounds of cell division.
(C) Left: the average percentage of total cells in each daughter generation. Each bar represents three independent experiments from one clonal line. The western blot inset illustrates the efficacy of the GATA-2 knockdown (*p < 0.05, **p < 0.01, and ***p < 0.001). Right: proliferation index of Ka-sumi-1/sh-luc cells and Kasumi-1/sh-G2 cells is shown. Each bar represents three independent experiments from one clonal line (***p < 0.001).
(D) Comparison of the growth rates of Kasumi-1 cells stably infected with sh-luc virus or sh-G2 virus with 100 ng/ml recombinant human CXCL2. The Kasumi-1/sh-luc cells and Kasumi-1/sh-G2 cells (1 × 105) were plated, and cells were counted every second day. Cells were passaged at a density of 1 × 105 cells at day 4 (n = 9, mean ± SE, *p < 0.05).
(E) Representative plots from proliferation analysis of Kasumi-1/sh-luc cells and Kasumi-1/sh-G2 cells treated with or without 100 ng/ml recombinant human CXCL2 using CellTrace Violet dye. A greater number of generations indicates the cells underwent more rounds of cell division.
(F) Proliferation index of Kasumi-1/sh-luc cells and Kasumi-1/sh-G2 cells. Cells were cultured with or without 100 ng/ml CXCL2 for 6 days (n = 3, mean ± SE, **p < 0.01 and ***p < 0.001).
See also Figure S5.
p38/ERK-GATA-2 Positive-Feedback Circuit in Human AML Patients
We tested whether GATA2 mRNA correlates with GATA-2 target gene mRNAs in AML patient samples. Comparison of the mRNAs of 19,798 genes in AML patient bone marrow data from The Cancer Genome Atlas (TCGA) (n = 196) (Figure 6A) revealed correlations among GATA2, IL1B, and CXCL2 mRNA levels in AML-M5 patients (Figures 6B and 6C). In AML-M5, high GATA2 expression correlates with poor prognosis (Luesink et al., 2012). IL1B and GATA2 mRNA levels also correlated in M1, M3, and M4 AML patients (Figure S6A). Similarly, IL1B and CXCL2 mRNA levels also correlated in M0, M3, and M4 AML (Figure S6A). IL-1β induces expression of pro-inflammatory genes including chemokines (Apte and Voronov, 2008). In Kasumi-1 cells, IL-1β induced CXCL2 expression (Figure 6D), indicating that GATA-2 increases CXCL2 expression directly via transcriptional activation and indirectly via IL-1β. These results conform to a type 1 coherent feedforward loop. CXCL2 also increased IL1B mRNA expression (Figure 6D). Analysis of additional GATA-2 target genes, HDC, RUNX1, and IRF8 (Gao et al., 2013; Katsumura et al., 2014), revealed GATA2 mRNA levels correlated with HDC and RUNX1 mRNAs, although IRF8 mRNA negatively correlated (Figures S6C–S6F).
Figure 6. GATA2, IL1B, and CXCL2 Expression in AML Patients.

(A) Heatmap depicts 19,798 genes, based on ranking of the correlation with GATA2 expression.
(B) Heatmap depicts GATA2, CXCL2, and IL1B mRNA correlations.
(C) Scatterplot depicts correlations among GATA2, CXCL2, and IL1B mRNA expression in M5-AML patients.
(D) Real-time RT-PCR analysis of CXCL2 and IL1B mRNA in Kasumi-1 cells treated with 10 ng/ml IL-1β or 100 ng/ml CXCL2 (n = 3, mean ± SE). Kasumi-1 cells were serum-starved overnight and treated with IL-1β or CXCL2 for 30 min (*p < 0.05).
(E) Kaplan-Meier plots compare overall survival of patients with high versus low GATA2, CXCL2, or IL1B mRNA.
(F) Kaplan-Meier plots depict overall survival of patients with high versus low GATA2/IL1B mRNA, GATA2/CXCL2 mRNA, and IL1B/CXCL2 mRNA. This analysis was based on the cohort of 163 patients with CN-AML from the GEO: GSE12417 dataset (Metzeler et al., 2008).
See also Figures S6 and S7.
Similar GATA2 and CXCL2 mRNA levels correlated in a distinct human AML patient dataset (Stirewalt et al., 2008). In this dataset, GATA2 and CXCL2 mRNA levels were significantly higher in AML patients (Figure S7B). Although the samples were bone marrow and peripheral blood mononuclear cell derived, correlations between GATA2 and CXCL2 mRNAs and between IL1B and CXCL2 mRNAs were most obvious with bone marrow (Figure S7C).
We asked whether GATA2, IL1B, and CXCL2 mRNA levels predict AML prognosis in the publicly available dataset GEO: GSE12417 (n = 163) (Metzeler et al., 2008). High CXCL2 expression correlated with shorter survival (Figure 6E). High GATA2 + CXCL2 expression and high IL1B and CXCL2 expression correlated with shorter survival in the GEO: GSE12417 cohort (Figure 6F). GATA2 and CXCL2 mRNA levels correlated, as well as CXCL2 and IL1B, in M5-AML patients, resembling the TCGA dataset (Figures 6C and S7D).
DISCUSSION
Many reports have described inhibitory GATA2 mutations and elevated GATA2 expression in AML (Bresnick et al., 2012; Dickinson et al., 2014; Ganapathi et al., 2015; Spinner et al., 2014; Wang et al., 2015a; Wlodarski et al., 2016). Herein we described a GATA-2-chemokine/cytokine circuit that regulates AML cell proliferation. p38/ERK signaling phosphorylated GATA-2 via a mechanism requiring a GATA-2 DEF motif. This mechanism increased GATA-2 chromatin occupancy and expression of target genes, which constitute a leukemia cell growth-regulatory circuit. By upregulating IL1B and CXCL2 expression, p38/ERK-GATA-2 instigates a positive-feedback mechanism that increases AML cell proliferation. Given the clinical correlations among GATA2, IL1B, and CXCL2 mRNA levels and the correlation between high GATA2/CXCL2 expression and poor prognosis of cytogenetically normal (CN)-AML patients, this circuit illustrates a mechanistic link between altered GATA-2 levels/activity and AML. It is reasonable to assume that the signal-dependent GATA-2 mechanism impacts a broader repertoire of functionally important targets that interface with the circuit described herein.
Although GATA-2 overexpression and GATA-2 loss are implicated in AML, the mechanisms by which GATA-2 deregulation contributes to leukemogenesis were unclear. As the p38/ERK-GATA-2 axis regulates IL1B and CXCL2, both implicated in leukemogenesis, it is attractive to consider the role of this axis in the link between high GATA-2 expression and poor-prognosis AML. We propose that GATA-2 stimulates AML cell proliferation, in part, through CXCL2 and IL1B induction (Figure 7). Can this circuit provide an avenue for developing AML therapeutics? p38 and ERK signaling are implicated in AML cell proliferation (Birkenkamp et al., 1999). p38 inhibition enhances the anti-leukemic activity of Birinapant, a smac mimetic (Lalaoui et al., 2016). Besides MAPK inhibitors, HSP90 inhibitors suppress ERK signaling (Zong et al., 2015) and Rac inhibitors suppress p38 signaling (Zhang et al., 1995). Both inhibitors exert anti-leukemic activity. CXCR2 is activated by CXCL1, 2, 3, 5, 6, 7, and 8 (Balkwill, 2004). As CXCR2 antagonism impairs AML cell growth in vivo (Schinke et al., 2015), it will be instructive to consider combining interventions upstream (anti-p38 or ERK) and downstream of GATA-2 (anti-CXCR2), as well as to develop CXCL2-selective antagonists, to target GATA-2-driven AML. Interestingly, while Schinke et al. (2015) focused on IL-8, CXCL2 was the second most highly expressed CXCR2 ligand in AML patients.
Figure 7. p38/ERK-GATA-2 Axis Function in AML Cells.

Ras-p38/ERK signaling increases GATA-2 phosphorylation. GATA-2 phos-phorylation facilitates GATA-2 chromatin occupancy at GATA-2 target genes. GATA-2 stimulates GATA2 transcription through positive autoregulation. Loci are differentially sensitive to the signal-dependent GATA-2 mechanism. GATA-2 upregulates IL1B and CXCL2 expression. These factors activate p38 and ERK signaling, enhancing GATA-2 activity through a positive-feedback circuit. GATA-2 increases CXCL2 transcription directly or indirectly (through IL-1β), which constitutes a type I coherent feedforward loop (FFL). This GATA-2-chemokine/cytokine circuit is predicted to be an important determinant of AML cell proliferation.
Consistent with our results, transformed B-lymphocytes from patients with MonoMAC syndrome have decreased CXCL2 expression (Hsu et al., 2013). Decreased production of GATA-2-regulated inflammatory mediators may have implications for MonoMAC immunodeficiency. In this regard, IL1B or CXCR2 mutations increase susceptibility to pathogens (Hang et al., 2000; Raupach et al., 2006).
The inverse correlation of GATA2 and IRF8 expression (Figures S6C and S6F) is of considerable interest, since Irf8 mutations cause leukemogenesis in mice (Holtschke et al., 1996) and decreased IRF8 expression can characterize AML and MDS patients (Otto et al., 2011; Qian et al., 2010). GATA-2-regulated leukemogenesis may, therefore, involve increased production of growth factors and tumor suppressor downregulation.
In addition to MDS and AML, GATA2 is implicated in non-small-cell lung cancer, prostate cancer, and glioma (Kumar et al., 2012; Vidal et al., 2015; Wang et al., 2015b). GATA2 confers chemotherapy resistance of prostate cancer cells and JNK and AKT are linked to resistance. GATA2 contributes to glioma cell proliferation in an ERK-dependent manner (Wang et al., 2015b). Although it is unclear whether GATA-2 is phosphorylated in these cancer cells, given the broad impact of p38/ERK signaling, the p38/ERK-GATA-2 axis also may function in these diseases and, therefore, constitute a foundation for developing therapies for GATA-2-linked solid tumors.
EXPERIMENTAL PROCEDURES
Cell Culture
Kasumi-1 and Kasumi-3 cells were maintained in RPMI 1640 medium containing 1% penicillin-streptomycin and 20% fetal bovine serum (FBS). Kasumi-1 cells stably infected with sh-luc or sh-GATA2 viruses were maintained in 2 μg/ml puromycin. MAE cells were maintained in Medium 200 (Gibco/Invitrogen) containing Low Serum Growth Supplement (Gibco/Invitrogen) and 1% penicillin-streptomycin. Cells were transfected with the Nucleofector II.
Real-Time RT-PCR
Total RNA was purified with TRIzol. The cDNA was synthesized with murine Moloney leukemia virus reverse transcriptase. PCR product accumulation was monitored by SYBR green fluorescence.
ChIP
Samples containing 3 × 106 cells were crosslinked with 1% formaldehyde for 10 min. Lysates were immunoprecipitated with anti-GATA-2 antibody or rabbit pre-immune serum. The accession numbers for the ChIP-seq datasets are GEO: GSE45144 and GSE73207.
FAIRE
Cells were fixed with 1% formaldehyde and sonicated to shear the DNA. Then 10% of the sonicated chromatin was used as the input.
Protein Analysis
Protein samples from 1 × 106 cells were resolved by SDS-PAGE and proteins were detected by western blotting.
Phosphatase Treatment
Total protein was prepared by lysing cells in radio-immunoprecipitation assay (RIPA) buffer. For phosphatase treatment, proteins were incubated with λ-phosphatase at 30°C for 90 min and analyzed by western blotting.
Proliferation Analysis
Cells (1 × 106) were labeled with 10 μM CellTrace Violet Dye. Then 6 days later, CellTrace Violet fluorescence intensity was measured.
Patient Sample Analysis
Using clinical data from TCGA for AML, 196 patients were identified for the analysis. The Spearman correlation coefficient was computed. Clinical data from GEO: GSE12417, with a cohort of 163 patients with CN-AML, was used for Kaplan-Meier survival analysis (Metzeler et al., 2008). The accession numbers for clinical datasets are GEO: GSE1159, GSE9476, and GSE12417.
Statistical Analysis
Statistical significance was determined by paired Student’s t test using web-based GraphPad (http://www.graphpad.com). Significance of Kaplan-Meier survival analysis was determined by log-rank test.
Supplementary Material
Highlights.
p38/ERK functions through a docking motif in GATA-2 to phosphorylate GATA-2 in AML cells
p38/ERK mechanism increases GATA-2 activity at select target genes in AML cells
GATA-2-regulated chemokine/cytokine circuit controls Kasumi-1 cell proliferation
GATA-2-chemokine/cytokine circuit predicts poor prognosis of an AML subtype
Acknowledgments
This work was supported by NIH DK68634 (E.H.B.), Midwest Athletes Against Childhood Cancer Organization (E.H.B.), and Cancer Center Support Grant P30 CA014520. K.R.K. was supported by the Kanae Foundation for the Promotion of Medical Science. A.W.D. was supported by a Cancer Biology Pre-doctoral NIH Training Grant from the NIH (T32CA009135) and an American Heart Association Predoctoral Fellowship. I.M.O. was supported by the Cancer Center Support Grant P30 CA014520 from the Carbone Cancer Center and NIH National Center for Advancing Translational Sciences (NCATS) grant UL1TR000427.
Footnotes
ACCESSION NUMBERS
The accession numbers for the ChIP-seq dataset of GATA-2 in Kasumi-3 cells reported in this paper is GEO: GSE84782.
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.058.
AUTHOR CONTRIBUTIONS
Conceptualization, K.R.K. and E.H.B.; Methodology, K.R.K., I.M.O., A.W.D., and R.S.; Investigation, K.R.K., I.M.O., A.W.D., and R.S.; Writing – Original Draft, K.R.K. and E.H.B.; Writing – Review & Editing, K.R.K., I.M.O., A.W.D., and E.H.B.; Funding Acquisition, E.H.B.; Resources, K.R.K. and E.H.B.; Supervision, E.H.B.
References
- Ågerstam H, Karlsson C, Hansen N, Sandén C, Askmyr M, von Palffy S, Högberg C, Rissler M, Wunderlich M, Juliusson G, et al. Antibodies targeting human IL1RAP (IL1R3) show therapeutic effects in xenograft models of acute myeloid leukemia. Proc Natl Acad Sci USA. 2015;112:10786–10791. doi: 10.1073/pnas.1422749112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte RN, Voronov E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol Rev. 2008;222:222–241. doi: 10.1111/j.1600-065X.2008.00615.x. [DOI] [PubMed] [Google Scholar]
- Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood. 1991;77:2031–2036. [PubMed] [Google Scholar]
- Asou H, Suzukawa K, Kita K, Nakase K, Ueda H, Morishita K, Kamada N. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn J Cancer Res. 1996;87:269–274. doi: 10.1111/j.1349-7006.1996.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, Ben-Neriah S, Montagna C, Parekh S, Pellagatti A, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120:1290–1298. doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck D, Thoms JA, Perera D, Schütte J, Unnikrishnan A, Knezevic K, Kinston SJ, Wilson NK, O’Brien TA, Göttgens B, et al. Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood. 2013;122:e12–e22. doi: 10.1182/blood-2013-03-490425. [DOI] [PubMed] [Google Scholar]
- Birkenkamp KU, Dokter WH, Esselink MT, Jonk LJ, Kruijer W, Vellenga E. A dual function for p38 MAP kinase in hematopoietic cells: involvement in apoptosis and cell activation. Leukemia. 1999;13:1037–1045. doi: 10.1038/sj.leu.2401447. [DOI] [PubMed] [Google Scholar]
- Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, Leblanc T, Thomas X, Hermine O, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006;20:965–970. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- Bradbury D, Bowen G, Kozlowski R, Reilly I, Russell N. Endogenous interleukin-1 can regulate the autonomous growth of the blast cells of acute myeloblastic leukemia by inducing autocrine secretion of GM-CSF. Leukemia. 1990;4:44–47. [PubMed] [Google Scholar]
- Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem. 2010;285:31087–31093. doi: 10.1074/jbc.R110.159079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick EH, Katsumura KR, Lee HY, Johnson KD, Perkins AS. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res. 2012;40:5819–5831. doi: 10.1093/nar/gks281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celton M, Forest A, Gosse G, Lemieux S, Hebert J, Sauvageau G, Wilhelm BT. Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia. 2014;28:1617–1626. doi: 10.1038/leu.2014.67. [DOI] [PubMed] [Google Scholar]
- Coombs CC, Tallman MS, Levine RL. Molecular therapy for acute myeloid leukaemia. Nat Rev Clin Oncol. 2016;13:305–318. doi: 10.1038/nrclinonc.2015.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delwel R, van Buitenen C, Salem M, Bot F, Gillis S, Kaushansky K, Altrock B, Löwenberg B. Interleukin-1 stimulates proliferation of acute myeloblastic leukemia cells by induction of granulocyte-macrophage colony-stimulating factor release. Blood. 1989;74:586–593. [PubMed] [Google Scholar]
- Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–2658. doi: 10.1182/blood-2011-06-360313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson RE, Milne P, Jardine L, Zandi S, Swierczek SI, McGovern N, Cookson S, Ferozepurwalla Z, Langridge A, Pagan S, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123:863–874. doi: 10.1182/blood-2013-07-517151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois C, Schlageter MH, de Gentile A, Balitrand N, Toubert ME, Krawice I, Fenaux P, Castaigne S, Najean Y, Degos L, et al. Modulation of IL-8, IL-1 beta, and G-CSF secretion by all-trans retinoic acid in acute promyelocytic leukemia. Leukemia. 1994;8:1750–1757. [PubMed] [Google Scholar]
- Estrov Z, Manna SK, Harris D, Van Q, Estey EH, Kantarjian HM, Talpaz M, Aggarwal BB. Phenylarsine oxide blocks interleukin-1beta-induced activation of the nuclear transcription factor NF-kappaB, inhibits proliferation, and induces apoptosis of acute myelogenous leukemia cells. Blood. 1999;94:2844–2853. [PubMed] [Google Scholar]
- Ganapathi KA, Townsley DM, Hsu AP, Arthur DC, Zerbe CS, Cuellar-Rodriguez J, Hickstein DD, Rosenzweig SD, Braylan RC, Young NS, et al. GATA2 deficiency-associated bone marrow disorder differs from idiopathic aplastic anemia. Blood. 2015;125:56–70. doi: 10.1182/blood-2014-06-580340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Johnson KD, Chang YI, Boyer ME, Dewey CN, Zhang J, Bresnick EH. Gata2 cis-element is required for hematopoietic stem cell generation in the mammalian embryo. J Exp Med. 2013;210:2833–2842. doi: 10.1084/jem.20130733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans BF, Zwaan CM, Miller M, Zimmermann M, Harlow A, Meshinchi S, Loonen AH, Hählen K, Reinhardt D, Creutzig U, et al. Mutations in KIT and RAS are frequent events in pediatric core-binding factor acute myeloid leukemia. Leukemia. 2005;19:1536–1542. doi: 10.1038/sj.leu.2403870. [DOI] [PubMed] [Google Scholar]
- Grass JA, Jing H, Kim S-I, Martowicz ML, Pal S, Blobel GA, Bresnick EH. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol. 2006;26:7056–7067. doi: 10.1128/MCB.01033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gröschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BA, Erpelinck C, van der Velden VH, Havermans M, Avellino R, van Lom K, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–381. doi: 10.1016/j.cell.2014.02.019. [DOI] [PubMed] [Google Scholar]
- Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. doi: 10.1038/ng.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang L, Frendéus B, Godaly G, Svanborg C. Interleukin-8 receptor knockout mice have subepithelial neutrophil entrapment and renal scarring following acute pyelonephritis. J Infect Dis. 2000;182:1738–1748. doi: 10.1086/317599. [DOI] [PubMed] [Google Scholar]
- Holtschke T, Löhler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch KP, Gabriele L, Waring JF, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, Patel SY, Frucht DM, Vinh DC, Auth RD, Freeman AF, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–2655. doi: 10.1182/blood-2011-05-356352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AP, Johnson KD, Falcone EL, Sanalkumar R, Sanchez L, Hickstein DD, Cuellar-Rodriguez J, Lemieux JE, Zerbe CS, Bresnick EH, Holland SM. GATA2 haploinsufficiency caused by mutations in a conserved intronic element leads to MonoMAC syndrome. Blood. 2013;121:3830–3837. S1–S7. doi: 10.1182/blood-2012-08-452763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999;13:163–175. [PMC free article] [PubMed] [Google Scholar]
- Johnson KD, Hsu AP, Ryu MJ, Wang J, Gao X, Boyer ME, Liu Y, Lee Y, Calvo KR, Keles S, et al. Cis-element mutated in GATA2-dependent immunodeficiency governs hematopoiesis and vascular integrity. J Clin Invest. 2012;122:3692–3704. doi: 10.1172/JCI61623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KD, Kong G, Gao X, Chang YI, Hewitt KJ, Sanalkumar R, Prathibha R, Ranheim EA, Dewey CN, Zhang J, Bresnick EH. Cis-regulatory mechanisms governing stem and progenitor cell transitions. Sci Adv. 2015;1:e1500503. doi: 10.1126/sciadv.1500503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumura KR, Yang C, Boyer ME, Li L, Bresnick EH. Molecular basis of crosstalk between oncogenic Ras and the master regulator of hematopoiesis GATA-2. EMBO Rep. 2014;15:938–947. doi: 10.15252/embr.201438808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepin HD, Rao AV, Pardee TS. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J Clin Oncol. 2014;32:2541–2552. doi: 10.1200/JCO.2014.55.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knall C, Young S, Nick JA, Buhl AM, Worthen GS, Johnson GL. Interleukin-8 regulation of the Ras/Raf/mitogen-activated protein kinase pathway in human neutrophils. J Biol Chem. 1996;271:2832–2838. doi: 10.1074/jbc.271.5.2832. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Hancock DC, Molina-Arcas M, Steckel M, East P, Diefenbacher M, Armenteros-Monterroso E, Lassailly F, Matthews N, Nye E, et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell. 2012;149:642–655. doi: 10.1016/j.cell.2012.02.059. [DOI] [PubMed] [Google Scholar]
- Lalaoui N, Hänggi K, Brumatti G, Chau D, Nguyen NY, Vasilikos L, Spilgies LM, Heckmann DA, Ma C, Ghisi M, et al. Targeting p38 or MK2 enhances the anti-leukemic activity of Smac-mimetics. Cancer Cell. 2016;29:145–158. doi: 10.1016/j.ccell.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Linnemann AK, O’Geen H, Keles S, Farnham PJ, Bresnick EH. Genetic framework for GATA factor function in vascular biology. Proc Natl Acad Sci USA. 2011;108:13641–13646. doi: 10.1073/pnas.1108440108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luesink M, Hollink IH, van der Velden VH, Knops RH, Boezeman JB, de Haas V, Trka J, Baruchel A, Reinhardt D, van der Reijden BA, et al. High GATA2 expression is a poor prognostic marker in pediatric acute myeloid leukemia. Blood. 2012;120:2064–2075. doi: 10.1182/blood-2011-12-397083. [DOI] [PubMed] [Google Scholar]
- Lugus JJ, Chung YS, Mills JC, Kim SI, Grass J, Kyba M, Doherty JM, Bresnick EH, Choi K. GATA2 functions at multiple steps in hemangioblast development and differentiation. Development. 2007;134:393–405. doi: 10.1242/dev.02731. [DOI] [PubMed] [Google Scholar]
- Mazumdar C, Shen Y, Xavy S, Zhao F, Reinisch A, Li R, Corces MR, Flynn RA, Buenrostro JD, Chan SM, et al. Leukemia-Associated Cohesin Mutants Dominantly Enforce Stem Cell Programs and Impair Human Hematopoietic Progenitor Differentiation. Cell Stem Cell. 2015;17:675–688. doi: 10.1016/j.stem.2015.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzeler KH, Hummel M, Bloomfield CD, Spiekermann K, Braess J, Sauerland MC, Heinecke A, Radmacher M, Marcucci G, Whitman SP, et al. Leukemia Group B; German AML Cooperative Group. An 86-probe-set gene-expression signature predicts survival in cyto-genetically normal acute myeloid leukemia. Blood. 2008;112:4193–4201. doi: 10.1182/blood-2008-02-134411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome) Nat Genet. 2011;43:929–931. doi: 10.1038/ng.923. [DOI] [PubMed] [Google Scholar]
- Otto N, Manukjan G, Göhring G, Hofmann W, Scherer R, Luna JC, Lehmann U, Ganser A, Welte K, Schlegelberger B, Steinemann D. ICSBP promoter methylation in myelodysplastic syndromes and acute myeloid leukaemia. Leukemia. 2011;25:1202–1207. doi: 10.1038/leu.2011.61. [DOI] [PubMed] [Google Scholar]
- Persons DA, Allay JA, Allay ER, Ashmun RA, Orlic D, Jane SM, Cunningham JM, Nienhuis AW. Enforced expression of the GATA-2 transcription factor blocks normal hematopoiesis. Blood. 1999;93:488–499. [PubMed] [Google Scholar]
- Qian Z, Joslin JM, Tennant TR, Reshmi SC, Young DJ, Stoddart A, Larson RA, Le Beau MM. Cytogenetic and genetic pathways in therapy-related acute myeloid leukemia. Chem Biol Interact. 2010;184:50–57. doi: 10.1016/j.cbi.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaldi A, Torcia M, Bettoni S, Vannier E, Barbui T, Shaw AR, Dinarello CA, Cozzolino F. Modulation of cell proliferation and cytokine production in acute myeloblastic leukemia by interleukin-1 receptor antagonist and lack of its expression by leukemic cells. Blood. 1991;78:3248–3253. [PubMed] [Google Scholar]
- Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanalkumar R, Johnson KD, Gao X, Boyer ME, Chang YI, Hewitt KJ, Zhang J, Bresnick EH. Mechanism governing a stem cell-generating cis-regulatory element. Proc Natl Acad Sci USA. 2014;111:E1091–E1100. doi: 10.1073/pnas.1400065111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinke C, Giricz O, Li W, Shastri A, Gordon S, Barreyro L, Bhagat T, Bhattacharyya S, Ramachandra N, Bartenstein M, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015;125:3144–3152. doi: 10.1182/blood-2015-01-621631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Mao CQ, Yang XZ, Du XJ, Liu Y, Zhu YH, Wang J. Cationic lipid-assisted polymeric nanoparticle mediated GATA2 siRNA delivery for synthetic lethal therapy of KRAS mutant non-small-cell lung carcinoma. Mol Pharm. 2014;11:2612–2622. doi: 10.1021/mp400714z. [DOI] [PubMed] [Google Scholar]
- Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, Arthur DC, Gu W, Gould CM, Brewer CC, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–821. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckel M, Molina-Arcas M, Weigelt B, Marani M, Warne PH, Kuznetsov H, Kelly G, Saunders B, Howell M, Downward J, Hancock DC. Determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells reveals novel therapeutic targeting strategies. Cell Res. 2012;22:1227–1245. doi: 10.1038/cr.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirewalt DL, Meshinchi S, Kopecky KJ, Fan W, Pogosova-Agadjanyan EL, Engel JH, Cronk MR, Dorcy KS, McQuary AR, Hockenbery D, et al. Identification of genes with abnormal expression changes in acute myeloid leukemia. Genes Chromosomes Cancer. 2008;47:8–20. doi: 10.1002/gcc.20500. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Hino M, Kutsuna H, Hato F, Sakamoto C, Takahashi T, Tatsumi N, Kitagawa S. Selective activation of p38 mitogen-activated protein kinase cascade in human neutrophils stimulated by IL-1beta. J Immunol. 2001;167:5940–5947. doi: 10.4049/jimmunol.167.10.5940. [DOI] [PubMed] [Google Scholar]
- Tsai FY, Keller G, Kuo FC, Weiss M, Chen J, Rosenblatt M, Alt FW, Orkin SH. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221–226. doi: 10.1038/371221a0. [DOI] [PubMed] [Google Scholar]
- Turzanski J, Grundy M, Russell NH, Pallis M. Interleukin-1beta maintains an apoptosis-resistant phenotype in the blast cells of acute myeloid leukaemia via multiple pathways. Leukemia. 2004;18:1662–1670. doi: 10.1038/sj.leu.2403457. [DOI] [PubMed] [Google Scholar]
- Tzarum N, Komornik N, Ben Chetrit D, Engelberg D, Livnah O. DEF pocket in p38α facilitates substrate selectivity and mediates auto-phosphorylation. J Biol Chem. 2013;288:19537–19547. doi: 10.1074/jbc.M113.464511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente C, Vazquez I, Conchillo A, García-Sánchez MA, Marcotegui N, Fuster O, González M, Calasanz MJ, Lahortiga I, Odero MD. Overexpression of GATA2 predicts an adverse prognosis for patients with acute myeloid leukemia and it is associated with distinct molecular abnormalities. Leukemia. 2012;26:550–554. doi: 10.1038/leu.2011.235. [DOI] [PubMed] [Google Scholar]
- Vidal SJ, Rodriguez-Bravo V, Quinn SA, Rodriguez-Barrueco R, Lujambio A, Williams E, Sun X, de la Iglesia-Vicente J, Lee A, Readhead B, et al. A targetable GATA2-IGF2 axis confers aggressiveness in lethal prostate cancer. Cancer Cell. 2015;27:223–239. doi: 10.1016/j.ccell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Muramatsu H, Okuno Y, Sakaguchi H, Yoshida K, Kawashima N, Xu Y, Shiraishi Y, Chiba K, Tanaka H, et al. GATA2 and secondary mutations in familial myelodysplastic syndromes and pediatric myeloid malignancies. Haematologica. 2015a;100:e398–e401. doi: 10.3324/haematol.2015.127092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Yuan H, Sun C, Xu L, Chen Y, Zhu Q, Zhao H, Huang Q, Dong J, Lan Q. GATA2 promotes glioma progression through EGFR/ERK/Elk-1 pathway. Med Oncol. 2015b;32:87. doi: 10.1007/s12032-015-0522-1. [DOI] [PubMed] [Google Scholar]
- Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120:3397–3406. doi: 10.1182/blood-2012-05-378596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarski MW, Hirabayashi S, Pastor V, Starý J, Hasle H, Masetti R, Dworzak M, Schmugge M, van den Heuvel-Eibrink M, Ussowicz M, et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood. 2016;127:1387–1397. doi: 10.1182/blood-2015-09-669937. quiz 1518. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, Yamamoto M. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25:415–427. doi: 10.1016/j.ccr.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Han J, Sells MA, Chernoff J, Knaus UG, Ulevitch RJ, Bokoch GM. Rho family GTPases regulate p38 mitogen-activated protein kinase through the downstream mediator Pak1. J Biol Chem. 1995;270:23934–23936. doi: 10.1074/jbc.270.41.23934. [DOI] [PubMed] [Google Scholar]
- Zhang SJ, Ma LY, Huang QH, Li G, Gu BW, Gao XD, Shi JY, Wang YY, Gao L, Cai X, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105:2076–2081. doi: 10.1073/pnas.0711824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Gozman A, Caldas-Lopes E, Taldone T, Sturgill E, Brennan S, Ochiana SO, Gomes-DaGama EM, Sen S, Rodina A, et al. A hyperactive signalosome in acute myeloid leukemia drives addiction to a tumor-specific Hsp90 species. Cell Rep. 2015;13:2159–2173. doi: 10.1016/j.celrep.2015.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwaan CM, Kolb EA, Reinhardt D, Abrahamsson J, Adachi S, Aplenc R, De Bont ES, De Moerloose B, Dworzak M, Gibson BE, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33:2949–2962. doi: 10.1200/JCO.2015.62.8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.