

Abstract
Isolates of the human pathogen Mycobacterium tuberculosis recovered from clinical samples exhibit genetic heterogeneity. Such variation may result from the stressful environment encountered by the pathogen inside the macrophage, which is the host cell tubercle bacilli parasitize. To study the evolution of the M. tuberculosis genome during growth inside macrophages, we developed a model of intracellular culture in which bacteria were serially passaged in macrophage-like THP-1 cells for about 80 bacterial generations. Genome sequencing of single bacterial colonies isolated before and after the infection cycles revealed that M. tuberculosis developed mutations at a rate of about 5.7 × 10−9 / bp/ generation, consistent with mutation rates calculated during in vivo infection. Analysis of mutant growth in macrophages and in mice showed that the mutations identified after the cyclic infection conferred no advantage to the mutants relative to wild-type. Furthermore, activity testing of the recombinant protein harboring one of these mutations showed that the presence of the mutation did not affect the enzymatic activity. The serial infection protocol developed in this work to study M. tuberculosis genome microevolution can be applied to exposure to stressors to determine their effect on genome remodeling during intra-macrophage growth.
Introduction
Tuberculosis remains a global health concern; the evolution of drug resistance represents a major impediment to anti-tuberculosis efforts. While Mycobacterium tuberculosis was long thought to be genetically monomorphic, recent studies have shown that heterogeneity can be found within the infecting bacterial population [1–4]. Indeed, many low-frequency genetic variants, most of which are transient, coexist in clinical samples [5]. M. tuberculosis genetic heterogeneity observed in samples collected from infected humans at multiple times post-infection [6, 7], in different clinical stages of disease [8], from different anatomical sites [9], or even within the same lesion [8], prompts the question of whether mutations developed during infection confer a bacterial phenotype more adapted to the host environment.
Since M. tuberculosis resides within host macrophages for much of the infection [10, 11], these phagocytes may represent a potent contributor to M. tuberculosis genetic heterogeneity. The role of macrophages as driving force for bacterial genome evolution has been suggested by macrophage infection experiments performed with other pathogens [12]. In addition, heterogeneity among intracellular mycobacteria has been observed in short-term macrophage infections in vitro [13]. Many factors in the macrophage environment may have a mutagenic effect [14]. For example, oxidative stress has been proposed as a major mutagenic agent encountered by M. tuberculosis during in vivo infection [8]. The macrophage environment may not only induce the development of new mutations, but also operate a selective pressure on M. tuberculosis, leading to selection and fixation of adaptive mutations in the bacterial population. Typical protocols of macrophage infection are not amenable to long-term bacterial genome evolution studies due to the short lifespan of in vitro infected cells.
To determine whether mutations occur in the M. tuberculosis DNA during macrophage infection, we developed a new model of serial macrophage infections. Tubercle bacilli were passaged in macrophages for multiple, consecutive cycles for a total of 80 bacterial generations; passaging was associated with bacterial mutations that were identified by whole genome sequencing.
Material and Methods
Bacterial strains and growth conditions
To establish the initial inoculum for the M. tuberculosis intracellular culture model, a single colony-derived bacterial culture of M. tuberculosis H37RvSiena strain (P007027.1) [15] was obtained by disruption of clumps by shaking with glass beads [16] followed by plating on solid medium. A single colony was subcultured to prepare bacterial stocks that were used for the first cycle of THP-1 infection.
M. tuberculosis cultures were grown in Middlebrook 7H9 broth (Becton Dickinson, Sparks, MD) (liquid medium) or 7H10 (solid medium) (Difco, Franklin Lakes, NJ) supplemented with 0.05% Tween 80, 0.2% glycerol and 10% ADN (2% glucose, 5% bovine serum albumin, 0.15 M NaCl). Liquid cultures of M. tuberculosis were grown in 25-ml tubes at 37°C with magnetic-bar stirring at 450 rpm and optical density was measured at 580 nm with a densitometer. Plates were incubated at 37°C in sealed plastic bags.
Macrophage culture and differentiation
The THP-1 human monocytic cell line was obtained from the American Type Culture Collection (ATCC TIB-202). Cells were grown in RPMI 1640 medium containing 2 mM L-glutamine (Corning Cellgro, Manassas, VA) and supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich, St. Louis, MO), maintaining a concentration of 1 × 105 to 5 × 105 cells/ml. Cell aliquots were frozen in FBS containing 10% dimethylsulfoxide. For each infection experiment, one aliquot was thawed and cells were grown for four passages and subsequently differentiated by treatment with 40 nM phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich, St. Louis, MO) for 72 h at the concentration of 5 × 105 cells/ml in 25 cm2 cell culture flasks (Costar, New York, NY). Cell differentiation following PMA stimulation was monitored over time by microscopic observation of cell morphology, plastic adherence proprieties, and analysis of CD11b and CD11c macrophage marker expression [17, 18].
To analyze marker expression, 1 × 106 cells were stained with Cy-Chrome-conjugated anti-human CD11b and PE-conjugated anti-human Cd11c (BD Biosciences, San Jose, CA) for 30 min at 4°C, washed, resuspended in 1 × PBS, and analyzed by flow cytometry with a FACSCalibur instrument (BD Biosciences, San Jose, CA). The number of total and viable adherent cells was determined in all experiments by detaching cells from the flasks, staining with 0.4% Trypan Blue solution (Sigma-Aldrich, St. Louis, MO) and counting in a TC10 automated cell counter (Biorad, Hercules, CA).
Serial passaging of M. tuberculosis in THP-1 cells and collection of single-colony isolates
Infections were performed as previously described [16]. For the first cycle of infection, an aliquot of the bacterial culture was defrosted and diluted in RPMI 1640 medium containing 2mM L-glutamine and 10% FBS. After clump disaggregation by shaking with 3mm-diameter glass beads, one aliquot of the bacterial culture was serially diluted and plated for CFU determination and a second aliquot was added to four 25 cm2 tissue culture flasks (2.1 × 104 CFU/flask), each containing 2.1 × 106 THP-1 cells to obtain an expected multiplicity of infection (MOI) of 1:100 (bacterial CFU:cells). After one day of incubation at 37°C in 5% CO2 atmosphere, medium was removed, and cell monolayers were washed with 1× PBS for three times to remove extracellular bacteria. One flask was used for THP-1 count while a second flask was used to determine the number of intracellular bacterial CFU. In this second flask, THP-1 cells were lysed by adding 0.01% sodium dodecyl sulphate (SDS) in ice-cold water and incubating at room temperature for 20 min. The cell lysate was centrifuged at 3,800 × g for 30 min and the sedimented bacteria were resuspended in liquid medium for CFU enumeration. Fresh medium was added to the remaining two flasks that were incubated for six more days. At this time, cell culture medium was removed and the cell monolayers were washed with 1 × PBS three times. One flask was used to check the viability of THP-1 cells. Cells from the second flask were lysed as described above to release intracellular bacteria for the next infection cycle. Bacteria were collected by centrifugation of cell lysates, resuspended in RPMI medium, and treated with glass beads (as described above) to infect a new THP-1 culture. An aliquot of this suspension was used for CFU count. Following this procedure, ten serial infection cycles were performed. The number of bacterial intracellular generations was calculated for each cycle by using the following formula: (log2 CFU day 7- log2 CFU 18 h)/log2 2.
Thirteen single colonies were collected from the bacterial inoculum and from the intracellular bacterial population harvested at the end of the ten infection cycles for genomic DNA extraction and whole genome Illumina sequencing.
DNA genomic extraction
Genomic DNA was extracted from heat-inactivated bacteria by using the cetyltrimethylammonium bromide (CTAB)-lysozyme protocol, essentially as previously described [19]. To increase the efficiency of mycobacterial disruption, mechanical lysis was performed utilizing 150–212 μm-diameter acid washed glass beads (Sigma-Aldrich, St. Louis, MO) and a Vortex Genie 2 disrupter (Stratech Scientific ltd, Oaks Drive, UK).
Illumina sequencing and identification of genetic variations
Whole genome sequencing was performed at IGA Technology Services S.r.l. (Udine, Italy) using Illumina 2 × 100 bp paired-end reads HiSeq 2000 system. Reads were trimmed, quality filtered, and mapped against H37RvSiena as a reference sequence (CP007027.1) using the Mosaik Assembler software [20]. Genome fraction coverage and read depth were analyzed by Qualimap software [21].
Single nucleotide substitutions, insertions, and deletions were retrieved with the VarScan software [22] by setting a minimum read depth of fifteen and a minimum variant frequency of 50% required to call a variant. This cutoff was selected to exclude variants located in genomic regions containing prophages and repetitive sequences, where most sequencing errors occur. The mutations identified were validated by PCR amplification and Sanger sequencing (Sanger sequencing was performed by Eurofins MWG Operon, Ebersberg, Germany). The primers used for amplification and sequencing are listed in S1 Table.
Large genomic fragment mutations relative to the sequence of the H37RvSiena reference strain were evaluated as follows: the filtered reads obtained from the sequencing of each genome were de novo assembled by using Ray 2.3.0 [23] and the alignment of the contigs to H37RvSiena genome sequence was visualized by using Mauve 2.3.1 [24]. For polymorphisms located in coding sequences of genes, variations in amino acid sequences were predicted using the translation Expasy tool (http://web.expasy.org/translate/). Presence and location of a signal peptide and conserved protein domains were evaluated by TatP 1.0 (http://cbs.dtu.dk/services/TatP/), SignalP 4.1 (http://cbs.dtu.dk/services/SignalP/), and Pfam (http://pfam.sanger.ac.uk/) searches.
Mutation frequency and rate
To determine the frequency of the identified mutations in bacterial populations, aliquots of the inoculum and the tenth cycle culture were subjected to glass bead treatment and plated on agar plates for single-colony isolation. 37 single-colony isolates were collected and bacteria were mechanically lysed by shaking with 150–212 μm acid washed glass beads (Sigma-Aldrich, St. Louis, MO) in Tissue Lyser instrument (Qiagen, Hilden, Germany). Beads and unbroken bacteria were sedimented by centrifugation, and supernatant was used as PCR template. Amplification and Sanger sequencing of the regions containing the mutations were performed using the primers indicated in S1 Table. M. tuberculosis mutation rate was estimated by using a standard formula [8].
THP-1 infection with M. tuberculosis cut3 Val223Ala and plcA His39Arg mutant isolates
THP-1 cells were differentiated with 40 nM PMA for 3 days in 24 well plates (Costar, New York, NY) at a concentration of 5 × 105 cells per well. For infection, aliquots of wild type, cut3 Val223Ala and plcA His39Arg mutant bacteria were defrosted and diluted in RPMI 1640 medium containing 2 mM L-glutamine and 10% FBS to obtain an MOI of 1:10 (bacterial CFU:cells). After clump disruption by glass bead treatment, the bacterial culture was added to THP-1 cells. After one day of infection, the cell monolayers were washed three times with 1 × PBS to remove extracellular bacteria. Fresh medium was added to the culture and incubation was continued for six additional days. Bacterial CFU and THP-1 viability were determined at days 1, 3 and 7. For these purposes, the adherent cells were detached by incubation with 5 mM EDTA in 1 × PBS for 20 minutes, counted, and lysed with 0.05% SDS. The lysates were serially diluted and plated on agar plates for CFU enumeration. The CFU counts were normalized to the number of adherent THP-1 cells. Each experiment was performed in triplicate.
Murine model of infection
Eight-week-old female BALB/c mice were infected with approximately 300 CFU via the aerosol route with wild type, cut3 Val223Ala, and plcA His39Arg mutant strains. At 3h (time zero) and 2, 4, 8 and 12 weeks post-infection, 3–4 mice per condition were sacrificed by cervical dislocation. For bacterial CFU enumeration, one lung per mouse was removed aseptically and homogenized, and serial dilutions of organ homogenates were plated on solid medium. At 12 weeks post-infection, one lung per mouse infected with wild type and cut3 Val223Ala mutant was fixed in neutral formalin and embedded in paraffin for hematoxylin and eosin, and Ziehl-Neelsen staining. Lung sections were evaluated in a blinded manner for semi-quantitative evaluation of pathology, which was based on the following scoring criteria: 0- intact lung (no granuloma); 1- increased cellularity; 2- granulomas occupying up to 25% of lung section; 3- granulomas occupying 25–50% of lung section; 4- granulomas occupying 50–75% of lung section, and 5- granulomas occupying >75% of lung section with necrosis. The morphometric analysis of involved lung area and granuloma size was performed using Sigmascan Pro Software (Systat Software Inc, San Jose, CA), as described [25].
Cloning, expression, and purification of the wild type and His39Arg mutant PlcA protein
Purified genomic DNA of M. tuberculosis H37RvSiena wild type and plcA His39Arg mutant strains was used as template for PCR amplification of plcA gene by using the primers shown in S2 Table. The PCR products were digested with BsphI and HindIII restriction enzymes, purified, and cloned into the pMyC plasmid at the Nco1 and HindIII cloning sites. The gene was under the control of the acetamidase promoter, as previously described [26]. Recombinant plasmid pMyC-plcA was analyzed by nucleotide sequencing (GATC biotech) and used to transform Mycobacterium smegmatis mc2 155 GroEL1ΔC [27]. Expression and purification of recombinant wild type and His39Arg mutant PlcA proteins were performed as described by Bakala N’ Goma et al. [26]. Briefly, transformed M. smegmatis mc2 155 GroEL1ΔC was grown in Middlebrook 7H9 medium supplemented with 10% ADN, and 50 μg/ml Hygromycin B at 37°C with shaking to OD600 of 3. Expression of recombinant proteins was induced by treatment with 0.2% acetamide for 16 h. After induction, the cells were sedimented by centrifugation and resuspended in 30 ml of ice cold buffer A (10 mM Tris/HCl pH 8.0, 150 mM NaCl) prior to mechanical lysis with a French Press device at 1100 psi. After centrifugation of the lysate at 17,000 × g, the pellet was resuspended in ice cold buffer A, sonicated as described previously [26], and incubated with 1% N-laurylsarcosine at 4°C for 16 h. After incubation, samples were centrifuged at 17,000 × g for 30 min and then at 27,000 × g for 20 min at 4°C. Supernatants were loaded onto a ProBond Ni2+-agarose column (Thermo Fisher Scientific, Waltham, MA) pre-equilibrated in buffer A supplemented with 0.5% N-laurylsarcosine. The column was washed with 60 ml of buffer A, and elution was performed with a 20 mM to 500 mM imidazole gradient. Recombinant proteins eluted at 100 mM and 250 mM imidazole were analyzed by SDS-PAGE electrophoresis onto a 12% polyacrylamide gel. Fractions were pooled, dialyzed overnight at 4°C against buffer A, concentrated to obtain a final concentration of approximately 1 mg/ml, and stored at -80°C.
Phospholipase C assay
Phospholipase C activity of the recombinant wild type and mutant PlcA protein was tested by using the Amplex Red Phosphatidylcholine-Specific PLC Assay Kit and a spectrofluorimeter (Thermo Fisher Scientific, Waltham, MA), according to the manufacturer’s instructions. Bacillus cereus phospholipase C and heat-inactivated recombinant PlcA were used as positive and negative controls, respectively. The enzymatic activity (EA) corresponds to the mole number of fluorescent fatty acid release from the phosphatidylcholine per minute in arbitrary unit (AU). Specific activity was expressed in pmol/min and per mg of protein.
Statistical analysis
Two tailed Student’s t-test was used to analyze the data; statistical significance was defined as P < 0.05.
Nucleotide sequencing data availability
The full genome sequence of the strain M. tuberculosis H37RvSiena is available from the NCBI GenBank database, with the accession number CP007027.1.
Ethics statement
The animal study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of Rutgers, the State University of New Jersey (permit number: 14020D0417).
Results and Discussion
A model of extended intracellular growth of M. tuberculosis
To obtain long-term intracellular growth of M. tuberculosis, we developed a protocol in which THP-1 cells were serially infected with M. tuberculosis for a total of 10 infection cycles (Fig 1). Infection was performed with fully differentiated macrophages, as demonstrated by microscopic analysis, plastic adherence proprieties, and expression of macrophage markers (S1 and S2 Figs).
Fig 1. Schematic representation of the long-term intracellular M. tuberculosis culture protocol.

For each infection cycle, a frozen stock of THP-1 cells was thawed and cells were cultured for 4 passages. Cells were differentiated by PMA stimulation and infected with M. tuberculosis. After 18 h of infection, medium was replaced, and infected cells were incubated for 6 days. At day 7, THP-1 cells were lysed to release the intracellular bacteria, which were diluted in cell culture medium and subjected to clump disaggregation as described in Methods. Approximately 1/10 to 1/20 of this bacterial suspension was used to infect a new THP-1 cell culture. A total of 10 serial infection cycles were performed, and bacterial CFU and THP-1 cells were counted at 18 h and 7 days of infection for each cycle. An aliquot of the initial bacterial inoculum and of the intracellular bacteria collected at the end of the tenth cycle were diluted and spread onto agar plates for single-colony isolation and whole genome sequencing.
Multiple parameters were evaluated in each cycle. These included bacterial CFU counts at 1 and 7 days of infection, number of bacterial generations, and number of total and viable adherent THP-1 cells (Fig 2). The bacterial growth rate did not change significantly across cycles (the slight variation observed in CFU at day 1 of infection can be presumably attributed to the variability of the MOI) (Fig 2A). The number of bacterial generations per cycle showed a moderate fluctuation, ranging from 5 to 10 (Fig 2B), for a total of 79.6 generations. Moreover, the proportion of viable cells in the cultures was relatively constant across cycles, with averages of ~80% and 60% viable cells at 1 and 7 days of infection, respectively (Fig 2C). Overall, no significant change was observed in host cell viability and bacterial growth across cycles.
Fig 2. Parameters of the continuous intracellular culture model.
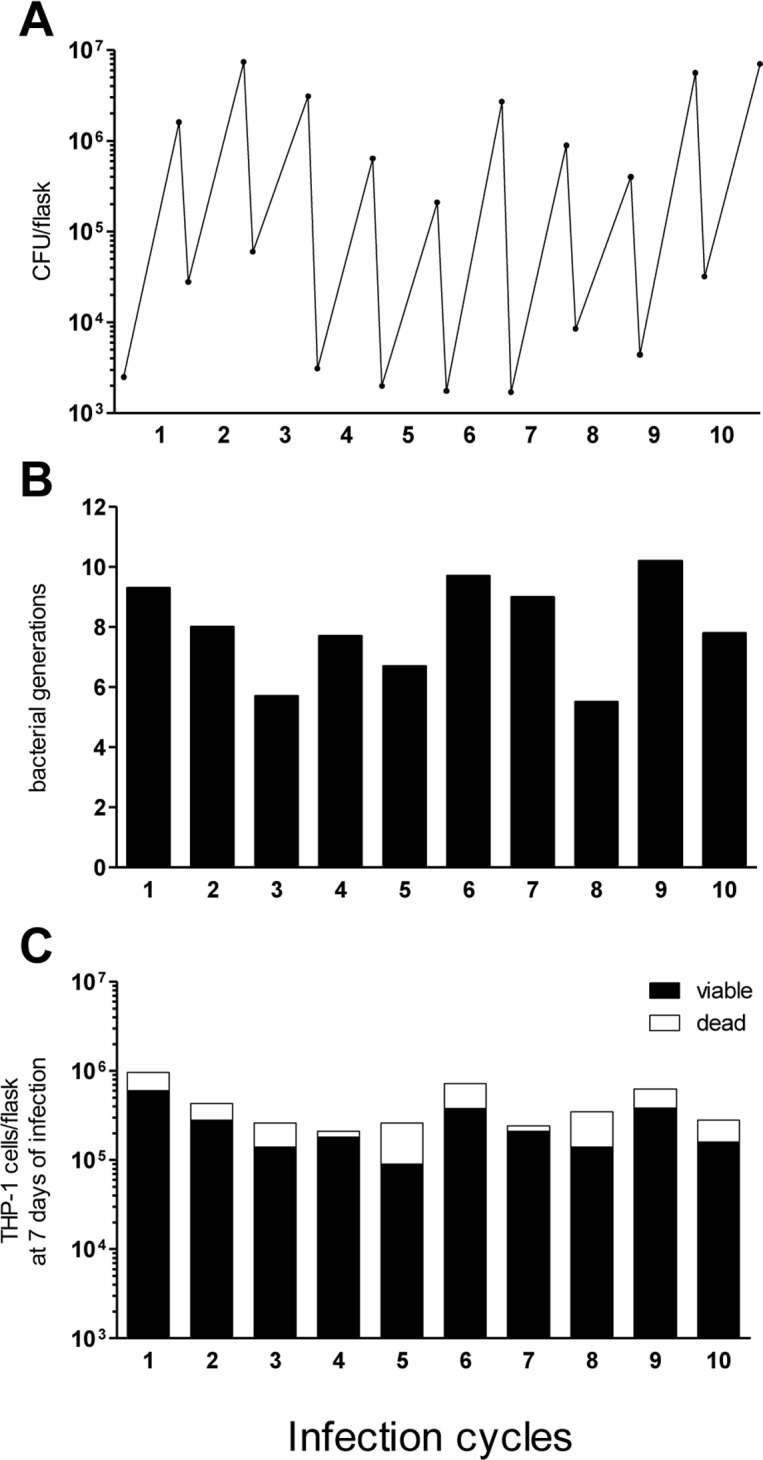
THP-1 cells were infected with M. tuberculosis H37RvSiena strain for 10 serial infection cycles. The graphs show the parameters evaluated for each cycle. (A) Bacterial CFU count. Bacterial CFU at day 1 and day 7 of infection. (B) Intracellular generations. Number of bacterial generations per cycle, calculated by using the formula (log2 CFU day 7- log2 CFU day 1)/log2 2. (C) Count of infected THP-1 cells. Number of total, viable adherent THP-1 cells (black bar), and dead cells (white bar) at day 7 of infection.
Identification and frequency of mutations
To assess the presence of genetic variants among isolates in the bacterial population, whole genome sequencing was performed for selected single-colony isolates obtained from the inoculum and from the tenth cycle of infection (Table 1). This comparison was necessary to distinguish mutations existing prior to infection from those that may have been acquired during infection. An average read depth of 325× and a mean genome coverage of 99.86% was obtained. Low (< 10×) read depth was obtained only for genomic regions containing prophages or highly repeated sequences such as Y980_0336 (13E12 repeat family gene), Y980_1575 (PhiRv1 prophage element), Y980_2933 (ppsC), Y980_0746 (pe_pgrs9), Y980_1450c (pe_pgrs27), Y980_3508 (pe_pgrs54) and Y980_3478 (ppe60) genes, as also observed in previous studies [28].
Table 1. Mutations identified in M. tuberculosis genome and their frequency.
| Frequencyb | ||||
|---|---|---|---|---|
| Mutationa | Gene (position or amino acid substitution) | Annotation/ Comments | Inoculum | 10th cycle |
| T-1216143-C | celA2b (3’ non coding region) | Putative cellulase/ Hydrolyzes barley β-glucan [30] | 9/50 (18%) | 5/50 (10%) |
| T-4284317-C | papA2 (Thr237Ala) | Polyketide synthase associated protein/ Involved in sulfolipid-I synthesis [31, 32] | 1/50 (2%) | 0/50 (0%) |
| T-2631348-C | plcA (His39Arg) | Phospholipase C 1/ Hydrolyzes macrophage phospholipids [26] Translocated by Tat system [33] | 0/50 (0%) | 1/50 (2%) |
| T-3872665-C | cut3 (Val223Ala) | Cutinase precursor/ Primary trehalose dimycolate hydrolase in M. tuberculosis [34] | 0/50 (0%) | 1/50 (2%) |
a All four mutations are thymine to cytosine transitions. The number between the nucleotides indicates the position of the mutation in the genome of M. tuberculosis H37RvSiena (CP007027.1).
b Number of single-colony isolates bearing the mutation relative to the total number of the isolates sequenced, and relative percent (in parenthesis).
The sequencing results showed that 9 out of 13 single-colony isolates obtained from the inoculum were identical to the H37RvSiena reference strain (wild type isolates). In addition, four thymine to cytosine mutations were found (Table 1): (i) T-1216143-C in the 3’ non coding region of celA2b (Y980_1090) [29] was present in three isolates (ii) T-4284317-C (Thr237Ala) in the coding region of papA2 (Y980_3820c) was present in one isolate. Sequencing of 13 single-colony isolates from the tenth cycle of intracellular growth yielded 10 isolates having the wild-type genotype and three isolates presenting a single-point mutation. All mutations were thymine to cytosine transitions: (i) T-1216143-C in the 3’ non coding region of celA2b (Y980_1090), as found in the inoculum, (ii) T-2631348-C (His39Arg) in the coding region of plcA gene (Y980_2351c), and (iii) T-3872665-C (Val223Ala) in the coding region of cut3 gene (Y980_3451) (Table 1). The latter two mutations were not found in the inoculum.
To determine the frequency of the four T→C transitions, an additional 37 single-colony isolates from the bacterial inoculum and from the tenth infection cycle were subjected to targeted re-sequencing (S1 Table). The transition T-1216143-C (3’ non coding celA2b) was present in the inoculum and at the tenth cycle at a similar frequency (18% and 10%, respectively). The mutations T-4284317-C (papA2), T-2631348-C (plcA) and T-3872665-C (cut3) were not found in additional isolates, indicating a very low frequency occurrence (≤ 2%). If we assume that T-2631348-C (plcA) and T-3872665-C (cut3) were acquired during intracellular growth, the estimated mutation rate was ≥ 5.7 × 10−9 / bp/ generation (since prophages and repetitive sequences were excluded from the analysis due to high probability of sequencing errors, mutations that may have been present in those regions are not included in this calculation). This number is in accordance with the predicted M. tuberculosis in vivo mutation rate [8].
In vitro and in vivo growth of cut3 Val223Ala and plcA His39Arg mutants
We asked whether the mutations identified at the tenth infection cycle but absent from the inoculum were associated with a fitness advantage. To test this possibility, we compared growth rates of mutant and wild type bacteria in axenic cultures and in infected THP-1 cells. No difference was observed (Fig 3A–3D). Moreover, determination of lung bacillary load at different time points after mouse infection showed comparable growth rates for mutant and wild type bacteria (Fig 3E). Thus, no differences were observed in the growth of both cut3 Val223Ala and plcA His39Arg mutants relative to wild type cells in vitro or during infection.
Fig 3. Growth curves of wild type, cut3 Val223Ala and plcA His39Arg mutant strains in liquid medium, THP-1 cells and murine lung.

(A and C) Growth curves in liquid medium. Growth curves in liquid medium over 16 days of incubation for cut3 Val223Ala mutant (A) and plcA His39Arg mutant (C) strains. (B and D) Growth curves in THP-1 cells. Growth curves of cut3 Val223Ala mutant (B) and plcA His39Arg mutant (D) strains in THP-1 cells over 6 days of infection. Data are expressed as CFU increase relative to day 0. (E) Growth curves in murine lung. Growth curves of wild type, cut3 Val223Ala mutant, and plcA His39Arg mutant strains in the murine lung up to 12 weeks post-infection. Panels A-D show the mean and standard deviation of results obtained from three replicates; panel E shows the mean and standard deviation of results obtained from 3–4 animals per time point.
Histopathological analysis of murine lungs infected with wild type and cut3 Val223Ala mutant
Gross inspection of lungs harvested at 12 weeks of infection from mice infected with wild type and cut3 Val223Ala mutant strains, revealed multiple, well-formed granulomas, characteristic of chronic M. tuberculosis infection (Fig 4A). No difference was observed between lung tissue infected with mutant and wild type bacteria in terms of proportion of lung parenchyma occupied by granulomas (involved lung) (Fig 4B) or size of lung granulomas (Fig 4C) (P > 0.05).
Fig 4. Morphometric analysis of mouse lungs infected with wild type and cut3 Val223Ala mutant strains at 12 weeks of infection.

(A) Representative image of mouse lung sections. The image represents 4× magnification of lung sections stained with hematoxylin and eosin. Arrows indicate granulomatous lesions. Scale = 1mm. (B-C) Proportion of involved lung and granuloma size. The graphs show the proportion of lung parenchyma occupied by granulomas (B) and the size of granulomas (C). Results represent the mean and standard deviation of data collected from 3 mice per infecting bacterial strain.
Accordingly, the disease score for these two strains were also similar (2.67+/-0.57 for the cut3 Val223Ala mutant strain and 2.33+/-0.57 for the wild type strain) (P > 0.05). Thus, infection of murine lungs with wild type and mutant bacteria induced similar histopathological effects.
Biochemical characterization of wild type and His39Arg mutant PlcA recombinant protein
In order to identify the effect of the plcA mutation on enzymatic activity, wild type and His39Arg mutant PlcA enzymes were purified from a recombinant M. smegmatis strain, and their activities were compared in vitro. No difference in rate of phosphatidylcholine hydrolysis was observed between the two enzymes: the enzymatic activity was 50.8 and 55.2 mAU/min and the specific activity was 16.32 and 17.74 pmol/min/mg for the wild type and mutant protein, respectively (Fig 5). Therefore, the presence of the His39Arg mutation had no effect on the enzymatic activity of PlcA.
Fig 5. Phospholipase C activity testing.

Time-course hydrolysis of phosphatidylcholine (PC) with 100μg of wild type (blue circles) and His39Arg mutant (red circles) recombinant PlcA. The release of phosphocholine was measured indirectly by the fluorescence measurement of resorufin (AU) released using the Amplex red phosphatidylcholine kit, and continuously monitored at λexc of 510 nm and λem of 590 nm. Blue and red curves represent the mean of three independent experiments (coefficient of variation was 0.0195 and 0.0293, respectively).
Conclusions
In conclusion, whole genome sequencing of M. tuberculosis grown in macrophages for ~80 generations showed genetic heterogeneity in the bacterial population that is not associated with fitness advantage. Since genetic variants of M. tuberculosis are also found in vivo at a frequency similar to that obtained in our experiments [1–4], our results suggest that most of the mycobacterial diversity arising during infection is likely associated with non-adaptive mutations. Our study does not exclude the possibility that phenotype-changing mutations of M. tuberculosis occur during infection. In our protocol, the frequency of such mutations may have been below the limit of detection of our study. Alternatively, the macrophage model used in this study might have imposed insufficient stress to M. tuberculosis to select for adaptive mutations. Refinements of the model will have to include use of activated macrophages to determine whether the frequency of adaptive mutations of M. tuberculosis increases when macrophages express higher antibacterial activity. The long-term macrophage infection system developed in the present study makes it possible to investigate bacterium-macrophage interactions over a time span that is much longer than conventional macrophage infections. Moreover, addition of drugs at various concentrations might make our cyclic infection protocol useful for the study of development of drug resistance.
Supporting Information
THP-1 cells were incubated with 40nM PMA for 72 h. At 24 h, 48 h and 72 h of PMA incubation, cell morphology was observed under a light microscope at a magnification of 60 × (A-B-C). At the same time points, culture medium was discarded, and three washes performed to remove non-adherent cells. Adherent cells were gently detached from the flask by using a cell scraper, and the number of viable cells was determined. The results are expressed as percentage of viable adherent cells relative to the number of initially stimulated cells (D). Each point represents the mean ± standard deviation of three replicates.
(PDF)
THP-1 cells were stimulated with 40nM PMA for 72 h. At 24 h (purple line), 48 h (green line) and 72 h (blue line) of PMA treatment, cells were stained with fluorophore-conjugated anti-human CD11b (B) and anti-human CD11c (D) antibodies, and analyzed by flow cytometry. Unstimulated cells were analyzed for CD11b (A) and CD11c (C) expression as controls. Cells not incubated with antibodies were included in each experiment (pink line).
(PDF)
(PDF)
(PDF)
Acknowledgments
We thank Karl Drlica for critical reading of the manuscript. We are also grateful to the staff of the PHRI animal facility for carrying out the mouse infections.
Data Availability
All relevant data are within the paper and its Supporting Information files. Additionally, genome sequence of M. tuberculosis H37RvSiena strain is available from NCBI GenBank database. The accession number is CP007027.1.
Funding Statement
The work was funded in part by NIH grants HL106788 and AI104615 to MLG and in part by European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement FP7-223681 (TB PAN-NET) to GP. PS’s PhD fellowship was financed by the Ministère Français de l’Enseignement Supérieur et de la Recherche. We are also grateful for the support from the Centre National de la Recherche Scientifique (CNRS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Garcia de Viedma D, Marin M, Ruiz MJ, Bouza E. Analysis of clonal composition of Mycobacterium tuberculosis isolates in primary infections in children. J Clin Microbiol. 2004;42(8):3415–8. Epub 2004/08/07. PubMed Central PMCID: PMC497594. 10.1128/JCM.42.8.3415-3418.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin A, Herranz M, Serrano MJ, Bouza E, Garcia de Viedma D. Rapid clonal analysis of recurrent tuberculosis by direct MIRU-VNTR typing on stored isolates. BMC Microbiol. 2007;7:73 Epub 2007/08/01. PubMed Central PMCID: PMC1988808. 10.1186/1471-2180-7-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ford C, Yusim K, Ioerger T, Feng S, Chase M, Greene M, et al. Mycobacterium tuberculosis—heterogeneity revealed through whole genome sequencing. Tuberculosis. 2012;92(3):194–201. Epub 2012/01/06. PubMed Central PMCID: PMC3323677. 10.1016/j.tube.2011.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun G, Luo T, Yang C, Dong X, Li J, Zhu Y, et al. Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis. 2012;206(11):1724–33. Epub 2012/09/18. PubMed Central PMCID: PMC3488197. 10.1093/infdis/jis601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Neill MB, Mortimer TD, Pepperell CS. Diversity of Mycobacterium tuberculosis across Evolutionary Scales. PLoS Pathog. 2015;11(11):e1005257 Epub 2015/11/13. PubMed Central PMCID: PMC4642946. 10.1371/journal.ppat.1005257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez-Lago L, Comas I, Navarro Y, Gonzalez-Candelas F, Herranz M, Bouza E, et al. Whole genome sequencing analysis of intrapatient microevolution in Mycobacterium tuberculosis: potential impact on the inference of tuberculosis transmission. J Infect Dis. 2014;209(1):98–108. Epub 2013/08/16. 10.1093/infdis/jit439 [DOI] [PubMed] [Google Scholar]
- 7.Eldholm V, Norheim G, von der Lippe B, Kinander W, Dahle UR, Caugant DA, et al. Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol. 2014;15(11):490 Epub 2014/11/25. PubMed Central PMCID: PMC4223161. 10.1186/s13059-014-0490-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, et al. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet. 2011;43(5):482–6. Epub 2011/04/26. PubMed Central PMCID: PMC3101871. 10.1038/ng.811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaplan G, Post FA, Moreira AL, Wainwright H, Kreiswirth BN, Tanverdi M, et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect Immun. 2003;71(12):7099–108. 10.1128/IAI.71.12.7099-7108.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2(8):569–77. Epub 2001/08/03. 10.1038/35085034 [DOI] [PubMed] [Google Scholar]
- 11.Russell DG, VanderVen BC, Lee W, Abramovitch RB, Kim MJ, Homolka S, et al. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe. 2010;8(1):68–76. Epub 2010/07/20. PubMed Central PMCID: PMC2929656. 10.1016/j.chom.2010.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ensminger AW, Yassin Y, Miron A, Isberg RR. Experimental evolution of Legionella pneumophila in mouse macrophages leads to strains with altered determinants of environmental survival. PLoS Pathog. 2012;8(5):e1002731 Epub 2012/06/14. PubMed Central PMCID: PMC3364954. 10.1371/journal.ppat.1002731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giovannini D, Cappelli G, Jiang L, Castilletti C, Colone A, Serafino A, et al. A new Mycobacterium tuberculosis smooth colony reduces growth inside human macrophages and represses PDIM Operon gene expression. Does an heterogeneous population exist in intracellular mycobacteria? Microb Pathog. 2012;53(3–4):135–46. Epub 2012/07/10. 10.1016/j.micpath.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 14.Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev. 2011;240(1):252–68. Epub 2011/02/26. PubMed Central PMCID: PMC3174472. 10.1111/j.1600-065X.2010.00984.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Santoro F., Guerrini V., Lazzeri E., Iannelli F., G. P. Genomic polymorphisms in a laboratory isolate of Mycobacterium tuberculosis reference strain H37Rv (ATCC27294). New Microbiol. 2016;39(4). [PubMed] [Google Scholar]
- 16.Guerrini V, De Rosa M, Pasquini S, Mugnaini C, Brizzi A, Cuppone AM, et al. New fluoroquinolones active against fluoroquinolones-resistant Mycobacterium tuberculosis strains. Tuberculosis. 2013;93(4):405–11. Epub 2013/03/26. 10.1016/j.tube.2013.02.017 [DOI] [PubMed] [Google Scholar]
- 17.Tsuchiya S, Kobayashi Y, Goto Y, Okumura H, Nakae S, Konno T, et al. Induction of maturation in cultured human monocytic leukemia cells by a phorbol diester. Cancer Res. 1982;42(4):1530–6. Epub 1982/04/01. [PubMed] [Google Scholar]
- 18.Michee S, Brignole-Baudouin F, Riancho L, Rostene W, Baudouin C, Labbe A. Effects of benzalkonium chloride on THP-1 differentiated macrophages in vitro. PLoS ONE. 2013;8(8):e72459 Epub 2013/08/31. PubMed Central PMCID: PMC3747170. 10.1371/journal.pone.0072459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR Jr. Genetic Manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007;Chapter 10:Unit 10A 2. Epub 2008/09/05. [DOI] [PubMed] [Google Scholar]
- 20.Lee WP, Stromberg MP, Ward A, Stewart C, Garrison EP, Marth GT. MOSAIK: a hash-based algorithm for accurate next-generation sequencing short-read mapping. PLoS ONE. 2014;9(3):e90581 Epub 2014/03/07. PubMed Central PMCID: PMC3944147. 10.1371/journal.pone.0090581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, Tarazona S, et al. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012;28(20):2678–9. Epub 2012/08/24. 10.1093/bioinformatics/bts503 [DOI] [PubMed] [Google Scholar]
- 22.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. Epub 2009/06/23. PubMed Central PMCID: PMC2734323. 10.1093/bioinformatics/btp373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boisvert S, Laviolette F, Corbeil J. Ray: simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol. 2010;17(11):1519–33. Epub 2010/10/21. PubMed Central PMCID: PMC3119603. 10.1089/cmb.2009.0238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403. Epub 2004/07/03. PubMed Central PMCID: PMC442156. 10.1101/gr.2289704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subbian S, Tsenova L, Yang G, O'Brien P, Parsons S, Peixoto B, et al. Chronic pulmonary cavitary tuberculosis in rabbits: a failed host immune response. Open Biol. 2011;1(4):110016 Epub 2012/05/31. PubMed Central PMCID: PMC3352086. 10.1098/rsob.110016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bakala N'goma J C, Schue M, Carriere F, Geerlof A, Canaan S. Evidence for the cytotoxic effects of Mycobacterium tuberculosis phospholipase C towards macrophages. Biochim Biophys Acta. 2010;1801(12):1305–13. Epub 2010/08/26. 10.1016/j.bbalip.2010.08.007 [DOI] [PubMed] [Google Scholar]
- 27.Noens EE, Williams C, Anandhakrishnan M, Poulsen C, Ehebauer MT, Wilmanns M. Improved mycobacterial protein production using a Mycobacterium smegmatis groEL1DeltaC expression strain. BMC Biotechnol. 2011;11:27 Epub 2011/03/29. PubMed Central PMCID: PMC3076238. 10.1186/1472-6750-11-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng H, Lu L, Wang B, Pu S, Zhang X, Zhu G, et al. Genetic basis of virulence attenuation revealed by comparative genomic analysis of Mycobacterium tuberculosis strain H37Ra versus H37Rv. PLoS ONE. 2008;3(6):e2375 Epub 2008/06/28. PubMed Central PMCID: PMC2440308. 10.1371/journal.pone.0002375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitra A, Angamuthu K, Nagaraja V. Genome-wide analysis of the intrinsic terminators of transcription across the genus Mycobacterium. Tuberculosis. 2008;88(6):566–75. Epub 2008/09/05. 10.1016/j.tube.2008.06.004 [DOI] [PubMed] [Google Scholar]
- 30.Mba Medie F, Vincentelli R, Drancourt M, Henrissat B. Mycobacterium tuberculosis Rv1090 and Rv1987 encode functional beta-glucan-targeting proteins. Protein Expr Purif. 2011;75(2):172–6. Epub 2010/09/10. 10.1016/j.pep.2010.08.015 [DOI] [PubMed] [Google Scholar]
- 31.Bhatt K, Gurcha SS, Bhatt A, Besra GS, Jacobs WR Jr. Two polyketide-synthase-associated acyltransferases are required for sulfolipid biosynthesis in Mycobacterium tuberculosis. Microbiology. 2007;153(Pt 2):513–20. Epub 2007/01/30. 10.1099/mic.0.2006/003103-0 [DOI] [PubMed] [Google Scholar]
- 32.Rezapov BR, Popov SV, Gimrikh EO, Chekhov AM, Mitiutnev AN. [The treatment of supraventricular tachyarrhythmias by the transvenous electrical destruction of the atrioventricular junction of the heart]. Ter Arkh. 1991;63(9):155–8. Epub 1991/01/01. [PubMed] [Google Scholar]
- 33.McDonough JA, McCann JR, Tekippe EM, Silverman JS, Rigel NW, Braunstein M. Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J Bacteriol. 2008;190(19):6428–38. Epub 2008/07/29. PubMed Central PMCID: PMC2566002. 10.1128/JB.00749-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Y, Kulka K, Montelaro RC, Reinhart TA, Sissons J, Aderem A, et al. A hydrolase of trehalose dimycolate induces nutrient influx and stress sensitivity to balance intracellular growth of Mycobacterium tuberculosis. Cell Host Microbe. 2014;15(2):153–63. Epub 2014/02/18. PubMed Central PMCID: PMC3974621. 10.1016/j.chom.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
THP-1 cells were incubated with 40nM PMA for 72 h. At 24 h, 48 h and 72 h of PMA incubation, cell morphology was observed under a light microscope at a magnification of 60 × (A-B-C). At the same time points, culture medium was discarded, and three washes performed to remove non-adherent cells. Adherent cells were gently detached from the flask by using a cell scraper, and the number of viable cells was determined. The results are expressed as percentage of viable adherent cells relative to the number of initially stimulated cells (D). Each point represents the mean ± standard deviation of three replicates.
(PDF)
THP-1 cells were stimulated with 40nM PMA for 72 h. At 24 h (purple line), 48 h (green line) and 72 h (blue line) of PMA treatment, cells were stained with fluorophore-conjugated anti-human CD11b (B) and anti-human CD11c (D) antibodies, and analyzed by flow cytometry. Unstimulated cells were analyzed for CD11b (A) and CD11c (C) expression as controls. Cells not incubated with antibodies were included in each experiment (pink line).
(PDF)
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. Additionally, genome sequence of M. tuberculosis H37RvSiena strain is available from NCBI GenBank database. The accession number is CP007027.1.