

Abstract
Synthetic nucleic acids offer rich potential to understand and engineer new cellular functions, yet an unresolved limitation in their production and usage is deleterious products, which restrict design complexity and add cost. Herein, we employ a solid-state nanopore to differentiate molecules of a gene synthesis reaction into categories of correct and incorrect assemblies. This new method offers a solution that provides information on gene synthesis reactions in near-real time with higher complexity and lower costs. This advance can permit insights into gene synthesis reactions such as kinetics monitoring, real-time tuning, and optimization of factors that drive reaction-to-reaction variations as well as open venues between nanopore-sensing, synthetic biology, and DNA nanotechnology.
Keywords: single molecule, synthetic biology, DNA, translocation, HIV protease
Graphical abstract
Synthetic genes1,2 and genomes3,4 offer a transformative ability to design and create new cellular functions and entire organisms.5 To leverage these programmable DNA elements into their cellular chassis, precise synthetic control of their properties (sequence, length) must be obtained. However, reliable construction of these DNA elements using polymerization,6 restriction enzymes,7 and/or ligation reactions8,9 can be challenging, especially for long formats. Every gene synthesis method faces the challenge of unwanted side products, with the purity of resultant mixture being a function of the assembly method, DNA sequence composition (length, complexity, repeats), quality of input oligonucleotide building blocks, and other factors.10,11
To further increase the complexity of producible synthetic genes and better understand their function, new methods are needed to assess and control their output diversity so that unwanted resultant product formation is minimized. Two major classes of deleterious products that arise during gene synthesis reactions are global structural defects (incomplete assemblies and misassemblies) and small local defects (point mutations, short insertions, and/or deletions). These errors can be identified using laborious procedures such as electrophoresis, cloning, and sequencing, which consequently become major components of the time and cost of gene synthesis. Moreover, as synthesis approaches continue to move toward miniaturization (employing microarrays12 and microfluidic chips13), where reactions are parallelized and costs are lowered, analytical quality-control tools that match smaller scales (μm sizes and nM/pM concentrations) become increasingly valuable.
Solid-state nanopores14 are a new class of sensors that can electronically detect the structure and conformation of single DNA molecules with high throughput (thousands of molecules per minute). A nanopore is a nanometer-scale aperture in a dielectric membrane that separates two ionic solutions. Application of a voltage bias across the membrane induces an ionic current through the nanopore (Figures 1a–b). As single DNA molecules in the ionic solution electrophoretically move through the pore, they temporarily block ion current through the pore. Transient changes in the ionic current can then be used to infer characteristics of the translocating molecules such as length differences,15–17 bound proteins,18,19 epigenetic modifications,20,21 and defects.22 The ability to detect small sample amounts of nucleic acids without amplification or labeling, as well as the potential of using nanopores to isolate a selected type of molecule, makes nanopores an attractive platform for direct analysis of gene synthesis reactions. Herein, we utilize ultrathin solid-state nanopore sensors23 to directly detect different types of synthetic gene products. We designed, assembled, and interrogated a set of DNA structures that represent major categories of expected defects (dsDNA molecules with single-base mismatches, overhangs, flaps, and Holliday junctions) to train our system and depict differentiation of these structures. We then monitor a gene synthesis reaction at different stages and discretize the reaction products into categories of correct and incorrect assemblies based on ensemble bunching of molecular transport signatures. With further refinement of nanopore-based discrimination among correctly and incorrectly assembled structures and real-time on-the-fly analysis, we envision a true molecular selection process in which correct structures can be identified and enriched.
Figure 1.
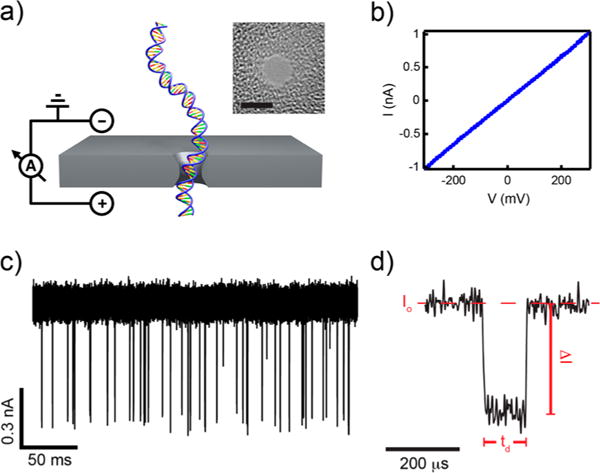
DNA translocation through a solid-state nanopore. (a) Illustration of a small transmembrane voltage causing dsDNA to pass through a nanoscale aperture. Inset: Transmission electron microscope image of a small SiN nanopore (scale bar is 2 nm). (b) Current–voltage curve in the range −300 to 300 mV for one of the nanopores used in this study. (c) Current trace for 70 bp DNA at an applied voltage of 200 mV, low-pass filtered at 200 kHz. Each deep spike corresponds to a single DNA molecule translocating the nanopore in head-to-tail fashion. (d) Analysis of DNA translocation data extracts three desired quantities: dwell time (td), current blockade (ΔI), and fractional current blockade (ΔI/Io).
RESULTS AND DISCUSSION
Rapid Detection of Small Defects
De novo gene synthesis reactions can produce a variety of unwanted products such as small sequence defects as well as structural misassemblies. To enable detection of these defects, we first aimed to show that the translocation signatures of correctly assembled, homogeneous DNA molecules would be fundamentally different from those of misassembled synthetic products. First, thin nanopores were fabricated in silicon nitride (SiN) windows as previously described24 with diameters of 2.4–2.6 nm and effective thickness of 4–8 nm (Figure 1a). Nanopores within this diameter range permit unfolded entry and single current blockade levels during electrophoretic translocation.25 The TEM-drilled nanopores were rinsed with hot piranha, cleansed thoroughly with deionized water, vacuum-dried, and mounted into a custom PTFE flow cell that was subsequently filled with an ionic solution (0.40 M KCl, 10 mM Tris, 1 mM EDTA, pH 8.0). Application of a voltage bias across the membrane drove an ionic current through the nanopore, which demonstrated Ohmic behavior (Figure 1b). Upon addition of a small concentration of 70 bp dsDNA homoduplexes (50 nM, IDT Technologies) and application of a voltage bias (200 mV), transient dips in current were observed, signifying translocation of individual DNA molecules through the nanopore (Figure 1c). DNA capture has been shown to be dominated by electrophoresis due to an exponential increase in the capture rate with voltage,26 despite a small negative surface charge present in SiN nanopores, which causes an electroosmotic flow in a direction opposite to DNA translocation.27,28 With the use of custom analysis software, two key parameters were extracted for each translocation event: the dwell time td and the current blockade ΔI (Figure 1d). Plotting the fractional current blockade (ΔI/Io) versus td for 1830 events showed a distribution with an average translocation speed (1 bp/μs) consistent with previous studies24,25 and a single current blockade level with mean amplitude of ΔI/Io = 0.79 ± 0.03 (Figure 2b). Next, we sought to determine the nanopore’s ability to detect small synthetic errors, such as point deletions or insertions, using one strand of the control oligo of “correct” sequence (70 nt) annealed to a “defective” DNA oligo (71 nt), which was synthesized with a single base insertion, as illustrated in Figure 2a. The defective site was located in the center of the mismatch oligo and is expected to cause a small kink in the short, otherwise stiff dsDNA, which we hypothesized would alter the entry kinetics and enable differentiation. Insertion of a small concentration of the mismatch dsDNA species (70/71 bp) into the nanopore setup and application of a voltage bias as before generated 1410 events with a highly similar distribution of ΔI/Io against td to the correctly matched oligos (Figure 2b). One subtle difference observed between the current blockade distributions of the correct and defective DNA was a small, additional population at lower ΔI/Io, which could be due to the translocation of misassembled DNA molecules or unsuccessful translocations (collisions) caused by the kink. The dwell time distributions yielded no statistically significant discrepancies between the two molecules, and as such, differentiation of single-base defects would be inherently difficult to detect directly.
Figure 2.
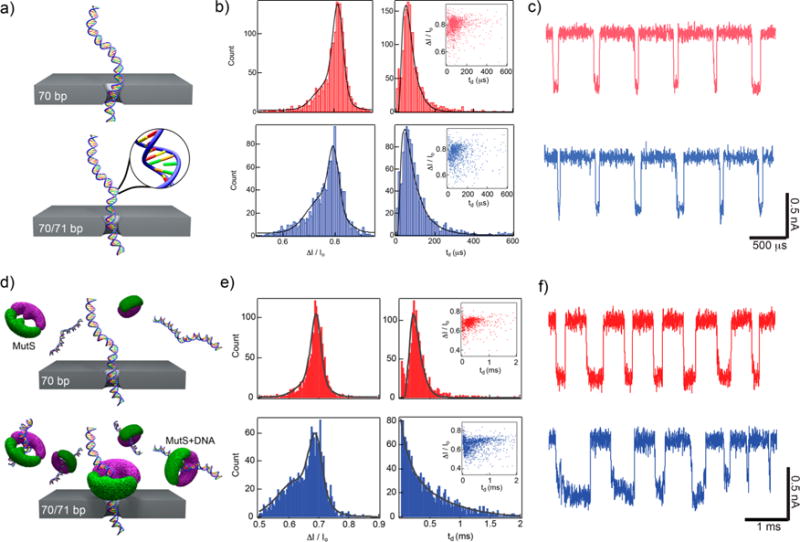
Single mismatch detection using nanopores. (a) Schematics of two different DNA molecules translocating a nanopore: a homologous 70 bp DNA and a 70/71 bp DNA that contains a single base mismatch at the center. (b) Fractional current blockade ΔI/Io and dwell time td histograms for transport through a 2.4 nm diameter nanopore at 200 mV. Insets: Scatter plots of ΔI/Io vs td show similar translocation populations for the two samples. (c) Concatenated, consecutive events of each analyte further demonstrates difficulty detecting the single mismatch (traces low-pass filtered at 200 kHz). (d) Illustration of each sample passing through a 2.6 nm pore after incubation with mismatch-binding protein MutS. (e) Histograms of current blockade (both fit to a double Gaussian model) show a clear shoulder develops at a lower current blockade in the 70/71 bp DNA when incubated with MutS. In addition, the dwell time histogram for 70/71 bp has a drastically different shape than that for 70 bp, indicating an altered transport process (fit using a single exponential function). Insets: Scatter plots of each sample show an increase in longer dwell time events in 70/71 bp, suggesting stripping of MutS during translocation. (f) Concatenated, consecutive events for each analyte exemplify differences between two samples (traces low-pass filtered at 200 kHz).
We validated the difference between the correctly matched and defective species by incubation of both products with a mismatch-binding protein from Thermus aquaticus named MutS29 (Taq MutS, 89 kDa), which is a part of the DNA repair pathway in a variety of organisms (Supporting Information Figure 1). Taq MutS possesses an elongated shape (longest dimension about 10–12 nm) and binds to all single base mismatches, insertions or deletions up to four bp long in double stranded DNA. Previously, we demonstrated the use of Taq MutS to discriminate all types of single base mismatches and insertions/deletions of various lengths30 (in one case as much as a 50 bp deletion), and gel shift analysis showed a clear shift for the defective products compared to the correctly matched samples (Supporting Information Figure 1). To further enable detection of small sequence defects on synthetic genes using a solid-state nanopore, the correctly matched 70 bp DNA and mismatch-containing 70/71 bp DNA species were each incubated with the MutS protein (ratio of 2 MutS/5 DNA) for 20 min at 60 °C in 0.40 M KCl, 8 mM MgCl2 buffer (Figure 2d). Both populations were individually measured using the nanopore setup as before, and the correctly matched dsDNA incubated with MutS demonstrated highly similar td and ΔI/Io distributions to the same correctly matched dsDNA population without MutS incubation, indicating little to no interaction between the two species (Figure 2e,f). This observation is consistent with gel shift analysis whereby incubation of the correctly matched dsDNA with MutS did not significantly alter correctly assembled product migration, even with increases in concentration of MutS (Supporting Information Figure 1). In contrast to the correctly matched dsDNA products incubated with MutS, the mismatch products + MutS displayed td distributions that could not be fit to a drift-diffusion model,31,32 and ΔI/Io distributions did not fit to a single Gaussian distribution (Figure 2e). The current blockade distribution for the mismatch products + MutS showed a similar peak as the 70 bp molecule with an additional shoulder at a lower current blockade, which may be attributed to collisions of the DNA/MutS complex with the pore mouth, or translocation of free oligos and misassembled DNA molecules that also bind MutS. In contrast to the correctly matched dsDNA + MutS, the mismatch dsDNA + MutS also displayed a distorted dwell time distribution that was fit to a single exponential with mean time scale of td = 829 ± 60 μs, compared to a mean td of 524 ± 47 μs for the correctly matched dsDNA (Figure 2e,f). The increase in dwell times can be attributed to weak DNA–protein interactions that stall the complex inside the pore until dissociation,33–35 which consequently enable the detection of single-base mismatches and enumeration of defective species among a population. To ensure that the increase in dwell time we observed is not due to MutS being trapped near the pore, we analyzed the power spectral densities of the open pore current for DNA translocations of each sample, with and without MutS, and found that all the spectra had similar noise characteristics that differed greatly from when MutS clogs the nanopore (Supporting Information Figure 2).
Differentiation of Structural Defects
In addition to small sequence defects, de novo gene synthesis reactions generate a range of incomplete or structurally misassembled products. We designed and assembled a set of DNA structures intended to represent the major categories of expected defects: overhangs, flaps, and Holliday junctions (Figure 3a). For ease of construction, we generated populations of each of these types of defects directly from synthetic oligonucleotides (35 and 70 nt) by thermal annealing, without any enzymatic steps, and verified their formation using gel electrophoresis (Supporting Information Figure 3). Each type of population was inserted into the nanopore setup and compared to a correctly paired 70 bp dsDNA reference molecule constructed from the same oligos. Figure 3b,c shows representative current traces of each type of molecule translocating through the nanopore and their corresponding td histograms compared to those of the 70 bp dsDNA control (see heat map contours of ΔI/Io vs td in Supporting Information Figure 3). The 70 bp dsDNA reference molecules showed relatively low scatter with highly repeatable transport dynamics when translocating through the nanopore, in stark contrast to the defective molecules. The overhang molecule (blue in Figure 3), which is composed of a 70 nt oligo annealed to a 35 nt oligo, forms a double-stranded structure with a single-stranded end. On average, the overhang molecules migrated through the pore faster and with a lower current blockade, which agrees with its smaller average diameter compared with the dsDNA. As seen in Figure 3c, the dwell time histogram contains two populations, which can be ascribed to unbound oligos (most likely the faster population) and correctly paired overhang complexes. This result is consistent with gel electrophoresis assays, where two populations were observed (Supporting Information Figure 3) and consisted of an unpaired ssDNA oligo and the correctly assembled overhang structure. The flap molecule (green in Figure 3), which is composed of a 70 nt oligo annealed to a 35 nt oligo and another 70 nt oligo, contains a 70 bp dsDNA body with a 35 nt ssDNA overhang in the center. The flap sample, similarly to the overhang, yielded two distinct dwell time populations, which is also in agreement with gel electrophoresis results, whereby free oligos and properly constructed molecules were detected. However, the flap molecules generated on average longer dwell times than the overhang molecule, which was also expected given its added bulk when compared to the nanopore diameter. The last structural defect studied was a synthetically constructed Holliday junction (red in Figure 3), which consists of four dsDNA strands that connect via Watson–Crick base-pairing (Supporting Information Figure 3) to form a four-armed molecule.36 Holliday junctions are the primary elements used to create DNA origami structures and typically act as scaffolds for assembly of more complex shapes and structures.37 Figure 3b,c highlights how Holliday junctions yield long dwell time, multilevel translocation events and a large breadth in dwell time, which can be ascribed to extreme bending or breaking of the molecule to transport through the small nanopore. Many translocation events of the Holliday junction sample, as seen in Figure 3b, begin with a moderate current blockade and terminate with a deeper current blockade, indicating the initial capture of one strand of the Holliday junction (moderate ΔI) followed by the rupturing of the complex leading to several DNA strands occupying the pore at once (deep ΔI). With the use of these unique translocation signatures, an algorithm could be developed to analyze and differentiate correctly assembled gene synthesis products from various misassembled products.
Figure 3.
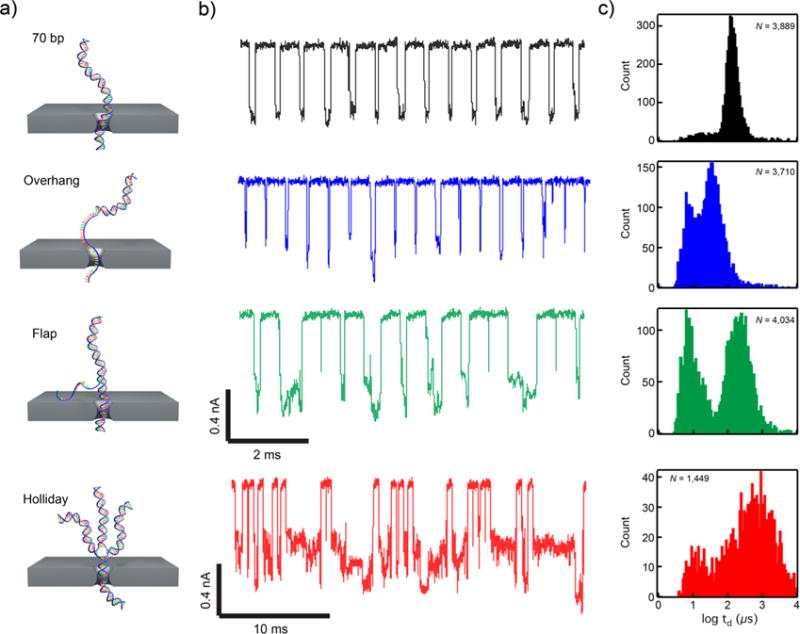
Gene synthesis misassemblies have unique translocation profiles. (a) Illustrations of four common products of gene synthesis by polymerase chain assembly: dsDNA (desired product), overhangs, flaps, and Holliday junctions. (b) Concatenated event traces of each product for translocation through a 2.5 nm nanopore (traces low-pass filtered at 100 kHz to show the complex number of current levels for the events). (c) Log-dwell time histograms for each product show distinct transport “fingerprints” for each species (N = number of events).
In addition to discriminating between these samples by dwell time differences, we quantified their capture rates using the time between successive events, or the interevent time. When fitting interevent time histograms of each sample to single exponential functions, we found that the normalized capture rate for the 70 bp sample was highest, the overhang and flap samples had slightly lower capture rates, and the Holliday junction sample had by far the lowest capture rate (see Table 1 and Supporting Information Figure 4). Notably, these capture rates were normalized using concentration units of ng/μL because each sample contains impurities in the form of free oligos or incomplete assemblies as seen in gel electrophoresis assays (Supporting Information Figure 3). These impurities, which skew the true capture rate of each misassembly, make quantifying the percentage purity of a gene synthesis product (containing a mixture of these misassemblies along with correct assemblies) very difficult. Despite this, recognizing the presence of the correct gene assembly is still possible, as described in detail below.
TABLE 1.
Capture Rates of Gene Synthesis Misassemblies
| Sample | RC (s−1 (ng/μL)−1) |
|---|---|
| 70 bp | 54.6 ± 0.9 |
| Overhang | 24.0 ± 0.4 |
| Flap | 18.2 ± 0.3 |
| Holliday | 3.4 ± 0.1 |
Discrimination between Correctly Assembled Gene Products and Misassembled Gene Products
As a case study, we synthesized a gene for HIV Protease (HIVPr) from small building blocks (oligonucleotides) using polymerase construction and amplification (PCA), wherein the oligonucleotides themselves act as both template and primers38 (see Methods). HIVPr is a protease that is required for the human immunodeficiency virion to remain infectious and sequence variants of the gene can confer resistance to drug inhibitors of the enzyme.39,40 The synthetic gene products were analyzed on a gel (Supporting Information Figure 5) and showed both correctly assembled products (524 bp) and incorrectly assembled products (such as oligos, intermediate assemblies of shorter length, as well as longer fragments). A fraction of the as-synthesized products (6 ng/μL) were analyzed using the nanopore setup and transient blockades in current were observed as before (Figure 4). Inspection of the distribution of the current blockade versus translocation time showed a significantly larger spread of ΔI/Io and td values compared to homogeneous products of similar size (500 bp, Fisher Scientific), indicating the solution contained a range of DNA sizes and structures (blue in Figure 4). This result agrees well with the gel that showed both smaller molecular weight species (assembly intermediates) below the product bands as well as larger products such as dimers and trimers (Supporting Information Figure 5). In contrast to larger diameter solid-state nanopores, where high translocation speed and DNA self-interaction/folding induce signal scatter that precludes definitive assignment of molecular features such as length, small-diameter nanopores (<3 nm) produce narrow distributions of td values,24 which enable accurate identification of errors. The increase in the range of the distribution of current blockades can be ascribed to fast oligo and incomplete assembly translocations, collisions with the pore, and multilevel translocation events (Figure 3b), such as flaps or Holliday junctions migrating through the nanopore. With the use of the signatures of homogeneous populations of synthetic defects (overhangs, flaps, and Holliday junctions) and reference dsDNA with similar size (500 bp) as classifiers for detection in the synthetic gene population, direct detection of different types of structural misassemblies could be accomplished. This result is in contrast to gel electrophoresis where complex structures such as overhangs and flaps are difficult to view directly.
Figure 4.
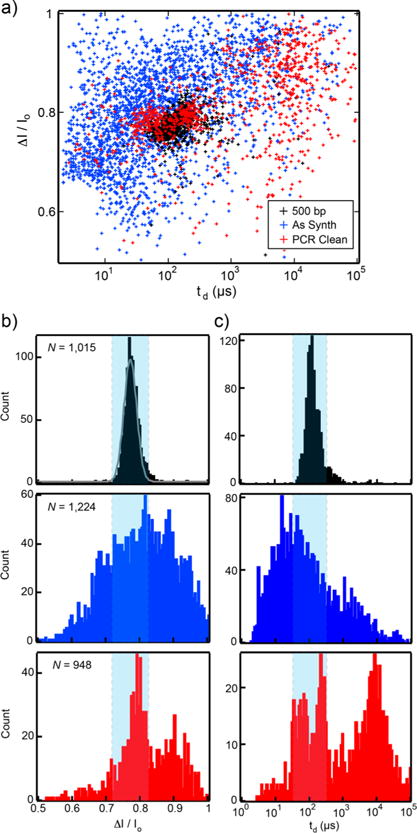
Nanopore-based monitoring of gene synthesis products for the HIV-Protease gene (524 bp) with and without additional PCR purification. (a) Scatter plot of current blockade ΔI/Io versus dwell time td for reference 500 bp dsDNA products (black), as-synthesized HIVPr synthetic genes (blue), and PCR-cleaned HIVPr synthetic gene products (red). (b) The HIV-Protease gene product as synthesized (blue) shows a wide spread in current blockade (ΔI/Io), when compared to homogeneous products (black). PCR-cleanup of the HIVPr synthetic genes (red) improved the fraction of events that resemble correctly assembled genes, but did not remove many incorrect or misassembled synthesis products. (c) Comparison of the dwell times td for the homogeneous products to the as-synthesized (blue) and PCR-purified samples (red) shows significant variation that can be ascribed to both pure and impure synthetic gene products. Light blue shaded regions depict potential thresholds for selecting correct gene assemblies using a nanopore-based on-the-fly “accept or reject” protocol.
A common strategy to remove unwanted gene synthesis products after assembly reactions is PCR amplification, whereby the full-length product can be selectively amplified with the typical exponential profile of PCR. Incomplete products (which contain only one of the two PCR primer sequences) amplify linearly and the end effect is that the incomplete products are “diluted” in the final population of DNA molecules. However, misassembled DNA containing both primer sites will also amplify exponentially, and this approach does nothing to deal with small sequence defects such as point mutations. The as-synthesized HIVPr gene was amplified by PCR and a fraction of PCR-cleaned up products (6 ng/μL) were loaded into the nanopore setup and measured as before. In contrast to the wide distribution of current blockades observed for the unpurified products, the purified product distribution (red data in Figure 4) contains a dominant peak that closely resembles the homogeneous 500 bp reference distribution as well as other smaller peaks that resemble defective species. In addition to the multipeak distribution of current blockades for the purified synthetic genes, a large variation in translocation times was also observed. This variation could be ascribed to different types of defective species such as unpaired oligos and assembly dimers and trimers, that were unable to be filtered out by PCR and were also visible on a gel (Supporting Information Figure 5).
Translocating our gene synthesis products through a nanopore after PCR clean up yields a clear population of properly assembled genes, but notably does not isolate a pure sample of the gene in the trans chamber of the nanopore setup. Using a selection protocol based upon stringent dwell time and current blockade thresholds defined by a control molecule (in this case 500 bp DNA), our nanopore system could be designed to reject any translocating molecule that does not fall within these thresholds. If this protocol is followed for many translocation events, it would be possible to isolate the correct gene assemblies, which could then be amplified by PCR with as few as a hundred correctly assembled molecules. Even though very short defective molecules may translocate too fast for our electronics to reject, these molecules can be effectively diluted by a post-nanopore PCR step, since they would not contain both primer sequences necessary for amplification.
CONCLUSIONS
De novo gene synthesis has tremendous potential to design and develop combinations of new or altered genes, gene regulatory networks and pathways, as well as chromosomes and genomes for new cellular functions. While gene synthesis is an incredibly powerful tool, a significant limitation preventing large-scale and low-cost production has been the removal of unwanted side products. Since gene synthesis techniques are rapidly moving in the direction of array-based synthesis41 where oligo costs and concentrations are lower, new tools need to be developed that can operate on these scales.42
An ultrathin, small-diameter nanopore system was employed to detect different types of synthetic gene products and characterize a conventional gene synthesis reaction at different stages (before and after PCR cleanup). Using this approach, we discretize the reaction products into categories of correct and incorrect assemblies and speculate that an algorithm could be devised for isolation and amplification of correct gene assemblies. The new method described herein offers a potential solution to provide information on gene synthesis reactions with higher complexity, lower costs, and during the reactions as opposed to after the reaction is complete. This advance may facilitate monitoring of reaction kinetics, real-time tuning, and understanding of factors that drive reaction-to-reaction variations, which will further our ability to engineer gene synthesis and DNA nanotechnology complexity.43 Since nanopore systems can easily be integrated with micro- and nanofluidic systems,44 which have been used for single-molecule sorting,45 the possibility of constructing12,13 and purifying molecules from a pool of mostly defective products in an automated fashion on-chip and with concentrations far below those required for current electrophoresis techniques, makes them attractive options for future research. Thus, using nanopore-based monitoring to enhance gene synthesis product purity can facilitate the fabrication of constructs that perform new cellular functions such as complex sensing, computation, and actuation, which will have a broad impact on biomedicine and biotechnology.
METHODS
Fabrication and Characterization of Ultrathin Nanopore Devices
Silicon nitride (SiN) membranes were fabricated using photolithography along with dry and wet etching steps as explained in previous work.24 An additional SF6 plasma-etch step was used to thin the SiN membranes to a total thickness of 15–20 nm for nanopore experiments. All experimental data was collected with a sampling rate of 4.167 MHz using the Chimera Instruments VC100 (New York, NY), which allows the detection of events as fast as 2.5 μs when low-pass filtering at 200 kHz. Nanopore data collected for the single mismatch molecules (Figure 2), isolated misassembled products (Figure 3), and gene synthesis products (Figure 4) were low-pass filtered at 200, 400, and 300 kHz, respectively, before event analysis. Prior to addition of DNA, pores were deemed fit for translocation experiments by ramping voltage from −300 to 300 mV and observing an Ohmic relationship between current and voltage (i.e., linear IV curve).
Gene Synthesis Reactions
The HIV protease (HIVpr) synthetic gene was assembled using 22 construction oligos (a table of oligo sequences is provided in Supporting Information Figure 3) in a 50 μL PCA reaction using KOD Hot Start polymerase reagents (EMD Millipore/Novagen). Reactions contained 1×KOD buffer, 1.5 mM MgSO4, 0.2 mM dNTP, 1 unit KOD polymerase, 0.5 uM HIVpr-B22 primer (CCGCCTCTCCCCGCGAGTTCCTCCTTTCAGCAAAAAACCCCTCAA), 0.5 μM HIVpr-T1 primer (ATGAATCGGCCAACGTCCGGCGTAGAGGCGAAATTAATACGACTCACTATAGGGAG), and 1 μM of each construction oligo. The initial PCA reaction was cycled 30 times (95 °C for 20 s/72 °C for 30 s) followed by a 5 min extension at 72 °C and cooling to 12 °C. An aliquot of the reaction was diluted 1:1000 prior to being used in a secondary PCR amplification. The secondary PCR amplification for the HIVpr DNA was performed in a 100 μL reaction using 1 μL of the initial PCA template, 0.5 uM of primers HIVpr-B22 and HIVpr-T1, and Phusion High-Fidelity PCR Master Mix (New England Biolabs, M0531). This reaction was heated to 98 °C for 45 s, then cycled 30 times (98 °C, 45 s/55 °C, 45 s/72 °C, 45 s), finishing with a 5 min extension at 72 °C and cooling to 4 °C. Aliquots of both reactions were purified using the Qiagen Gel Extraction Kit as per manufacturers protocol, eluting in water. DNA was quantitated via Nanodrop spectrophotometer (Thermo Scientific). Primers were synthesized at 100 nM scale (IDT), and HPLC purified. Primer annealing for different DNA structures and mismatches was performed by mixing 2 μM of each primer, 25 mM NaCl, 5 mM Tris pH 8.0, 1 mM EDTA, and 8 mM MgCl2. Solutions were heated to 95 °C for 1 min, followed by decreasing the temperature 5 °C each minute to room temperature to anneal the primer pairs. The initial PCA reaction was Sanger sequenced and found to have a defect rate of 0.0018 per base (1 error per 555 bp). With the use of this error rate, the percentage of defect free strands could be predicted as (1–0.0018)524 = 39% and double-stranded duplexes to be 15% (or 0.389 × 0.389).
Gel Electrophoresis Assays
A 75 ng aliquot of each annealed primer mixture and individual primers was analyzed by electrophoresis on both a 20% TBE–acrylamide gel and a 15% TBE–urea acrylamide gel (Life Technologies). Samples analyzed on TBE–urea gels were heated to 70 °C for 3 min in 2× TBE–urea sample buffer (Life Technologies) prior to loading. After electrophoresis, gels were rinsed for 30 s in water prior to immersion staining for 10 min in water and SYBR-Gold DNA stain (1:10 000 dilution; Life Technologies). Gels were rinsed for 30 s in water prior to exposure to UV light and photo documentation with Gel Doc camera and software.
Analysis of DNA Translocations
All nanopore data was analyzed using Pythion, a custom Python-based analysis software designed by Robert Y. Henley in the Wanunu lab (github.com/rhenley/Pyth-Ion). Gaussian and 1D drift-diffusion fits of histograms were computed using Igor Pro (Wavemetrics).
Supplementary Material
Acknowledgments
The authors thank Chester Beal for assistance with artwork. This work was sponsored by the Assistant Secretary of Defense for Research and Engineering under Air Force Contract No. FA8721-05-C-0002. Opinions, interpretations, recommendations and conclusions are those of the authors and are not necessarily endorsed by the United States Government. This work was also supported by grant from the National Institutes of Health [R21-HG006873 to M.W.].
Footnotes
Conflict of Interest: The authors declare the following competing financial interest(s): C.A.A. and P.A.C. are inventors of U.S. provisional patent application No.: 14/433,471.
Supporting Information Available: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.5b05782.
REFERENCES AND NOTES
- 1.Ellis T, Wang X, Collins JJ. Diversity-Based, Model-Guided Construction of Synthetic Gene Networks With Predicted Functions. Nat Biotechnol. 2009;27:465–471. doi: 10.1038/nbt.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu TK, Khalil AS, Collins JJ. Next-Generation Synthetic Gene Networks. Nat Biotechnol. 2009;27:1139–1150. doi: 10.1038/nbt.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carr PA, Church GM. Genome Engineering. Nat Biotechnol. 2009;27:1151–1162. doi: 10.1038/nbt.1590. [DOI] [PubMed] [Google Scholar]
- 4.Gibson DG, Smith HO, Hutchison CA, Venter JG, Merryman C. Chemical Synthesis of the Mouse Mitochondrial Genome. Nat Methods. 2010;7:901–903. doi: 10.1038/nmeth.1515. [DOI] [PubMed] [Google Scholar]
- 5.Gibson DG, Benders GA, Andrews-Pfannkoch C, Denisova EA, Baden-Tillson H, Zaveri J, Stockwell TB, Brownley A, Thomas DW, Algire MA, et al. Complete Chemical Synthesis, Sssembly, and Cloning of a Mycoplasma Genitalium Genome. Science. 2008;319:1215–1220. doi: 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- 6.Gibson DG, Young L, Chuang RY, Venter JG, Hutchison CA, Smith HO. Enzymatic Assembly of DNA Molecules Up to Several Hundred Kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 7.Shetty RP, Endy D, Knight TF. Engineering Biobrick Vectors From Biobrick Parts. J Biol Eng. 2008;2:5. doi: 10.1186/1754-1611-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoover DM, Lubkowski J. DNAWorks: An Automated Method For Designing Oligonucleotides for PCR-based Gene Synthesis. Nucleic Acids Res. 2002;30:e43. doi: 10.1093/nar/30.10.e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bang D, Church GM. Gene Synthesis By Circular Assembly Amplification. Nat Methods. 2008;5:37–39. doi: 10.1038/nmeth1136. [DOI] [PubMed] [Google Scholar]
- 10.Czar MJ, Anderson JC, Bader JS, Peccoud J. Gene Synthesis Demystified. Trends Biotechnol. 2009;27:63–72. doi: 10.1016/j.tibtech.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 11.Tian JD, Ma KS, Saaem I. Advancing High-Throughput Gene Synthesis Technology. Mol BioSyst. 2009;5:714–722. doi: 10.1039/b822268c. [DOI] [PubMed] [Google Scholar]
- 12.Quan J, Saaem I, Tang N, Ma S, Negre N, Gong H, White KP, Tian J. Parallel On-Chip Gene Synthesis And Application To Optimization of Protein Expression. Nat Biotechnol. 2011;29:449–452. doi: 10.1038/nbt.1847. [DOI] [PubMed] [Google Scholar]
- 13.Lee CC, Synder TM, Quake SR. A Microfluidic Oligonucleotide Synthesizer. Nucleic Acids Res. 2010;38:2514–2521. doi: 10.1093/nar/gkq092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatesan BM, Bashir R. Nanopore Sensors For Nucleic Acid Analysis. Nat Nanotechnol. 2011;6:615–624. doi: 10.1038/nnano.2011.129. [DOI] [PubMed] [Google Scholar]
- 15.Fologea D, Brandin E, Uplinger J, Branton D, Li S. DNA Conformation And Base Number Simultaneously Determined In A Nanopore. Electrophoresis. 2007;28:3186–3192. doi: 10.1002/elps.200700047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gershow M, Golovchenko JA. Recapturing And Trapping Single Molecules With A Solid-State Nanopore. Nat Nanotechnol. 2007;2:775–779. doi: 10.1038/nnano.2007.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen YH, Jain T, Aguilar CA, Karnik R. Enhanced Discrimination of DNA Molecules In Nanofluidic Channels Through Multiple Measurements. Lab Chip. 2012;12:1094–1101. doi: 10.1039/c2lc20771k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kowalczyk SW, Hall AR, Dekker C. Detection Of Local Protein Structures Along DNA Using Solid-State Nanopores. Nano Lett. 2010;10:324–328. doi: 10.1021/nl903631m. [DOI] [PubMed] [Google Scholar]
- 19.Plesa C, Ruitenberg JW, Witteveen MJ, Dekker C. Detection Of Individual Proteins Bound Along DNA Using Solid-State Nanopores. Nano Lett. 2015;15:3153–3158. doi: 10.1021/acs.nanolett.5b00249. [DOI] [PubMed] [Google Scholar]
- 20.Wanunu M, Cohen-Karni D, Johnson RR, Fields L, Benner J, Peterman N, Zheng Y, Klein ML, Drndic M. Discrimination Of Methylcytosine From Hydroxmethylcytosine In DNA Molecules. J Am Chem Soc. 2011;133:486–492. doi: 10.1021/ja107836t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguilar CA, Craighead HG. Micro And Nanoscale Devices For The Investigation Of Epigenetics And Chromatin Dynamics. Nat Nanotechnol. 2013;8:709–718. doi: 10.1038/nnano.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plesa C, van Loo N, Ketter P, Dietz H, Dekker C. Velocity Of DNA During Translocation Through A Solid-State Nanopore. Nano Lett. 2015;15:732–737. doi: 10.1021/nl504375c. [DOI] [PubMed] [Google Scholar]
- 23.Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M. Rapid Electronic Detection of Probe-Specific MicroRNAs Using Thin Nanopore Sensors. Nat Nanotechnol. 2010;5:807–814. doi: 10.1038/nnano.2010.202. [DOI] [PubMed] [Google Scholar]
- 24.Carson S, Wilson J, Aksimentiev A, Wanunu M. Smooth, D. N. A.; Transport Through, A. Narrowed Pore Geometry. Biophys J. 2014;107:2381–2393. doi: 10.1016/j.bpj.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wanunu M, Sutin J, McNally B, Chow A, Meller A. DNA Translocation Governed by Interactions With Solid State Nanopores. Biophys J. 2008;95:4716–4725. doi: 10.1529/biophysj.108.140475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wanunu M, Morrison W, Rabin Y, Grosberg AY, Meller A. Electrostatic Focusing of Unlabelled DNA Into Nanoscale Pores Using A Salt Gradient. Nat Nanotechnol. 2010;5:160–165. doi: 10.1038/nnano.2009.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Firnkes M, Pedone D, Knezevic J, Döblinger M, Rant U. Electrically Facilitated Translocations of Proteins Through Silicon Nitride Nanopores: Conjoint And Competitive Action of Diffusion, Electrophoresis, And Electroosmosis. Nano Lett. 2010;10:2162–2167. doi: 10.1021/nl100861c. [DOI] [PubMed] [Google Scholar]
- 28.Di Fiori N, Squires A, Bar D, Gilboa T, Moustakas TD, Meller A. Optoelectronic Control Of Surface Charge And Translocation Dynamics In Solid-State Nanopores. Nat Nanotechnol. 2013;8:946–951. doi: 10.1038/nnano.2013.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu H, Yang W, Seeman NC. DNA Scissors Device Used To Measure MutS Binding To DNA Mis-Pairs. J Am Chem Soc. 2010;132:4352–4357. doi: 10.1021/ja910188p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carr PA, Park JS, Lee YJ, Yu T, Zhang SG, Jacobson JM. Protein-Mediated Error Correction For De Novo DNA Synthesis. Nucleic Acids Res. 2004;32(20):e162. doi: 10.1093/nar/gnh160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muthukumar M. Polymer Translocation. CRC Press; Boca Raton, FL: 2011. [Google Scholar]
- 32.Ling DY, Ling XS. On The Distribution of DNA Translocation Times in Solid-State Nanopores: An Analysis Using Schrödinger’s First-Passage-Time Theory. J Phys: Condens Matter. 2013;25:375102. doi: 10.1088/0953-8984/25/37/375102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hornblower B, Coombs A, Whitaker RD, Kolomeisky A, Picone SJ, Meller A, Akeson M. Single-Molecule Analysis of DNA-Protein Complexes Using Nanopores. Nat Methods. 2007;4:315–317. doi: 10.1038/nmeth1021. [DOI] [PubMed] [Google Scholar]
- 34.Ivankin A, Carson S, Kinney SR, Wanunu M. Fast, Label-Free Force Spectroscopy of Histone DNA–Interactions in Individual Nucleosomes Using Nanopores. J Am Chem Soc. 2013;135:15350–15352. doi: 10.1021/ja408354s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Squires A, Atas E, Meller A. Nanopore Sensing of Individual Transcription Factors Bound to DNA. Sci Rep. 2015;5:11643. doi: 10.1038/srep11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao C, Sun W, Seeman NC. Designed Two-Dimensional DNA Holliday Junction Arrays Visualized by Atomic Force Microscopy. J Am Chem Soc. 1999;121:5437–5443. [Google Scholar]
- 37.Pinheiro AV, Han D, Shih WM, Yan H. Challenges and Opportunities For Structural DNA Nanotechnology. Nat Nanotechnol. 2011;6:763–772. doi: 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-Step Assembly of a Gene and Entire Plasmid From Large Numbers of Oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]
- 39.Condra JH, Schleif WA, Blahy OM, Gabryelski LJ, Graham DJ, Quintero J, Rhodes A, Robbins HL, Roth E, Shivaprakash M, et al. In Vivo Emergence of HIV-1 Variants Resistant to Multiple Protease Inhibitors. Nature. 1994;374:569–571. doi: 10.1038/374569a0. [DOI] [PubMed] [Google Scholar]
- 40.Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchinson CA. Complete Mutagenesis Of The HIV-1 Protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- 41.Kosuri S, Eroshenko N, LeProust EM, Super M, Way J, Li JB, Church GM. Scalable Gene Synthesis By Selective Amplification Of DNA Pools From High-Fidelity Microchips. Nat Biotechnol. 2010;28:1295–1299. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kosuri S, Church GM. Large-Scale De Novo DNA Synthesis: Technologies And Applications. Nat Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castro CE, Kilchherr F, Kim DN, Shiao EL, Wauer T, Wortmann P, Bathe M, Dietz H. A Primer to Scaffolded DNA Origami. Nat Methods. 2011;8:221–229. doi: 10.1038/nmeth.1570. [DOI] [PubMed] [Google Scholar]
- 44.Jain T, Guerrero RG, Aguilar CA, Karnik R. Integration of Solid-State Nanopores in Microfluidic Networks via Transfer Printing of Suspended Membranes. Anal Chem. 2013;85:3871–3878. doi: 10.1021/ac302972c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cipriany BR, Murphy PJ, Hagarman JA, Cerf A, Latulippe D, Levy SL, Benitez JJ, Tan CP, Topolancik J, Soloway PD, et al. Real-Time Analysis And Selection Of Methylated DNA By Fluorescence-Activated Single Molecule Sorting In A Nanofluidic Channel. Proc Natl Acad Sci U S A. 2012;109:8477–8482. doi: 10.1073/pnas.1117549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.