

Abstract
Alzheimer’s disease (AD) is a progressive dementia disorder characterized by synaptic degeneration and amyloid-β (Aβ) accumulation in the brain. Through whole-genome sequencing of 1345 individuals from 410 families with late-onset AD (LOAD), we identified three highly penetrant variants in PRKCA, the gene that encodes protein kinase Cα (PKCα), in five of the families. All three variants linked with LOAD displayed increased catalytic activity relative to wild-type PKCα as assessed in live-cell imaging experiments using a genetically encoded PKC activity reporter. Deleting PRKCA in mice or adding PKC antagonists to mouse hippocampal slices infected with a virus expressing the Aβ precursor CT100 revealed that PKCα was required for the reduced synaptic activity caused by Aβ. In PRKCA−/− neurons expressing CT100, introduction of PKCα, but not PKCα lacking a PDZ interaction moiety, rescued synaptic depression, suggesting that a scaffolding interaction bringing PKCα to the synapse is required for its mediation of the effects of Aβ. Thus, enhanced PKCα activity may contribute to AD, possibly by mediating the actions of Aβ on synapses. In contrast, reduced PKCα activity is implicated in cancer. Hence, these findings reinforce the importance of maintaining a careful balance in the activity of this enzyme.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by progressive cognitive decline. Synaptic degeneration is among the earliest manifestations of AD in humans (1, 2) and is an early event in rodent models of AD (3–5). The loss of synapses may result from increased abundance of amyloid-β (Aβ) (6–8), a peptide that is associated with the development of AD (9). Familial forms of AD (hereditary early-onset AD), which account for less than 10% of AD cases, are caused by mutations in genes encoding amyloid precursor protein (APP) or β- and γ-secretases (enzymes that cleave APP) that result in increased abundance of the most toxic form of Aβ (9). The risk for late-onset AD (LOAD), which accounts for most AD cases, is increased in individuals expressing a variant of apolipoprotein E (APOE) called APOE-ε4 (10), which may be less effective than other APOE alleles at promoting the removal of Aβ (9). Additional factors, such as innate immunity, namely, the AD-associated genes CD33 and TREM2, also play a role (11, 12).
We sought to identify other genes that contribute to LOAD because their gene products may serve as therapeutic targets. We reasoned that variants of proteins participating in the detrimental effects of Aβ may contribute to the disease. We thus examined the signaling underlying Aβ-induced synaptic depression. We focused on the multi-isozyme protein kinase C (PKC) family (13) because an interacting partner of the isozyme PKCα, PKC-binding protein (PICK1), is required for the synaptic effects of Aβ (14), and additionally, PKCα is required for a plasticity form of synaptic depression (15). The role of PKC isozymes in cancer is complex, but the finding that many cancer-associated mutations in PKC isozymes are actually loss of function indicates that reduced PKC activity contributes to increased cell survival (16). Therefore, we speculated that increased PKC activity could participate in the synaptic loss seen in AD.
RESULTS
PKCα is necessary for the synaptic effects of Aβ
To test whether the activity of PKC contributes to Aβ-induced synaptic depression, we added PKC antagonists to rodent organotypic hippocampal slices infected sparsely with a virus expressing CT100, which increases Aβ production by neurons (6, 8, 17). CT100 is the product of APP cleavage by β-secretase; CT100 is subsequently cleaved by γ-secretase to produce Aβ (18). After 16 to 24 hours, we evoked synaptic transmission by electrically stimulating presynaptic axons, and recordings were simultaneously obtained from neighboring infected and uninfected postsynaptic hippocampal CA1 neurons. Infected neurons (that expressed CT100) exhibited depressed excitatory transmission relative to that in uninfected neurons (Fig. 1A). Previous studies have shown that the effects of CT100 expression are due to the subsequent production of Aβ that induces the endocytosis of glutamate receptors and loss of synapses (6, 8, 17). To examine the role of PKC in Aβ-dependent synaptic depression, we incubated hippocampal slices (for ~12 hours before recordings) with the PKC inhibitors Gö 6983, an adenosine 5′-triphosphate (ATP) competitive inhibitor, or bisindolylmaleimide IV (Bis IV), an uncompetitive inhibitor; these inhibitors inhibit conventional (such as PKCα) and novel PKC isozymes. Synaptic depression by CT100 expression was effectively blocked by Bis IV but not by Gö 6983 (Fig. 1, A and B). Because PKC is refractory to active-site inhibitors when it is bound to a scaffold protein near its substrates (19), these data suggest that scaffold protein–bound PKC may mediate the synaptic effects of Aβ. Of the PKC isozymes that are modulated by these inhibitors, only PKCα has a PDZ ligand, a three–amino acid segment at its C terminus that binds the PDZ domains of scaffolding proteins [notably PSD95, SAP97, and PICK1 (20, 21)]. Expression of CKAR, a genetically encoded fluorescence-based reporter of bulk PKC activity (22), in COS7 cells revealed that endogenous PKC activity at the PSD95 scaffold was effectively inhibited by Bis IV but not Gö 6983, whereas both inhibitors were equally effective at inhibiting bulk PKC activity measured at plasma membrane (Fig. 1C).
Fig. 1. Synaptic depression by Aβ blocked by uncompetitive PKC antagonist.
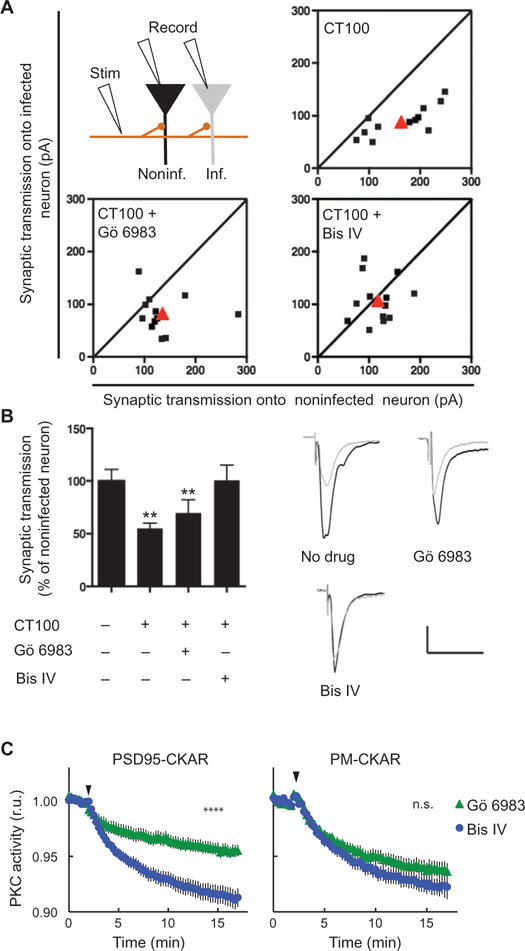
(A and B) Top left: Experimental design (see Materials and Methods). Plots (A) and bar graphs (B) of evoked synaptic response amplitudes recorded in CT100-infected versus noninfected neurons. Shown are individual (black) and average (red) of cell pair responses; sloped line, x = y. Sample traces (B, right) obtained from indicated conditions. Scale bars, 50 ms, 50 pA. Data are means ± SEM from 12 to 13 pairs of cells (one infected, the other not); **P < 0.03, by bootstrap test (see Materials and Methods). (C) Normalized average of PKC activity in COS7 cells expressing PKC activity reporter CKAR (C kinase activity reporter) fused to PSD95 (left) or targeted to plasma membrane (right) in response to the competitive (Gö 6983) or uncompetitive (Bis IV) PKC inhibitor, added at the time indicated by the arrow head. Data are means ± SEM from >16 cells; ****P < 0.0001, by bootstrap test (see Materials and Methods); n.s., not significant.
To test whether PKCα specifically mediates the synaptic effects of Aβ, organotypic hippocampal slices were prepared from wild-type mice and mice lacking PKCα (PRKCA−/−) (23). Sixteen to 24 hours after infection, neurons expressing CT100 displayed depressed synaptic transmission compared to nearby neurons in slices from wild-type mice but showed no difference in PRKCA−/− slices (Fig. 2, A and B). Western blot analysis revealed comparable levels of Aβ peptide in lysates from slices from PRKCA−/− or wild-type animals infected with virus expressing CT100 (fig. S1). To determine whether absence of PKCα, rather than some developmental alteration in PRKCA−/− mice, was responsible for the lack of effect by CT100 in neurons from PRKCA−/− mice, we used a dual promoter virus to coexpress CT100 and PKCα in PRKCA−/− organotypic slices. After expression, transmission onto infected neurons was depressed compared to noninfected neurons, indicating rescue of the effect of CT100 on synapses (Fig. 2A). To ensure the rescue of CT100-induced depression by PKCα coexpression was not due to potentiation of transmission by PKCα expression (which when added to the CT100-induced synaptic depression could normalize transmission), we coexpressed PKCα with CT84, the α-secretase product of APP that neither generates Aβ (18) nor causes synaptic depression (24). In this case, no significant potentiation was observed (Fig. 2A), indicating that PKCα coexpression rescued the effects of CT100 by enabling CT100-driven signaling. To test whether the effects of Aβ on synapses require that PKCα act at a scaffold, as suggested by the pharmacological results above, we coexpressed CT100 and a mutant form of PKCα that lacked the last three amino acids [PKCα (ΔPDZ)], which prevents its binding to PDZ domain proteins. In contrast to the coexpression of PKCα, coexpression of PKCα (ΔPDZ) did not rescue the effects of CT100 expression (Fig. 2A). These results indicate that PKCα, likely acting at a scaffold, is required for the depressive effects of Aβ on rodent synapses.
Fig. 2. PKCα is required for the effects of Aβ on synaptic transmission.
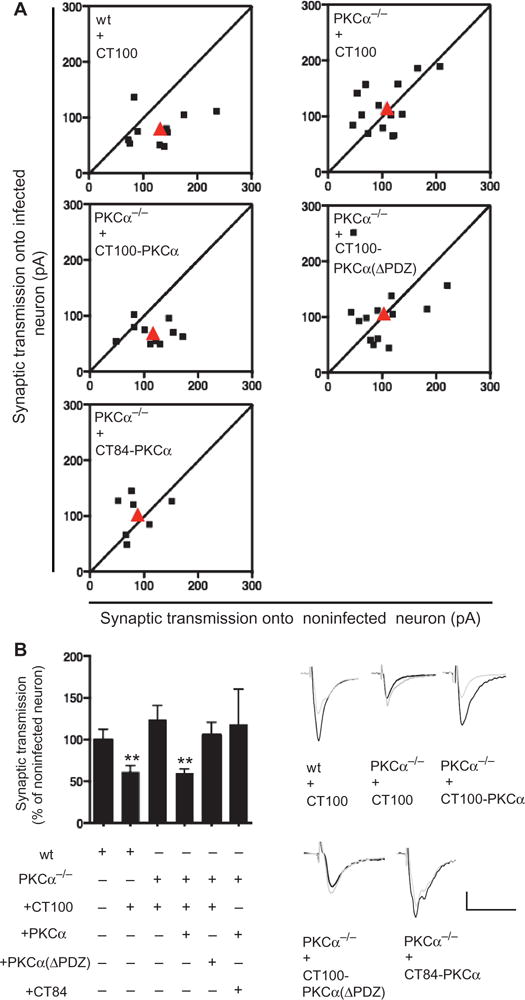
(A and B) Plot (A), and example traces (bottom right), of evoked synaptic response amplitudes recorded in infected versus noninfected neurons; genotype and infection indicated. Bar graph (B, left) of same data. **P < 0.03, by bootstrap test (see Materials and Methods). wt, wild-type.
Whole-genome sequencing reveals PKCα variants in AD
To test whether PKCα plays a role in human AD, we searched whole-genome sequencing data for rare variants in PRKCA that were present in any of the 410 pedigrees from the National Institute of Mental Health (NIMH) Alzheimer’s Disease Genetics Initiative (Fig. 3 and fig. S2). We identified three rare variants found in LOAD patients in these families. M489V [single nucleotide polymorphism: rs34406842; codon substitution: Atg/Gtg; minor allele frequency = 0.0005 in 1000 genomes of the general population (www.1000genomes.org)] was present in seven of nine affected individuals and was absent in the single unaffected subject in these four families. V636I (rs141376042; Gtc/Atc; minor allele frequency = 0.002 in 1000 genomes) was present in two of three affected subjects in one family in which no data from unaffected subjects were available. R324W (minor allele frequency = 0.00 in 1000 genomes) was present in one of two affected individuals and missing in the unaffected individual in a single LOAD pedigree. Thus, all individuals carrying a PRKCA variant developed AD, and all the individuals in these families who did not carry a PRKCA variant did not develop AD. Thus, the PRKCA variants identified in the NIMH data set appear to cosegregate with LOAD patients. A simple bootstrap test (see Materials and Methods) suggests that the probability of finding such linkage in this population is less than 0.03.
Fig. 3. Human genetics of rare PKCα variants.

Diagrams indicating number of families, along with phenotype and genotype of individuals, carrying M489V, V636I, or R324W PKCα variants. All PKCα variant carriers (yellow) displayed AD, and both individuals without AD (blue) lacked a PKCα variant.
AD-linked PKCα variants display increased cellular activity
Given that PKCα activity mediated the synaptic effects of increased abundance of Aβ (Figs. 1 and 2), we hypothesized that the rare PKCα variants found in LOAD families would have increased activity. This view is also suggested by the findings that reduced PKC function enhances survival in some cancer cells (16), whereas neuronal loss is seen in AD. Two of the rare variants observed in LOAD patients occur in key regulatory segments of the PKC family (13): M489V is in the activation loop near the entrance to the active site, and V636I is on the C-terminal segment that clamps over the kinase domain (Fig. 4A). Both segments interface with the Ca2+-sensing C2 domain, which maintains PKCα in an autoinhibited conformation (25). Thus, both mutations have the potential to destabilize autoinhibition. For example, molecular modeling reveals that replacement of the bulky Met at position 489 on the activation loop with the smaller Val loosens the packing of this key regulatory segment (Fig. 4A), consistent with reduced autoinhibition. Additionally, this residue is close to a segment in the kinase architecture, referred to as DFG +1, that is a key specificity determinant (26), and mutations in this region have been shown to alter substrate specificity in another PKC, PKCγ (27). The third variant, R324W, is also in a segment that controls autoinhibition; it is present in a flexible hinge, connecting the kinase domain with the regulatory moiety, that becomes exposed upon rupture of the C2-kinase domain contacts accompanying activation (25).
Fig. 4. AD-associated rare variants in PKCα.
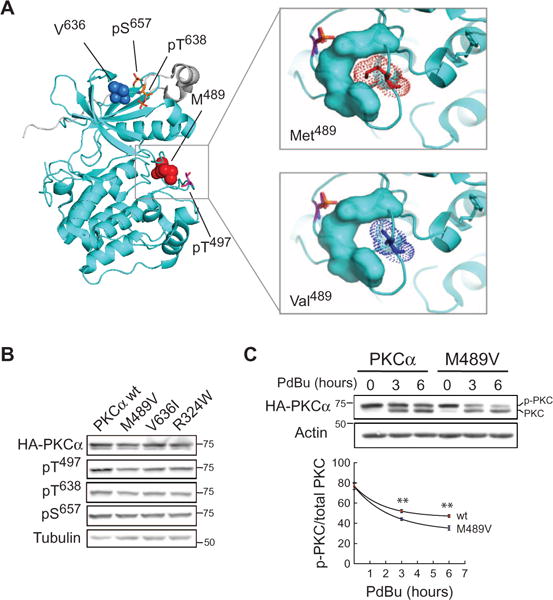
(A) PKCα kinase domain structure (53) showing two residues altered in AD:Met489 and Val636. Both are near key regulatory phosphorylation sites (stick representation). Enlargement of activation loop segment (right panels) showing that substitution of Met489 with Val loosens the structural packing of this segment. (B) Western blot showing phosphorylation of the indicated hemagglutinin (HA)–tagged PKCα proteins. (C) Western blot of COS7 cells expressing wild-type or M489V PKCα and treated with phorbol 12,13-dibutyrate (PDBu) for the indicated times and probed for HA. Quantitative analysis of phosphorylated/total PKC from five independent experiments. **P < 0.01, by bootstrap test (see Materials and Methods).
To examine the functional impact of amino acid changes, we assessed the effect of introducing these LOAD-associated rare variants on the function of PKCα. When expressed in COS7 cells, none of the three rare variants affected the phosphorylation of PKCα at priming sites that are indicative of proper protein folding (28), as assessed by Western blot analysis using phosphospecific antibodies (Fig. 4B). Further analysis of the M489V mutant revealed an increased rate of dephosphorylation after cells were stimulated with phorbol esters (Fig. 4C), potent activators that force PKCs into an open phosphatase-sensitive conformation (29). This enhanced rate of de-phosphorylation supports the looser packing of the kinase domain resulting from replacement of Met with Val, favoring a more open (meaning active) conformation of the kinase when challenged with phorbol esters (30). The V636I mutant did not display the same enhanced rate of dephosphorylation, indicating that there may be other structural mechanisms through which this mutation destabilizes PKC autoinhibition.
To assess the effect of the amino acid changes on the signaling output of PKCα, we coexpressed PKCα variants or wild-type PKCα with cytosolic CKAR (22) in COS7 cells. All three rare variants showed enhanced agonist-evoked activity relative to wild-type PKCα (Fig. 5). This assay measures the phosphorylation of CKAR and reflects the allosteric activation of PKC induced by second messenger binding. This acute stimulation by natural agonists does not cause the dephosphorylation or down-regulation of PKC that is caused by chronic activation by phorbol esters. These results indicate that all three rare PKCα variants found to cosegregate with LOAD display increased agonist-driven PKCα function.
Fig. 5. Live-cell imaging reveals higher signaling output of all three AD-associated rare variants.
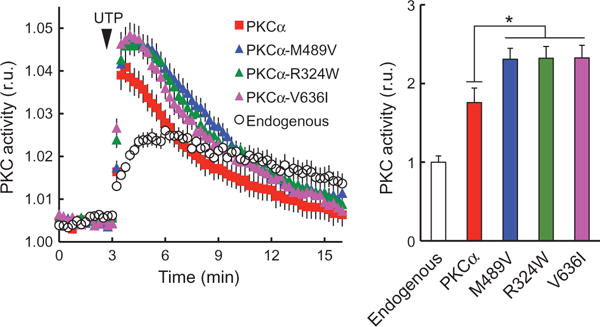
Left: Normalized FRET ratios (mean ± SEM) representing PKC activity in COS7 cells coexpressing PKC activity reporter, CKAR, (22) and indicated PKCα. Addition of uridine 5′-triphosphate (UTP) (100 μM) where indicated (arrow head). n > 25 cells for each construct. Data are means ± SEM from at least three independent experiments. Right: Area under the curve from 3 to 6 min; *P < 0.05, by bootstrap test (see Materials and Methods). r.u., relative units.
DISCUSSION
AD is a lethal neurodegenerative disorder with increasing prevalence and no effective treatment (9). Elucidating the cellular signaling events that underlie the initiation and progression of LOAD can provide potential targets amenable to therapeutic intervention. Human genetic studies (31), particularly family-based studies (10) of rare variants (32), can identify potentially important molecules in LOAD. However, delineation of LOAD pathophysiology primarily from genetic information from smaller family studies remains challenging [see published comments and responses to (32)]. We therefore sought to combine a large genetic family study with neurophysiological and biochemical analyses to elucidate the signaling underlying LOAD pathophysiology.
PKCα and AD pathophysiology
Although the cellular events that initiate LOAD remain unknown (9), increased accumulation of pathogenic forms of Aβ and the associated synaptic loss are strongly implicated in its early pathophysiology (1, 2, 33, 34). Various PKC isozymes have been implicated in AD in a number of studies (35–38). Supporting a role for enhanced signaling by PKC in AD, a recent phosphoproteomics study of postmortem brains identified increased phosphorylation of PKC substrates as a primary early event in AD (39). However, the role of PKC is not yet well understood. Understanding the biological functions of PKC in disease in general has presented a challenge given the presence of multiple isozymes and the opposing effects of phorbol esters in mediating short-term activation and long-term down-regulation. By combining the use of pharmacology, mouse genetics, and acute rescue experiments in this study, we can conclude the role of a specific PKC isozyme: our neurophysiological studies indicate that PKCα and its interaction with a protein through a PDZ domain interaction are required for Aβ to produce synaptic depression [consistent with a previous study demonstrating a requirement of PICK1 (14), a PDZ domain protein that binds PKCα]. Given the evidence supporting Aβ and synaptic loss in AD, our results suggest an important role for PKCα in AD.
Because conclusions regarding human disease based on physiology from immature rodent brain slices are tenuous, we turned to human genetics, which can provide insight into the function of aberrant PKCα signaling in disease. By scanning the genomes of individuals from a large LOAD family study, we identified three rare variants of PKCα that cosegregated with LOAD in six families. When examined in cell-based assays, all three variants displayed increased signaling output relative to wild-type PKCα. Notably, of the 46 PKC variants observed in cancers and tested for changed activity using the same assays, none displayed increased activity (16). Furthermore, the more strongly linked M489V mutation, found in seven of nine patients and not in an unaffected subject in four LOAD families, also elicited an enhanced rate of dephosphorylation after stimulation of cells with phorbol esters, consistent with a more open and signaling competent conformation. Thus, PKCα is required for deleterious synaptic effects of Aβ, observed early in LOAD, and increased PKCα activity can contribute to human LOAD.
PKCα activity: Increased in neurodegeneration and decreased in cancer
It is notable that we found activating PKC mutations in LOAD patients, whereas functional human cancer-associated PKC mutations are inactivating (16). The neurodegenerative disease, spinocerebellar ataxia, is caused by mutations in PKCγ that promote the open active conformation (40). Thus, our results are consistent with PKC gain-of-function mutations driving neurodegenerative diseases and loss-of-function mutations driving cell survival diseases. Supporting opposing roles of PKC in cancer versus AD, a recent meta-analysis of nine independent studies reveals that AD patients exhibit an overall 45% decreased risk of cancer compared with the general population (41), consistent with earlier reports that AD and cancer display an inverse association (42, 43). It is relevant that inhibitors of PKC have failed in cancer clinical trials (44), likely because PKC activity should be restored, rather than inhibited, in cancer therapy (16). In contrast, repurposing PKC inhibitors for LOAD may prevent the effects of Aβ on synapses and thereby mitigate loss of cognitive function. Together, our findings support inhibition of PKCα as a therapy early in LOAD and suggest that mutations in PKCα can serve as a diagnostic for disease susceptibility.
MATERIALS AND METHODS
Tissue preparation
Experiments were conducted in accordance with and received approval from the Institutional Animal Care and Use Committees at the University of California, San Diego. The experiments were carried out in accordance with guidelines laid down by the National Institutes of Health regarding the care and use of animals for experimental procedures.
Hippocampal slice cultures and Sindbis virus infection
Organotypic hippocampal slice cultures were made from postnatal day 6 or 7 rat pups as described (45). Slice cultures were maintained in culture for 6 to 8 days and then infected using a Sindbis virus (pSinRep5 dp APP-CT100 tdTomato). Cells were recorded 16 to 30 hours after infection. For Fig. 2 experiments, slices were made as described above, but from either wild-type or PRKCA−/− mouse pups and infected with the indicated Sindbis viruses.
Electrophysiology and pharmacological treatments
Slices were maintained in a solution of artificial cerebrospinal fluid containing the following: 119 mM NaCl, 26 mM NaHCO3, 1 mM NaH2PO4, 11 mM D-glucose, 2.5 mM KCl, 4 mM CaCl2, 4 mM MgCl2, and 1.25 mM NaHPO4 and gassed with 95% O2 5% CO2. In addition, the following drugs were included: 4 μM 2-chloroadenosine (to prevent stimulus induced bursting) and 100 μM picrotoxin (to block inhibitory transmission). For Figs. 1 and 2, simultaneous whole-cell recordings were obtained from pairs of neighboring (<50 μm) control and infected CA1 pyramidal neurons using 3- to 5-megohm glass pipettes with an internal solution containing the following: 115 mM cesium methanesulfonate, 20 mM CsCl, 10 mM Hepes, 2.5 mM MgCl2, 4 mM Na2ATP, 0.4 mM Na3GTP, 10 mM sodium phosphocreatine, 0.6 mM EGTA, and 0.1 mM spermine, at pH 7.25. All recordings were done by stimulating two independent synaptic inputs; EPSCs were recorded while holding the cells at −60 mV, alternating pathways every 8.4 s. Results from each pathway were averaged and counted as n = 1. For pharmacological experiments, slices were incubated overnight before recordings with either 0.3 μM Gö 6983 or 3 μM Bis IV; the drug was also added to the recording chamber at the same concentration. In addition, 10 μM gabazine (to block inhibitory transmission) was added to the perfusion chamber. All data are reported as means ± SEM. Statistical analysis used bootstrap (resampling) methods (46). For instance, in Fig. 1, we calculated the probability by bootstrap resampling (100,000 times) the data from groups a = (CT100−,Gö 6983−, Bis IV−),b=(CT100+,Gö6983−,BisIV−),c=(CT100+,Gö6983+,Bis IV−), and d = (CT100+,Gö 6983−,Bis IV+) and measuring the frequency with which A > C or B > C is true (where caps indicate mean); for Fig. 2, we bootstrap-resampled (100,000 times) data groups, measuring the frequency with which A > B or C > B or A > D is true. For Fig. 4, we bootstrap-resampled (100,000 times) data groups, measuring the frequency with which (MV at t = 3 hours > wild-type at t = 3 hours) or (MV at t = 6 hours > wild-type at t = 6 hours) is true.
Plasmid constructs
The CKAR and plasma membrane–localized PKC reporter were described previously (22). The PSD95-specific PKC reporter (PSD95-CKAR) contains CKAR with PSD95 fused to its N terminus via a four–amino acid linker (EPGQ) in a pcDNA3 vector (Life Technologies). PKC constructs were prepared as described previously (16). For HA-PKCα, human PKCα was N-terminally HA-tagged via Gateway cloning into pDEST-HA generated from ligating the reading frame cassette C into the EcoRV site of pcDNA3-HA. All mutants were generated using QuikChange site-directed mutagenesis (Agilent Technologies).
Antibodies and reagents
The pan antibody against the phosphorylated PKC activation loop (pT497) was previously described (47). The antibody to phosphorylated PKCα/βII (pT638/641; 9375S) and pan antibody to the phosphorylated PKC hydrophobic motif (βII pS660; 9371S) were purchased from Cell Signaling Technology. The α-HA antibody (anti-HA; 11867423001, clone 3F10) was purchased from Roche. PDBu, UTP trisodium salt, Gö 6983, and Bis IV were obtained from Calbiochem.
Cell culture, transfection, and immunoblotting
All cells were maintained in Dulbecco’s modified Eagle’s medium (Corning) containing 10% fetal bovine serum (Atlanta Biologicals) and 1% penicillin/streptomycin (Corning) at 37°C, in 5% CO2. Transient transfection of COS7 was carried out using FuGENE 6 transfection reagent (Roche) for ~24 hours. Cells were lysed in 50 mM tris (pH7.4), 1% Triton X-100, 50mM NaF, 10 mM Na4P2O7, 100 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, leupeptin (50 μg/ml), 1 μM microcystin, 1 mM dithiothreitol, and 2 mM benzamidine. For PKC immunoblotting, whole-cell lysates were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting via chemiluminescence on a FluorChem Q imaging system (ProteinSimple). For cellular dephosphorylation experiments, cells were treated with 200 nM PDBu for the indicated times at 37°C before lysis. For Aβ immunoblotting, 10 μg of protein per sample was electrophoresed on 10 to 20% SDS–polyacrylamide tris-tricine gels (Bio-Rad). Proteins were transferred to nitrocellulose, and membranes were boiled in phosphate-buffered saline for 5 min. Membranes were blocked in TBST(tris-buffered saline– Tween 20) containing 5% bovine serum albumin (#A3803, Sigma; >98% grade) for 1 hour at room temperature and probed with biotinylated 6E10 (BioLegend; 1:2500) or APPCter (#A8717, Sigma; 1:5000) in blocking solution. Primary antibodies were detected using anti–immunoglobulin G conjugated with either biotin or infrared dyes (LI-COR Biosciences). When biotin-conjugated antibodies were used, DyLight 800–conjugated NeutrAvidin (#22853, Thermo Scientific) was added to amplify the signal. Blots were imaged with an Odyssey detection system (LI-COR Biosciences).
Immunoprecipitation
Protein extract (200 μg) was diluted to 1 ml with dilution buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl] and incubated with 25 μl of MagG beads (Roche) cross-linked with 5 μg of 4G8 overnight at 4°C. The following day, immune complexes were briefly washed with immunoprecipitation buffers A and B for 5 min (48). The beads were then washed twice in 1 ml of dilution buffer, and proteins were eluted by boiling in 20 μl of SDS-PAGE loading buffer.
FRET imaging and analysis
Cells were imaged as described previously (49). COS7 cells were cotransfected with the indicated mCherry-tagged PKC and either CKAR, plasma membrane–targeted CKAR, or PSD95-CKAR, as specified. Cells were rinsed once with and imaged in Hanks’ balanced salt solution containing 1 mM CaCl2. Images were acquired on a Zeiss Axiovert microscope (Carl Zeiss Microimaging Inc.) using a MicroMax digital camera (Roper-Princeton Instruments) controlled by MetaFluor software (Universal Imaging Corp.). Using a 10% neutral density filter, cyan fluorescent protein, yellow fluorescent protein (YFP), fluorescence resonance energy transfer (FRET), and mCherry images were obtained every 15 s. YFP emission was monitored as a control for photobleaching, and mCherry was measured to ensure that overexpressed PKC levels were equal in all experiments. Baseline images were acquired for ≥2 min before ligand addition, and data were normalized to the baseline FRET ratios. The normalized average FRET ratio is the average of these normalized values ± SE. Area under the curve from 3 to 6 min was quantified and plotted in the bar graph in Fig. 5, and statistical significance was determined as indicated above.
Three-dimensional PKC structure modeling: The PKCα structure was visualized using the PyMOL Molecular Graphics System (version 1.7.4.1, Schrödinger LLC.).
Genetics, family cohort
The NIMH Alzheimer’s Disease Genetics Initiative Study (50), originally ascertained for the study of genetic risk factors in AD with family-based methods, was used in the whole-genome shotgun (WGS) analyses in this study. The basis for ascertainment in the NIMH collection was the existence of at least two affected individuals within a family, typically siblings. The complete NIMH study cohort contains a total of 1536 subjects from 457 families. For the purpose of this analysis, only subjects of self-reported European ancestry were included, consisting of 1376 participants (941 definitely affected and 404 definitely unaffected, and the remainder could not be determined as definitely unaffected or definitely affected) from 410 families.
To test the likelihood of finding the observed linkage in the mutant carrier families by chance, we conducted the following bootstrap analysis. We generated a “parent” set containing 941 ones (indicating affected) and 404 zeros (indicating unaffected), which is the nature of the definitely assessed population in this cohort. We wished to test the likelihood that choosing a set of 16 individuals (based on being in a family with a mutant protein) would show the observed linkage, that is, the likelihood that in choosing 10 individuals (variant carriers) all would have AD, and that in choosing 6 individuals (variant noncarriers) at least 2 would not have AD. We conducted the following sampling (allowing resampling) procedure: we chose 10 and 6 elements from the parent set to generate two subsets, y(1) and y(2). We then tested if all values in y(1) were 1 (test 1) and at least two values in y(2) were 0 (test2). If both tests were true, the result of the procedure was 1 (meaning chance could account for observed linkage); if any of the tests was false, the result of the procedure was 0. This procedure was conducted 100,000 times (run four times). The number of times the result of the procedure was 1 in the four runs was 2750, 2809, 2920, and 2861. Thus (dividing these values by 100,000), the likelihood of finding the observed linkage distribution by chance in this population is <0.03.
Standard protocol approvals, registrations, and patient consents
Diagnosis of AD dementia was established according to NINCDA-ADRDA criteria. Informed consent was provided by all participants, and research approval was established by the relevant institutional review boards in the study cohort.
WGS data generation
Three micrograms of total genomic DNA (150 ng/μl) obtained from Rutgers repository was sequenced at Illumina Inc. using their latest HiSeq 2500 paired-end sequencing platform. An average of 48× coverage of 98% of the genome was observed in the resulting 120-gigabyte data from each sample. Genomic variants were called in-house using FreeBayes (v0.9.9.2-18) and GATK best practices method (51), resulting in close to 400 terabytes of high-quality sequencing data in a fully annotated GEMINI database (52).
Supplementary Material
Fig. S1. Aβ abundance in brain slices infected with virus producing CT100 is similar in wild-type and PRKCA−/− slices.
Fig. S2. Pedigree charts of the NIMH families found to carry rare PKCα variants.
Acknowledgments
We thank L. Gallegos for making the PSD95-CKAR construct, T. Baffi for helping with Fig. 4A, A. Fields for providing PKCα mice, B. Monk for helping in bootstrap analysis, and S. Taylor and A. Kornov for helpful discussions.
Funding: This work was supported by NIH GM43154 (A.C.N.), NIH MH060009 (R.E.T.), NIH AG032132 (R.M.), GM007752 (C.E.A.), DGE1144086 (C.E.A.), and Cure Alzheimer’s Fund (R.E.T., A.C.N., and R.M.).
Footnotes
Author contributions: S.I.A. conducted electrophysiological experiments; M.A.S. and S.E.L. analyzed expression levels of Aβ; J.A.C. and C.E.A. conducted PKC activity experiments and modeling studies; B.H. and K.M. conducted sequence study; M.L. provided PKCA−/− mice; A.C.N., R.E.T., and R.M. designed the study; and all contributed to writing of the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data and materials presented in the study will be made available.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 3.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 6.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 7.Wilcox KC, Lacor PN, Pitt J, Klein WL. Aβ oligomer-induced synapse degeneration in Alzheimer’s disease. Cell Mol Neurobiol. 2011;31:939–948. doi: 10.1007/s10571-011-9691-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holtzman DM, Mandelkow E, Selkoe DJ. Alzheimer disease in 2020. Cold Spring Harb Perspect Med. 2012;2:a011585. doi: 10.1101/cshperspect.a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J Neurosci. 2010;30:14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen J. Function and dysfunction of presenilin. Neurodegener Dis. 2014;13:61–63. doi: 10.1159/000354971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newton AC. Protein kinase C: Poised to signal. Am J Physiol Endocrinol Metab. 2010;298:E395–E402. doi: 10.1152/ajpendo.00477.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alfonso S, Kessels HW, Banos CC, Chan TR, Lin ET, Kumaravel G, Scannevin RH, Rhodes KJ, Huganir R, Guckian KM, Dunah AW, Malinow R. Synapto-depressive effects of amyloid beta require PICK1. Eur J Neurosci. 2014;39:1225–1233. doi: 10.1111/ejn.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leitges M, Kovac J, Plomann M, Linden DJ. A unique PDZ ligand in PKCα confers induction of cerebellar long-term synaptic depression. Neuron. 2004;44:585–594. doi: 10.1016/j.neuron.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 16.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei W, Nguyen LN, Kessels HW, Hagiwara H, Sisodia S, Malinow R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat Neurosci. 2010;13:190–196. doi: 10.1038/nn.2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD. Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol Cell. 2010;37:541–550. doi: 10.1016/j.molcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill AK, Gallegos LL, Justilien V, Garcia EL, Leitges M, Fields AP, Hall RA, Newton AC. Protein kinase Ca promotes cell migration through a PDZ-dependent interaction with its novel substrate discs large homolog 1 (DLG1) J Biol Chem. 2011;286:43559–43568. doi: 10.1074/jbc.M111.294603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-a. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]
- 22.Violin JD, Zhang J, Tsien RY, Newton AC. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol. 2003;161:899–909. doi: 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV. Knockout of PKCα enhances insulin signaling through PI3K. Mol Endocrinol. 2002;16:847–858. doi: 10.1210/mend.16.4.0809. [DOI] [PubMed] [Google Scholar]
- 24.Kessels HW, Nabavi S, Malinow R. Metabotropic NMDA receptor function is required for b-amyloid-induced synaptic depression. Proc Natl Acad Sci USA. 2013;110:4033–4038. doi: 10.1073/pnas.1219605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antal CE, Callender JA, Kornev AP, Taylor SS, Newton AC. Intramolecular C2 domain-mediated autoinhibition of protein kinase C βII. Cell Rep. 2015;12:1252–1260. doi: 10.1016/j.celrep.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen C, Ha BH, Thévenin AF, Lou HJ, Zhang R, Yip KY, Peterson JR, Gerstein M, Kim PM, Filippakopoulos P, Knapp S, Boggon TJ, Turk BE. Identification of a major determinant for serine-threonine kinase phosphoacceptor specificity. Mol Cell. 2014;53:140–147. doi: 10.1016/j.molcel.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Creixell P, Schoof EM, Simpson CD, Longden J, Miller CJ, Lou HJ, Perryman L, Cox TR, Zivanovic N, Palmeri A, Wesolowska-Andersen A, Helmer-Citterich M, Ferkinghoff-Borg J, Itamochi H, Bodenmiller B, Erler JT, Turk BE, Linding R. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell. 2015;163:1–16. doi: 10.1016/j.cell.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keranen LM, Dutil EM, Newton AC. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol. 1995;5:1394–1403. doi: 10.1016/s0960-9822(95)00277-6. [DOI] [PubMed] [Google Scholar]
- 29.Dutil EM, Keranen LM, DePaoli-Roach AA, Newton AC. In vivo regulation of protein kinase C by trans-phosphorylation followed by autophosphorylation. J Biol Chem. 1994;269:29359–29362. [PubMed] [Google Scholar]
- 30.Antal CE, Violin JD, Kunkel MT, Skovsø S, Newton AC. Intramolecular conformational changes optimize protein kinase C signaling. Chem Biol. 2014;21:459–469. doi: 10.1016/j.chembiol.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JSK, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert J-C, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cruchaga C, Karch CM, Jin SC, Benitez BA, Cai Y, Guerreiro R, Harari O, Norton J, Budde J, Bertelsen S, Jeng AT, Cooper B, Skorupa T, Carrell D, Levitch D, Hsu S, Choi J, Ryten M, UK Brain Expression Consortium (UKBEC) Sassi C, Bras J, Gibbs JR, Hernandez DG, Lupton MK, Powell J, Forabosco P, Ridge PG, Corcoran CD, Tschanz JT, Norton MC, Munger RG, Schmutz C, Leary M, Demirci FY, Bamne MN, Wang X, Lopez OL, Ganguli M, Medway C, Turton J, Lord J, Braae A, Barber I, Brown K, Alzheimer’s Research UK (ARUK) Consortium. Pastor P, Lorenzo-Betancor O, Brkanac Z, Scott E, Topol E, Morgan K, Rogaeva E, Singleton AB, Hardy J, Kamboh MI, St George-Hyslop P, Cairns N, Morris JC, Kauwe JSK, Goate AM. Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mucke L, Selkoe DJ. Neurotoxicity of amyloid b-protein: Synaptic and network dysfunction. Cold Spring Harb Perspect Med. 2012;2:a006338. doi: 10.1101/cshperspect.a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 35.Da Cruz e Silva OAB, Rebelo S, Vieira SI, Gandy S, Da Cruz e Silva EF, Greengard P. Enhanced generation of Alzheimer’s amyloid-b following chronic exposure to phorbol ester correlates with differential effects on alpha and epsilon isozymes of protein kinase. C J Neurochem. 2009;108:319–330. doi: 10.1111/j.1471-4159.2008.05770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lane RF, Gatson JW, Small SA, Ehrlich ME, Gandy S. Protein kinase C and rho activated coiled coil protein kinase 2 (ROCK2) modulate Alzheimer’s APP metabolism and phosphorylation of the Vps10-domain protein, SorL1. Mol Neurodegener. 2010;5:62. doi: 10.1186/1750-1326-5-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kinouchi T, Sorimachi H, Maruyama K, Mizuno K, Ohno S, Ishiura S, Suzuki K. Conventional protein kinase C (PKC)-a and novel PKCε, but not -δ, increase the secretion of an N-terminal fragment of Alzheimer’s disease amyloid precursor protein from PKC cDNA transfected 3Y1 fibroblasts. FEBS Lett. 1995;364:203–206. doi: 10.1016/0014-5793(95)00392-m. [DOI] [PubMed] [Google Scholar]
- 38.Kim T, Hinton DJ, Choi DS. Protein kinase C-regulated aβ production and clearance. Int J Alzheimers Dis. 2011;2011:857368. doi: 10.4061/2011/857368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tagawa K, Homma H, Saito A, Fujita K, Chen X, Imoto S, Oka T, Ito H, Motoki K, Yoshida C, Hatsuta H, Murayama S, Iwatsubo T, Miyano S, Okazawa H. Comprehensive phosphoproteome analysis unravels the core signaling network that initiates the earliest synapse pathology in preclinical Alzheimer’s disease brain. Hum Mol Genet. 2015;24:540–558. doi: 10.1093/hmg/ddu475. [DOI] [PubMed] [Google Scholar]
- 40.Verbeek DS, Goedhart J, Bruinsma L, Sinke RJ, Reits EA. PKCγ mutations in spinocerebellar ataxia type 14 affect C1 domain accessibility and kinase activity leading to aberrant MAPK signaling. J Cell Sci. 2008;121:2339–2349. doi: 10.1242/jcs.027698. [DOI] [PubMed] [Google Scholar]
- 41.Shi H, Tang B, Liu Y-W, Wang X-F, Chen G-J. Alzheimer disease and cancer risk: A meta-analysis. J Cancer Res Clin Oncol. 2015;141:485–494. doi: 10.1007/s00432-014-1773-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roe CM, Behrens MI, Xiong C, Miller JP, Morris JC. Alzheimer disease and cancer. Neurology. 2005;64:895–898. doi: 10.1212/01.WNL.0000152889.94785.51. [DOI] [PubMed] [Google Scholar]
- 43.Driver JA, Beiser A, Au R, Kreger BE, Splansky GL, Kurth T, Kiel DP, Lu KP, Seshadri S, Wolf PA. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ. 2012;344:e1442. doi: 10.1136/bmj.e1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 46.Efron B. Bootstrap methods: Another look at the jackknife. Ann Stat. 1979;7:1–26. [Google Scholar]
- 47.Dutil EM, Toker A, Newton AC. Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1) Curr Biol. 1998;8:1366–1375. doi: 10.1016/s0960-9822(98)00017-7. [DOI] [PubMed] [Google Scholar]
- 48.Sherman MA, Lesné SE. Detecting aβ*56 oligomers in brain tissues. Methods Mol Biol. 2011;670:45–56. doi: 10.1007/978-1-60761-744-0_4. [DOI] [PubMed] [Google Scholar]
- 49.Gallegos LL, Kunkel MT, Newton AC. Targeting protein kinase C activity reporter to discrete intracellular regions reveals spatiotemporal differences in agonist-dependent signaling. J Biol Chem. 2006;281:30947–30956. doi: 10.1074/jbc.M603741200. [DOI] [PubMed] [Google Scholar]
- 50.Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RCP, Mahoney A, Beaty T, Fallin MD, Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ, Pugh EW, Tanzi RE, NIMH Genetics Initiative Alzheimer’s Disease Study Group Results of a high-resolution genome screen of 437 Alzheimer’s disease families. Hum Mol Genet. 2003;12:23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- 51.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paila U, Chapman BA, Kirchner R, Quinlan AR. GEMINI: Integrative exploration of genetic variation and genome annotations. PLOS Comput Biol. 2013;9:e1003153. doi: 10.1371/journal.pcbi.1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wagner J, von Matt P, Sedrani R, Albert R, Cooke N, Ehrhardt C, Geiser M, Rummel G, Stark W, Strauss A, Cowan-Jacob SW, Beerli C, Weckbecker G, Evenou JP, Zenke G, Cottens S. Discovery of 3-(1H-indol-3-yl)-4-[2-(4-methylpiperazin-1-yl)quinazolin-4-yl]pyrrole-2,5-dione (AEB071), a potent and selective inhibitor of protein kinase C isotypes. J Med Chem. 2009;52:6193–6196. doi: 10.1021/jm901108b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Aβ abundance in brain slices infected with virus producing CT100 is similar in wild-type and PRKCA−/− slices.
Fig. S2. Pedigree charts of the NIMH families found to carry rare PKCα variants.