

Abstract
Two mutational mechanisms are known to underlie Ullrich congenital muscular dystrophy (UCMD): heterozygous dominant negatively acting mutations and recessively acting loss of function mutations. We describe large genomic deletions on chromosome 21q22.3 as a novel type of mutation underlying recessively inherited UCMD in two families. Clinically unaffected parents carrying large genomic deletions of COL6A1 and COL6A2 also provide conclusive evidence that haploinsufficiency for COL6A1 and COL6A2 is not a disease mechanism for Bethlem myopathy. Our findings have important implications for the genetic evaluation of patients with collagen VI related myopathies as well as for potential therapeutic interventions for this patient population.
Introduction
Ullrich congenital muscular dystrophy (UCMD [MIM 254090]), Bethlem myopathy (BM [MIM 158810]) and phenotypes intermediate to UCMD and BM form a group of congenital muscular dystrophies known as the collagen VI related myopathies.1 Underlying these conditions is a decrease, absence or dysfunction of the extracellular matrix protein collagen VI. The collagen VI heterotrimeric monomer is composed of three alpha chains: α1(VI), α2(VI) and α3(VI)2,3 each containing a short triple helical domain flanked by globular domains. Assembly of collagen VI proceeds intracellularly with monomers aligning in an antiparallel fashion to form dimers, which then align laterally to form tetramers. The tetramers are secreted and align extracellularly in an end-to-end fashion, forming beaded microfilaments as the final product of collagen VI assembly.4,5,6
Mutations in any of the three genes coding for the three collagen VI alpha chains, COL6A1, COL6A2 or COL6A3, can affect the complex assembly and secretion of collagen VI, resulting in the phenotype of a collagen VI related myopathy. COL6A1 and COL6A2 are located on chromosome 21q22.3,7 and COL6A3 is located on chromosome 2q37.8 Ullrich congenital muscular dystrophy results from either recessive or dominantly acting mutations9,10 and is characterized by a combination of early onset muscle weakness, congenital contractures of the proximal joints and hyperlaxity of the distal joints.11 Bethlem myopathy typically follows autosomal dominant inheritance; however, autosomal recessive inheritance has recently been described as well.12,13 BM is characterized by slowly progressive muscle weakness and joint contractures.14
Here we describe a novel type of mutation underlying recessively inherited Ullrich congenital muscular dystrophy by delineating large genomic deletions on chromosome 21q22.3 resulting in loss of COL6A2 or both COL6A1 and COL6A2 and occurring in combination with a mutation in COL6A2 or a deletion of COL6A2 on the other allele to cause disease. We also conclusively demonstrate that haploinsufficiency for COL6A1 and COL6A2 is associated with decreased collagen VI deposition but is not associated with clinical neuromuscular disease.
Patients and Methods
Clinical details were collected according to a protocol approved by the institutional review board and are summarized in Table 1.
Table 1.
Clinical details
| Patients
|
|||
|---|---|---|---|
| P1 (Ullrich) | P2 (Ullrich) | P3 | |
|
| |||
| Sex | Male | Male | Male |
| Congenital hypotonia | Y | Y | N |
| Congenital hip dislocation | N | Y | N |
| Congenital abnormal position of hands and/or feet | Y | Y | N |
| Congenital torticollis | N | Y | N |
| Age at evaluation (years) | 3.25 | 3.25 | 1.17 |
| Weakness | Y | Y | N |
| proximal | Y (moderate) | Y (severe) | N |
| distal | Y (moderate) | Y (severe) | N |
| Contractures | Y | Y | N |
| elbows | N | N | N |
| hips | ++ | ++ | N |
| knees | + | + | N |
| ankles | + | + | N |
| Distal joint hyperlaxity | Y | Y | (Y) (mild) |
| Scoliosis | N | N | N |
| Abnormal skin findings | N | N | N |
| hypertrophic scars | N | N | N |
| keratosis pilaris | N | N | N |
| Achieved independent ambulation | Y | N | N |
| age achieved | 3 | – | – |
| age lost | – | – | – |
| Pulmonary compromise | N | Y (severe) | N |
| CK | 1.5 × normal | 1.4 × normal | normal |
Y = Yes
N = No
+: moderate
++: severe
CK = creatine kinase
Genomic and Biochemical Analyses
Exonic sequencing of COL6A1, COL6A2 and COL6A3 was performed at Prevention Genetics by extracting genomic DNA from patient blood cells followed by PCR amplification of individual exons and sequencing of PCR products on an ABI 3130x1 capillary sequencer in the forward and reverse directions.
SNP array analysis was performed using an Illumina Quad 610 at the Center for Applied Genomics at the Children’s Hospital of Philadelphia as per previously described protocols.15,16
Dermal fibroblasts from skin biopsies obtained from patient 1, patient 2, patient 2’s mother and patient 2’s brother were cultured and mutational analysis by RT-PCR and DNA sequencing completed as previously described.10 Immunostaining of fibroblasts as well as Western blot analysis on fibroblasts were performed using polyclonal and monoclonal antibodies as previously described.10, 17
Results
Patient 1 has a phenotype of moderately severe UCMD, and patient 2 has a phenotype of severe UCMD (Table 1). The parents of patients 1 and 2 have no neuromuscular complaints and normal neuromuscular examinations. The brother of patient 2 has global developmental delays and epilepsy of unclear etiology but no symptoms suggestive of a congenital muscular dystrophy. Patient 3 does not have a phenotype consistent with UCMD or BM but was evaluated for a clinical picture of global developmental delays and axial hypotonia. Patient 3’s father had a completely normal neuromuscular examination. Both patient 3 and his father had muscle ultrasounds performed which revealed normal-appearing muscles.
Mutational analysis
Genomic DNA sequencing revealed that patient 1 was heterozygous for an intronic nucleotide change (G>A) at position c.1970-9 at the intron 25–exon 26 junction of COL6A2 (Fig 1). On COL6A2 sequencing using cDNA extracted from patient 1’s fibroblasts we detected a 7 base pair insertion resulting from the use of a novel splice acceptor site in intron 25 created by the mutation (Fig 2). This insertion causes a frameshift, resulting in a premature stop codon (G656AfsX17). In addition, SNP analysis revealed a 69 kb genomic deletion at 21q22.3 encompassing at least the first 18 exons of the COL6A2 gene. Patient 1’s asymptomatic mother was found to be heterozygous for the nucleotide change (G>A) at c.1970-9 of COL6A2; his asymptomatic father was found to be heterozygous for the 69 kb deletion.
Figure 1.
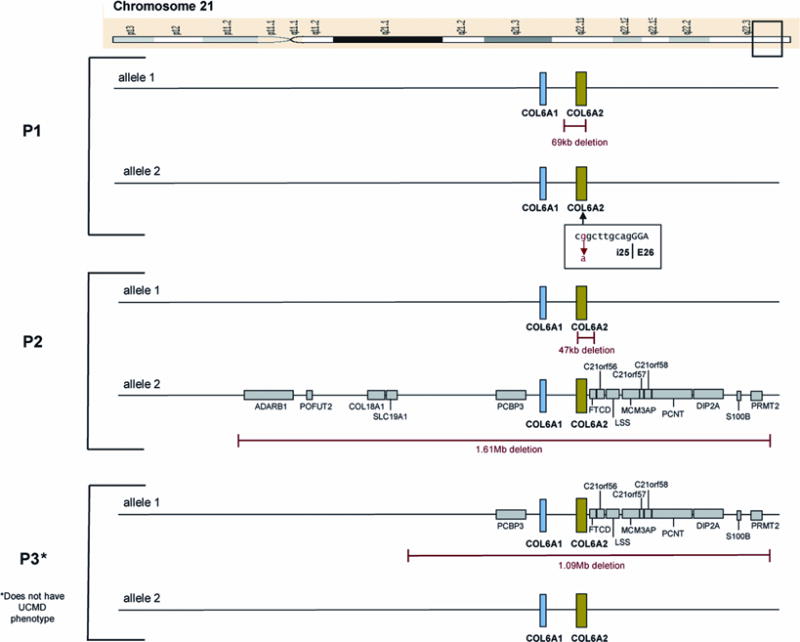
Figure 2.
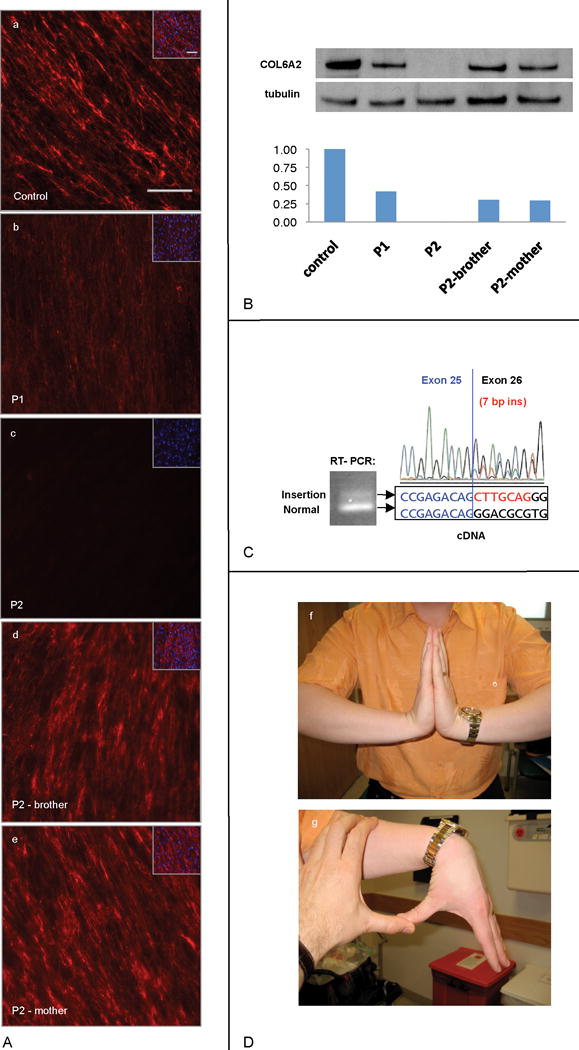
SNP based genome array analysis performed on patient 2 revealed evidence of two deletions at 21q22.3: a deletion of 1.61 Mb encompassing the entire COL6A1 and COL6A2 genes plus surrounding genes on one allele and a smaller deletion of 47 kb encompassing the entire COL6A2 gene on the other allele (Fig 1). Patient 2’s asymptomatic mother was found to be heterozygous for the 1.61 Mb deletion; his asymptomatic father was found to be heterozygous for the 47 kb deletion. Genomic DNA sequencing of patient 2’s mother and father did not reveal any mutation in either COL6A1 or COL6A2 in the non-deleted alleles. Patient 2’s brother was found to be heterozygous for the 1.61 Mb deletion inclusive of COL6A1 and COL6A2. Dermal fibroblasts cultured from skin biopsies of patient 2, his mother and his brother were stained for collagen VI and revealed a complete absence of collagen VI immunoreactivity in patient 2’s fibroblasts and evidence of a slightly reduced collagen VI matrix intensity in the fibroblasts of patient 2’s mother and brother (Fig 2). (Patient 2’s mother is clinically asymptomatic; his brother has a history of global developmental delays and epilepsy of unclear etiology)
SNP based genomic array analysis performed on patient 3 for developmental delays revealed a heterozygous 1.09 Mb deletion encompassing the entire COL6A1 and the COL6A2 genes as well as adjacent genes (Fig 1). The same deletion was detected in patient 3’s asymptomatic father (Fig 2).
Discussion
Two principle mutational mechanisms are known to underlie classic Ullrich congenital muscular dystrophy: heterozygous dominant negatively acting mutations and recessively acting loss of function mutations. Recessive null mutations have included nonsense mutations as well as exon skipping mutations precluding assembly. Small heterozygous in-frame intragenic deletions in the COL6A1 gene exerting a dominant negative effect have been described.10,18 The large genomic deletions on chromosome 21 described here involving COL6A2 or both COL6A1 and COL6A2 have not yet been described and establish a novel type of mutation in the collagen VI related myopathies. This finding also adds to the complexity of genetic evaluations in the collagen VI related myopathies, as this type of mutation will not be detected by single exon amplification and sequencing (unless done quantitatively). In addition, a hemizygous change detected on the non-deleted allele will seemingly appear homozygous, potentially obscuring the true genetic causation of the patient’s disease.
Large genomic deletions in recessive disorders are only pathogenic if they unmask a pathogenic mutation on the second non-deleted allele. In severe UCMD patients the mutation on the non-deleted allele will most likely be another loss of function mutation, although the precise nature of the second mutation will influence the severity of disease. It is notable that patient 1 has acquired ambulation, whereas patient 2, with a complete deletion of both COL6A2 and COL6A1 on the other allele, has not. Patient 1’s second mutation is an intronic mutation leading to an erroneous splice acceptor, adding a frameshifting 7 bp to the transcript. Based on RT-PCR evidence in patient 1’s fibroblasts (Fig 2), there also appears to be some normally spliced transcript from this allele which likely is responsible for ameliorating patient 1’s phenotype to a degree. We and others recently reported patients with classical Bethlem myopathy who were compound heterozygous for a functional null mutation and a missense mutation in the COL6A2 gene.12,13 Thus, large genomic deletions of the COL6A1 and COL6A2 loci in compound heterozygosity with a milder missense mutation would be predicted to result in a phenotype consistent with Bethlem myopathy and, therefore, would have to be considered in that patient population also.
The increased use of chromosomal micro-array platforms including those based on single nucleotide polymorphism (SNP) leads to an increasing catalogue of genomic deletion and duplication variants of unknown significance (http://projects.tcag.ca/variation/). It remains to be seen whether deletions within and around the COL6A1 and COL6A2 loci will be identified more often in asymptomatic carriers as a copy number variation (CNV) which would be benign but confer carrier status for a collagen VI related myopathy. It is to be anticipated that additional deletions encompassing neuromuscular disease loci will be found with more widespread use of this molecular technology, and vice versa, that chromosomal micro-array platforms have a place in the diagnostic repertoire for neuromuscular disorders.
Our observation that the carrier status in the parents for the large deletions found in patients 1, 2 and 3 are clinically asymptomatic is the most conclusive evidence yet that haploinsufficiency for collagen type VI is not a disease mechanism for Bethlem myopathy, even though it is associated with a reduction in the deposited collagen VI matrix evidenced by patient 2’s mother’s cells in culture (Fig 2). These findings suggest that the COL6A1 mutation reported in the literature as causing haploinsufficiency as a mechanism for Bethlem myopathy must in fact have more complex consequences.19 This observation is also of great translational importance, as therapeutic strategies directed at the elimination of a dominant negatively acting mutation are conceivable and would create a functional state of haploinsufficiency which, as demonstrated here, would not be associated with clinical manifestations of neuromuscular disease.
Acknowledgments
We would like to thank the patients and their families for their participation. C.G.B. is supported by grants from NIH/NIAMS (R01AR051999) and from MDA USA (MDA3896). A.R.F. is a MDC (UK) clinical research fellow (MC3/1057/2). T.H.S is supported by an NIH/NIGMS grant (GM081519).
References
- 1.Bertini E, Pepe G. Collagen type VI and related disorders: Bethlem myopathy and Ullrich scleroatonic muscular dystrophy. Eur J Paediatr Neurol. 2002;6:193–198. doi: 10.1053/ejpn.2002.0593. [DOI] [PubMed] [Google Scholar]
- 2.Timpl R, Chu ML. Microfibrillar collagen type VI. In: Mecham RP, editor. Extracellular matrix assembly and structure. Orlando: Academic Press; 1994. pp. 207–242. [Google Scholar]
- 3.Timpl R, Engel J. Type VI collagen. In: Mayne R, Burgeson R, editors. Structure and function of collagen types. Orlando: Academic Press; 1987. pp. 105–143. [Google Scholar]
- 4.Chu ML, Conway D, Pan TC, et al. Amino acid sequence of the triple-helical domain of human collagen type VI. J Biol Chem. 1988;263:18601–18606. [PubMed] [Google Scholar]
- 5.Chu ML, Pan TC, Conway D, et al. Sequence analysis of alpha 1(VI) and alpha 2(VI) chains of human type VI collagen reveals internal triplication of globular domains similar to the A domains of von Willebrand factor and two alpha 2(VI) chain variants that differ in the carboxy terminus. Embo J. 1989;8:1939–1946. doi: 10.1002/j.1460-2075.1989.tb03598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chu ML, Zhang RZ, Pan TC, et al. Mosaic structure of globular domains in the human type VI collagen alpha 3 chain: similarity to von Willebrand factor, fibronectin, actin, salivary proteins and aprotinin type protease inhibitors. Embo J. 1990;9:385–393. doi: 10.1002/j.1460-2075.1990.tb08122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heiskanen M, Saitta B, Palotie A, Chu ML. Head to tail organization of the human COL6A1 and COL6A2 genes by fiber-FISH. Genomics. 1995;29:801–803. doi: 10.1006/geno.1995.9008. [DOI] [PubMed] [Google Scholar]
- 8.Weil D, Mattei MG, Passage E, et al. Cloning and chromosomal localization of human genes encoding the three chains of type VI collagen. Am J Hum Genet. 1988;42:435–445. [PMC free article] [PubMed] [Google Scholar]
- 9.Camacho Vanegas O, Bertini E, Zhang RZ, et al. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc Natl Acad Sci USA. 2001;98:7516–7521. doi: 10.1073/pnas.121027598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan TC, Zhang RZ, Sudano DG, et al. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73:355–369. doi: 10.1086/377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ullrich O. Kongenitale atonisch-sklerotische Muskeldystrophie, ein weiterer Typus der heredodegeneration Erkrankungen des neuromuskulären Systems. Z Ges Neurol Psychiat. 1930;126:171–201. [Google Scholar]
- 12.Foley AR, Hu Y, Zou Y, et al. Autosomal recessive inheritance of classic Bethlem myopathy. Neuromuscul Disord. 2009;19:813–817. doi: 10.1016/j.nmd.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gualandi F, Urciuolo A, Martoni E, et al. Autosomal recessive Bethlem myopathy. Neurology. 2009;73:1883–1891. doi: 10.1212/WNL.0b013e3181c3fd2a. [DOI] [PubMed] [Google Scholar]
- 14.Bethlem J, van Wijnaarden GK. Benign myopathy, wth autosomal dominant inheritance: a report on three pedigrees. Brain. 1976;99:91–100. doi: 10.1093/brain/99.1.91. [DOI] [PubMed] [Google Scholar]
- 15.Gunderson KL, Steemers FJ, Lee G, et al. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 16.Shaikh TH, Gai X, Perin JC, et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pepe G, Bertini E, Giusti B, et al. A novel de novo mutation in the triple helix of the COL6A3 gene in a two-generation Italian family affected by Bethlem myopathy. A diagnostic approach in the mutations’ screening of type VI collagen. Neuromuscul Disord. 1999;9:264–271. doi: 10.1016/s0960-8966(99)00014-0. [DOI] [PubMed] [Google Scholar]
- 18.Pepe G, Lucarini L, Zhang RZ, et al. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann Neurol. 2006;59:190–195. doi: 10.1002/ana.20705. [DOI] [PubMed] [Google Scholar]
- 19.Lamande SR, Bateman JF, Hutchison W, et al. Reduced collagen VI causes Bethlem myopathy: a heterozygous COL6A1 nonsense mutation results in mRNA decay and functional haploinsufficiency. Hum Mol Genet. 1998;7:981–989. doi: 10.1093/hmg/7.6.981. [DOI] [PubMed] [Google Scholar]