

Abstract
High-valent terminal copper-nitrene species are postulated as key intermediates in copper-catalyzed aziridination and amination reactions. The high reactivity of these intermediates has eluded their characterization for decades, thereby making the mechanism ambiguous. Very recently, the Lewis acid adduct of a copper-nitrene intermediate has been trapped at -90°C and shown to be active in various oxidation reactions. Herein, we describe for the first time the synthesis and spectroscopic characterization of a room temperature stable terminal copper(II)-nitrene radical species (3) in the absence of any Lewis acid. The azide derivative of a triazamacrocyclic ligand has been employed as an ancillary ligand in the study, which has previously been utilized in the stabilization of aryl-CuIII intermediates. Compound 3 is able to perform nitrene-transfer to phosphines and H-atom abstraction from weak C-H bonds leading to the formation of oxidized products in modest yields.
Keywords: copper-nitrene, spectroscopic characterization, nitrene transfer, hydrogen atom abstraction, N-N bond formation
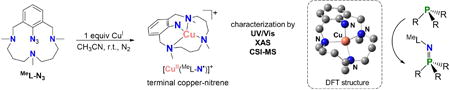
A room temperature stable mononuclear copper-nitrene species has been synthesized and spectroscopically characterized. This compound can perform nitrene-transfer to phosphines and H-abstraction from weak C-H bonds.
Terminal high-valent copper-nitrene intermediates have long been proposed as key intermediates in aziridination and amination reactions.[1-3] Obtaining mechanistic insight into these transformations and developing selective catalytic reagents and processes are only possible if the structure and properties of these key species are fully understood. While metal-nitrene species based on iron, cobalt and nickel have been previously isolated,[4-9] examples of terminal copper-nitrene intermediates have eluded detection for decades. Warren and co-workers indirectly proposed the involvement of terminal copper-nitrene intermediates derived from dicopper-nitrenes by NMR exchange experiments and kinetic measurements of C-H functionalization reactions.[2, 10] More recently, the involvement of such species in copper mediated oxidation reactions has been corroborated by the spectroscopic trapping of Lewis acid adducts of a copper-nitrene intermediate at -90°C.[11, 12] Furthermore, Bertrand and co-workers recently reported isolated examples of copper(II)-bis(nitrene) and dicopper(II)-nitrene species by reaction of a bulky phosphinonitrene ligand with 0.5 and 2 equiv. of copper(I) triflate, respectively. However, no terminal copper-nitrene intermediates could be isolated in these reactions, although their formation was suggested based on 31P-NMR studies.[13] Herein we report the unprecedented spectroscopic and theoretical characterization of terminal copper-nitrene species (1 and 3) in the absence of any Lewis acid by employing azide derivatives of triazamacrocyclic ligands (HL-N3 and MeL-N3). Such ligand architectures have previously been utilized in the successful stabilization of aryl-CuIII intermediates by enforcing a square planar geometry at the copper centre.[14, 15] Moreover, the room temperature stable copper-nitrene species 3 participates in a number of nitrene transfer and C-H activation reactions, thus providing a key precedent for the possible involvement of such species in oxidation catalysis.
During the ongoing studies of some of us regarding the relevance of aryl-CuIII species in model copper-catalyzed Caryl-Csp,[16] Caryl-Csp,[17] and Caryl-heteroatom[14, 15, 18, 19] bond formation reactions, we envisaged that compound HL-N3 (Scheme 1) could be a good candidate as a starting material for the preparation of elusive copper-nitrene complexes (see SI for HL-N3 synthesis). Accordingly, the reaction of HL-N3 with 1.5 equiv [CuI(CH3CN)4] (CF3SO3) in acetonitrile at room temperature under argon was monitored by UV-vis spectroscopy (Figure S3). Formation of a green-colored transient species (1, λmax = 380 and 790 nm) was observed during the first stages of the reaction. However, this compound quickly evolved to a purple species (2, λmax = 550 nm, ε = 1000 M-1cm-1) that proved to be stable for several hours at room temperature. Violet crystals (56% isolated yield) were obtained by slow diethyl ether diffusion into an acetonitrile solution of 2 at -30°C.
Scheme 1.
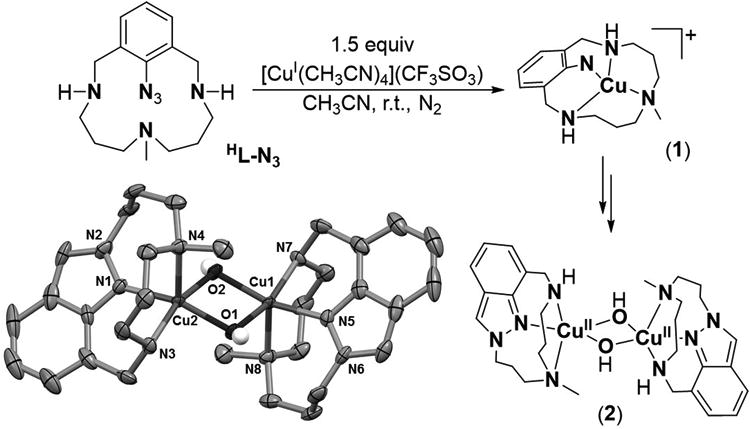
Schematic representation of 1 and 2. Thermal ellipsoid plot (50% probability) of 2. Hydrogen atoms (except for those belonging to the OH groups) and triflate anions have been omitted for clarity.
The X-ray structure of 2 reveals a dimeric core with significant modification of the initial HL-N3 structure (Scheme 1). Loss of a N2 molecule from the azide unit is accompanied by N-N bond formation giving rise to the formation of an indazole ring. Each Cu centre exists in a distorted square pyramidal geometry (τ = 0.1)[20] and binds to the N-CH3, NH, and Naryl groups of the resulting indazole-based ligand (Lin), as well as to two additional hydroxo ligands, thereby forming a bis(μ-hydroxo)dicopper(II) core in 2 (Scheme 1). The hydroxo groups in 2 presumably originate from adventitious water present in the anhydrous solvents used in the glovebox;[21] they also enforce a strong antiferromagnetic coupling between the two copper centers resulting in an S=0 ground state.[22] The integrity of this dimeric structure in solution was confirmed by 1H-NMR studies, which showed a diamagnetic spectrum in which the indazole protons could be clearly assigned at 8.2, 7.7, 7.2 and 7.1 ppm (Figure S4). Although unknown in Cu chemistry, the N-N bond formation reaction associated with the formation of 2 is typically exhibited by high-valent terminal metal −imido or −nitride complexes.[5, 23-25] A similar involvement of a terminal copper-nitrene intermediate can also be playing a key role during the Cu mediated N-N bond formation reaction that leads to the formation of 2.
Although UV-vis studies clearly showed the presence of a transient intermediate (1) along the reaction of HL-N3 with CuI at 25°C to form 2, spectroscopic assignment of the electronic structure 1 proved to be difficult because of its instability at 25°C. However, the stability of 1 could be significantly increased by generating it in acetone at -50°C (λmax [ε, M-1cm-1] =380 [>1300], 650 [>150] and 790 nm [>200]; Figure S7). Notably, CSI-MS of 1 (Figure S8) showed a major peak centered at m/z 323.1284 with a mass and isotope distribution pattern consistent with a terminal copper-nitrene species [Cu(HL-N)]+ where HL-N is the ligand formed after N2 release from HL-N3. The low temperatures required to prevent the decomposition of 1 into 2, however, prevented us from investigating the oxidizing capabilities of 1.
In order to increase the thermal stability of the putative copper-nitrene intermediate, efforts were made to block the facile decomposition of 1 to 2 by introducing methyl groups in the secondary amine units of the HL-N3 ligand (see SI for synthetic procedures). UV-vis monitoring of the stoichiometric reaction between the methylated version of the azide-derived ligand (MeL-N3, Scheme 2) and [CuI(CH3CN)4] (CF3SO3) in acetonitrile at 25°C showed the formation of a stable green species (3; t1/2 ∼ 45 h) with a distinct absorption spectrum (λmax [ε, M-1cm-1] = 360 [>1200], 710 nm [>200] and 980 nm [>150]; Figure 1a) relative to 1.[26] CSI-MS analysis revealed a predominant peak at m/z 351.1597 with a mass value and isotopic pattern fully consistent with [Cu(MeL-N)]+, that partially shifted to m/z 352.1601 when 50% 15N-labeled 3[27] was used (Figure 1b). Moreover, the EPR spectrum of 3 only exhibited a small signal of CuII that accounted for less than 5% of the sample; the majority of the species in solution was not detected by X-band EPR. Importantly, FT-IR analysis of a concentrated solution of 3 did not exhibit any azide vibration at ∼2200 cm-1 further confirming N2 release in line with the MS experiments. Furthermore, a low-intensity band at 426 cm-1 could be observed in the IR spectrum, which can be attributed to a Cu-N vibration based on the 15N/14N downshift of 8 cm-1 upon 15N labeling (Figure 1a, inset). Finally, an effective magnetic moment (μeff) of 2.14 μB, as determined by the Evans' method,[28, 29] confirms an S=1 ground state for 3. Note that the μeff of 3 is significantly lower than that expected for the spin-only value of a typical S=1 system (μeff =2.83 μB); this can be attributed to the presence of unreacted copper(I) or copper(II) impurities in solutions of 3.
Scheme 2.
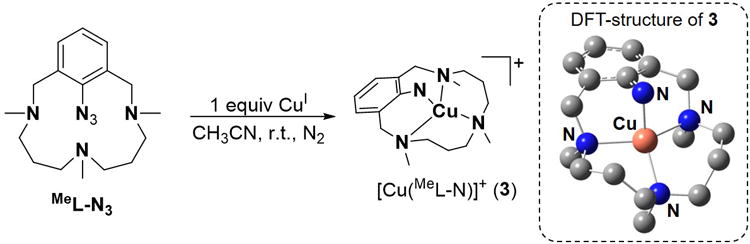
Schematic representation of 3 together with its DFT optimized structure.
Figure 1.
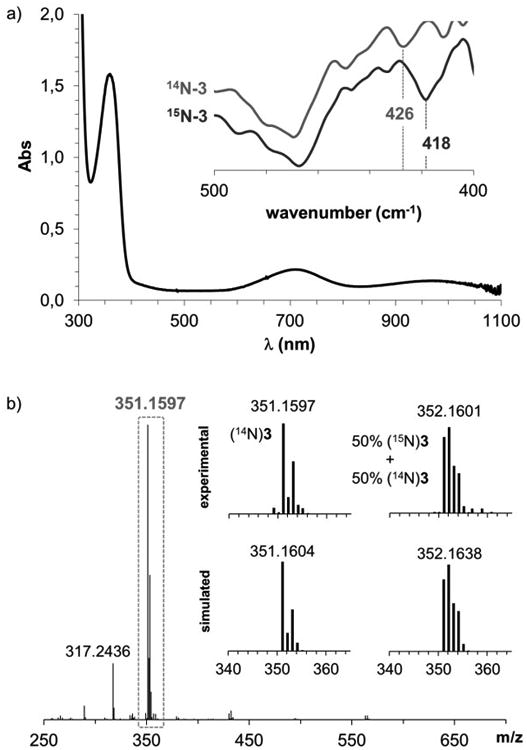
a) UV-vis spectrum of 3 in acetonitrile at 25°C. Inset: FT-IR spectrum of 3 (blue line) and its 50% 15N-labeled analogue (red line). b) ESI-MS spectrum of 3 in acetonitrile at 25°C under N2.
We then turned to X-ray absorption spectroscopy (XAS) to probe the oxidation state of copper in 3 directly. Notably, 3 exhibited X-ray absorption near-edge structure (XANES) features near identical to those of the dinuclear CuII complex 2. This strongly suggested that 3 also contains a CuII center (Figure S9); together with the experimentally determined μeff of 2.14 μB this may indicate an [CuII(MeL-N•-)]+ electronic structure for 3. Extended X-ray absorption fine structure (EXAFS) analysis revealed further structural details. For 3, the first coordination sphere could be satisfactorily fitted by four nitrogen scatterers at a distance of 2.04 Å (Table S3, Figure S10). The outer-shell features could be accounted for by single scattering paths arising from 2C at 2.44 Å and 12C at 3.23 Å. EXAFS analysis of 2 was also performed in order to confirm whether the solid-state dimeric structure is retained in a frozen acetonitrile solution. The best fit (Table S4) of the Cu EXAFS of 2 consists of two subshells of four short N/O scatterer at 1.99 Å and one long N/O scatterer at 2.13 Å, supporting a five-coordinate Cu center in 2, in agreement with the XRD studies (see above). Fits to a single shell of 5-6 nitrogen scatterers produced a significant decrease in fit quality. Cu EXAFS of 2 also shows a shell at 2.93 Å corresponding to the Cu scatterer, which is in reasonable agreement with the Cu-Cu distance of 3.03 Å determined by XRD.
DFT calculations at the B3LYP/TZVP level of theory in acetonitrile at 298K predict a triplet (S = 1; J=+3.2 cm-1) ground state for 3. The excited open and closed-shell singlet electronic states were determined to be 3 and 18 kcal·mol-1 higher in energy, respectively. The copper centre in the calculated structure of 3 (Scheme 2) presents a strongly distorted tetrahedral geometry involving the four coordinated nitrogen atoms of MeL-N. Notably, the calculated metrical parameters are in reasonable agreement with experiment (Table S3). Furthermore, the theoretically predicted Cu-Naryl vibration at 427 cm-1 with a -6 cm-1 shift upon 15N-labeling is fully congruent with the experimental IR spectrum (see above). Interestingly, a spin density of 1.20 is obtained for the Naryl atom indicating that the excess unpaired electron density on nitrogen is larger than what is expected for a classical metal-nitrene radical system (Figures S18-S19). Such a non-classical electronic structure has been reported previously for a cobalt-oxo species[30] and may be understood by considering the contribution of the CuI-nitrene biradical resonance form, where two electrons from the Naryl atom moiety are transferred to the Cu center. The calculated spin density of 0.4 on the copper atom, which is significantly lower than the expected value of 1 for a CuII center further confirms the involvement of both [CuII(MeL-N•-)]+ and [CuI(MeL-N••)]+ resonance forms in the electronic structure of s.
The reactivity of 3 in various oxidation reactions was also investigated. For example, transfer of the MeL-N unit of the copper-nitrene species 3 to phosphines was evaluated. The green colour of a solution of 3 in acetonitrile disappeared upon addition of triphenylphosphine (PPh3). ESI-MS analysis of the final reaction mixture showed the presence of a mass peak at m/z 551.35 consistent with nitrogen insertion to the phosphorous atom to form MeL-N=PPh3, which shifted by 1 mass unit when 50% 15N-labeled 3 (Figure S13) was used in the reaction. 31P-NMR analysis showed the presence of a peak at 25.6 ppm, which falls in the typical region for N=P bonds,[31] which accounted for a 41% yield with respect to 3 (NMR quantification using OPPh3 as an internal standard). Interestingly, 31P-NMR of a sample prepared by reacting 50% 15N-labeled 3 with PPh3 afforded the peak at 25.6 ppm together with a doublet at 25.3 ppm, corresponding to MeL-15N=PPh3, where coupling of 31P with 15N (S = 1/2) occurs (Figure S14). The measured coupling constant JN-P = 38.2Hz is in accordance with those typically measured for N-P bonds, further supporting the formation of the nitrene transfer product.[32] UV-vis monitoring of this reaction using excess substrate showed that the decay of the bands at 710 and 980 nm associated to 3 followed a pseudo-first order behaviour, so that kinetic traces could be fitted with single exponentials (Figure S12). The observed reaction rate (kobs) was in turn found to be linearly dependent on substrate concentration, affording a second order constant (k2) of 7.5 M-1s-1 for the reaction towards PPh3. Interestingly, k2 values were found to be highly dependent on the substituents at the phosphorous atom. Thus, reaction of 3 with the sterically bulkier tri(o-tolyl)phosphine was two orders of magnitude slower (0.052 M-1s-1) relative to PPh3, while reaction with tri(n-butyl)phosphine was too fast and reaction rate could not be determined. Finally, reaction rates were found to be dependent on the substituent at the para position of the phenyl groups attached to the phosphine. The logarithm of the second-order rate constants of a series of para-X-triphenylphospine (X = Me, H and Cl) showed a negative correlation with the Hammett parameter (σp) with a reaction constant (ρ) of -1.9 indicative of an electrophilic character of 3 (Figure S15).
Reaction of 3 with hydrocarbons bearing C-H bonds with low dissociation energies (BDEs) was also evaluated and xanthene was used as a model substrate. Strikingly, the k2 value measured for the reaction with xanthene (k2 = 0.009 M-1s-1, BDE = 75.5 kcal·mol-1) was lower than that determined in the reaction towards 1,4-cyclohexadiene (k2 = 0.020 M-1s-1, BDE = 78 kcal·mol-1), which does not correlate with the C-H bond strength. Again, steric hindrance can reason this result, so that the smaller 1,4-cyclohexadiene reacts faster despite its increased C-H bond strength (Table S5). Furthermore, a kinetic isotope effect of 5.2 was obtained when d2-xanthene was used as a substrate at 25°C (Figure S16).
In summary, in this work we report for the first time the synthesis and spectroscopic characterization of a terminal copper-nitrene species (3), without the need of using redox innocent cations such as Sc3+. The apparently simple methylation of the secondary amines in HL-N3 has been crucial to slow down the N-N bond formation event, thus retarding the main decomposition pathway for the copper-nitrene species in this system. Interestingly, this species can be trapped at room temperature and it can perform nitrogen-transfer reactions to organic substrates such as phosphines or weak C-H bonds.
Experimental Section
Full experimental and theoretical details, synthetic procedure and X-ray crystallographic data of 2 are available in Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by the European Commission (2011-CIG-303522 to A.C.), the MINECO of Spain (“Ramón y Cajal” contract and CTQ2013-43012-P to A.C. and X.R.), the Clara Immerwahr award of UniCat (to A.C.), the European Research Council (Starting Grant Project ERC-2011-StG-277801 to X.R.), and Generalitat de Catalunya (2014 SGR 862). K.R. thanks the German funding agency (Deutsche Forschungsgemeinschaft) for Heisenberg Professorship. X.R. also thanks ICREA-Acadèmia award. XAS measurements at SSRL BL 2-2 were made possible by the US Department of Energy Office of Science (contracts DE-AC02-76SF00515 and DE-SC0012704 to SSRL and NSLS-II, respectively) and the US National Institutes of Health (P30-EB-009998 to CWRU Center for Synchrotron Biosciences).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Dr. Xavi Ribas, Email: xavi.ribas@udg.edu.
Prof. Dr. Kallol Ray, Email: kallol.ray@chemie.hu-berlin.de.
Dr. Anna Company, Email: anna.company@udg.edu.
References
- 1.Gephart RT, Warren TH. Organometallics. 2012;31:7728–7752. [Google Scholar]
- 2.Badiei YM, Krishnaswamy A, Melzer MM, Warren TH. J Am Chem Soc. 2006;128:15056–15057. doi: 10.1021/ja065299l. [DOI] [PubMed] [Google Scholar]
- 3.Badiei YM, Dinescu A, Dai X, Palomino RM, Heinemann FW, Cundari TR, Warren TH. Angew Chem Int Ed. 2008;47:9961–9964. doi: 10.1002/anie.200804304. [DOI] [PubMed] [Google Scholar]
- 4.Ray K, Heims F, Pfaff FF. Eur J Inorg Chem. 2013;2013:3784–3807. [Google Scholar]
- 5.Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:720. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
- 6.Hu X, Meyer K. J Am Chem Soc. 2004;126:16322–16323. doi: 10.1021/ja044271b. [DOI] [PubMed] [Google Scholar]
- 7.Iovan DA, Betley TA. J Am Chem Soc. 2016;138:1983–1993. doi: 10.1021/jacs.5b12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskowski CA, Miller AJM, Hillhouse GL, Cundari TR. J Am Chem Soc. 2011;133:771–773. doi: 10.1021/ja1101213. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Liu Y, Deng L. J Am Chem Soc. 2014;136:15525–15528. doi: 10.1021/ja509731z. [DOI] [PubMed] [Google Scholar]
- 10.Aguila MJB, Badiei YM, Warren TH. J Am Chem Soc. 2013;135:9399–9406. doi: 10.1021/ja400879m. [DOI] [PubMed] [Google Scholar]
- 11.Kundu S, Miceli E, Farquhar E, Pfaff FF, Kuhlmann U, Hildebrandt P, Braun B, Greco C, Ray K. J Am Chem Soc. 2012;134:14710–14713. doi: 10.1021/ja306674h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monte-Pérez I, Kundu S, Ray K, Anorg Z. Allg Chem. 2015;641:78–82. [Google Scholar]
- 13.Dielmann F, Andrada DM, Frenking G, Bertrand G. J Am Chem Soc. 2014;136:3800–3802. doi: 10.1021/ja5007355. [DOI] [PubMed] [Google Scholar]
- 14.Casitas A, Canta M, Solà M, Costas M, Ribas X. J Am Chem Soc. 2011;133:19386–19392. doi: 10.1021/ja2058567. [DOI] [PubMed] [Google Scholar]
- 15.Casitas A, King AE, Parella T, Costas M, Stahl SS, Ribas X. Chem Sci. 2010;1:326–330. [Google Scholar]
- 16.Rovira M, Font M, Ribas X. ChemCatChem. 2013;5:687–691. [Google Scholar]
- 17.Rovira M, Font M, Acuña-Parés F, Parella T, Luis JM, Lloret-Fillol J, Ribas X. Chem Eur J. 2014;20:10005–10010. doi: 10.1002/chem.201402711. [DOI] [PubMed] [Google Scholar]
- 18.Huffman LM, Casitas A, Font M, Canta M, Costas M, Ribas X, Stahl SS. Chem Eur J. 2011;17:10643–10650. doi: 10.1002/chem.201100608. [DOI] [PubMed] [Google Scholar]
- 19.Font M, Parella T, Costas M, Ribas X. Organometallics. 2012;31:7976–7982. [Google Scholar]
- 20.Marlin DS, Olmstead MM, Mascharak PK. Inorg Chem. 2001;40:7003–7008. doi: 10.1021/ic010523n. [DOI] [PubMed] [Google Scholar]
- 21.The hydroxo ligands in the structure of 2 were found to exchange with H218O, as determined by ESI-MS (see Figure S6).
- 22.Costas M, Ribas X, Poater A, López Valbuena JM, Xifra R, Company A, Duran M, Solà M, Llobet A, Corbella M, Usón MA, Mahía J, Solans X, Shan X, Benet-Buchholz J. Inorg Chem. 2006;45:3569–3581. doi: 10.1021/ic051800j. [DOI] [PubMed] [Google Scholar]
- 23.Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]
- 24.MacLeod KC, Vinyard DJ, Holland PL. J Am Chem Soc. 2014;136:10226–10229. doi: 10.1021/ja505193z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harrold ND, Waterman R, Hillhouse GL, Cundari TR. J Am Chem Soc. 2009;131:12872–12873. doi: 10.1021/ja904370h. [DOI] [PubMed] [Google Scholar]
- 26.Analogously to 1, self-decomposition of 3 occurred by N2 release and N-N bond formation to form an indazole ring (70% yield) accompained by methane release (see SI).
- 27.50% 15N-labeled 2 or 3 were formed by reaction of [CuI(CH3CN)4](CF3SO3) with singly 15N-labeled HL-N3 or MeL-N3 (synthesized by reaction of HL-Br or MeL-Br with singly terminal labelled azide, 15N-14N-14N).
- 28.Evans DF. J Chem Soc. 1959:2003–2005. [Google Scholar]
- 29.Naklicki ML, White CA, Plante LL, Evans CEB, Crutchley RJ. Inorg Chem. 1998;37:1880–1885. [Google Scholar]
- 30.Crandell DW, Ghosh S, Berlinguette CP, Baik MH. ChemSusChem. 2015;8:844–852. doi: 10.1002/cssc.201403024. [DOI] [PubMed] [Google Scholar]
- 31.Kundu S. PhD Thesis. Department of Chemistry, Humboldt Universität zu Berlin; Germany: 2013. [Google Scholar]
- 32.Gombler W, Kinas RW, Stec WJ. Z Naturforsch. 1983;38:815–818. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.