

Abstract
Acute Intermittent Porphyria (AIP), an autosomal dominant inborn error of heme metabolism, typically presents in adulthood, most often in women in the reproductive age group. There are limited reports on the clinical presentation in children, and in contrast to the adults, most of the reported pediatric cases are male. While acute abdominal pain is the most common presenting symptom in children, seizures are commonly seen and may precede the diagnosis of AIP. As an example, we report a 9 year old developmentally normal pre-pubertal boy who presented with acute abdominal pain, vomiting and constipation followed by hyponatremia, seizures, weakness and neuropathy. After a diagnostic odyssey, his urine porphobilinogen was found to be significantly elevated and genetic testing showed a previously unreported consensus splice-site mutation IVS4-1G>A in the HMBS gene confirming the diagnosis of AIP. Here, we discuss the clinical presentation in this case, and 15 reported pediatric cases since the last review 30 years ago and discuss the differential diagnosis and challenges in making the diagnosis in children. We review the childhood-onset cases reported in the Longitudinal Study of the Porphyrias Consortium. Of these, genetically and biochemically confirmed patients, 11 of 204 (5%) reported onset of attacks in childhood. Most of these patients (91%) reported recurrent attacks following the initial presentation. Thus, AIP should be considered in the differential diagnosis of children presenting with unexplained abdominal pain, seizures, weakness and neuropathy.
Keywords: Acute Porphyria, seizures, metabolic, pediatric
INTRODUCTION
Acute Intermittent Porphyria (AIP, OMIM# #176000), an autosomal dominant disorder of heme biosynthesis, rarely presents in childhood. AIP is due to mutations in the hydroxymethylbilane synthase (HMBS) gene that result in half-normal HMBS enzymatic activity, the third enzyme in the heme biosynthetic pathway [1,2,3] (HMBS, EC 2.5.1.61, also known as porphobilinogen deaminase). This enzymatic deficiency predisposes heterozygous patients to life-threatening acute neurovisceral attacks that are precipitated by various factors, including porphyrinogenic drugs (e.g., P450 inducers), alcohol, infection, stress, prolonged fasting and chronic under nutrition, and steroid hormones. These factors induce the synthesis of aminolevulinic acid synthase 1 (ALAS1), the first and rate-limiting enzyme in the heme biosynthetic pathway. When hepatic ALAS1 is induced, the partial HMBS enzyme deficiency becomes limiting, resulting in the marked accumulation of the neurotoxic porphyrin precursors, aminolevulinic acid (ALA), and porphobilinogen (PBG). These porphyrin precursors act on the central and peripheral nervous systems to produce acute neurovisceral and psychiatric symptoms. The diagnosis of AIP is typically made by demonstrating markedly elevated urinary PBG levels during an acute attack, and by identifying a pathogenic HMBS gene mutation in the patient [1,4].
It is estimated that 10–20% of AIP heterozygotes experience acute attacks, while the majority are clinically asymptomatic throughout their lives [2, 3, 4]. The most common clinical presentation of an acute porphyric attack is severe abdominal pain, accompanied by vomiting, constipation, and abdominal distention, which can masquerade as an acute abdomen. Behavioral changes such as irritability, insomnia, emotional lability and hypertension and tachycardia due to sympathetic over-activity are important clues for the diagnosis [4]. Hyponatremia often occurs during severe attacks and can lead to seizures or an altered sensorium or even coma. The acute onset of progressive limb weakness due to motor axonopathy accompanied by myalgia can be extremely debilitating for patients. If these symptoms are not recognized and treated early, they can lead to residual morbidity and mortality due to bulbar and respiratory muscle paralysis [1, 2, 4]. Physical examination is usually unremarkable and patients may lie in the fetal position in response to extreme pain and debility. Due to the variable clinical presentation and non-specific symptoms of the neurovisceral attacks, and the fact that attacks are rare in children, a high index of suspicion is required to make the diagnosis in children.
Here, we 1) provide an instructive case that highlights the difficulty in diagnosis and attack progression in a pre-pubertal boy in whom porphyrinogenic drugs were used to treat an infection and seizures which further exacerbated, prolonged, and increased the severity of his acute attack 2) review the literature describing AIP in children since the previous review 30 years ago by Kaplan et al (1986) [5], 3) report the frequency of childhood AIP manifestations among the 204 AIP patients enrolled in the Longitudinal Study of the Porphyrias Consortium of the NIH-sponsored Rare Diseases Clinical Research Network.
Case Summary
The patient was a 9-year-old developmentally normal, pre-pubertal Indian boy (Tanner stage G1PH1), with no prior medical history. He was the son of unrelated parents. He was essentially well until he presented with acute abdominal pain, vomiting and constipation. He had a fever without an obvious localizing cause, which responded to paracetamol a day prior to the acute episode. The pain was severe and he was hospitalized for a presumed subacute intestinal obstruction, and fluids were restricted (NPO). He received intravenous antibiotics including ceftriaxone and metronidazole. An abdominal CT scan showed mild ascites, but no other cause of his symptoms. On Day 3 of illness, he developed a tonic–clonic seizure that was treated using intravenous midazolam and phenytoin loading. His clinical condition remained unchanged and he was transferred to another hospital where he was treated conservatively with IV fluids and different antibiotics, including sulbactam and amikacin.
Laboratory studies showed leukocytosis and hyponatremia (serum sodium levels of 111 meq/L and 114 meq/L on Day 4 and 5 of illness). Hyponatremia was treated with intravenous saline. On day 7 of illness, his abdominal pain and vomiting improved and he was discharged on multivitamins and an appetite stimulant. He was stable for a few days, but on day 12 he developed progressive weakness and pain of the lower extremities, and he was unable to support himself. On Day 15 of illness, the weakness advanced to the proximal muscles of the upper limb and he continued to have severe pain in his extremities and back.
He was hospitalized again on day 20 of illness with truncal weakness and was unable to sit without support. He complained of headache, palpitations, and excruciating pain in the limbs that was not relieved by analgesics (Ibuprofen and Diclofenac). His appetite was poor and he was irritable, restless and had insomnia. His physical examination revealed a thin boy with a BMI of 14.5 kg/m2 who was irritable and lethargic. He had tachycardia and hypertension (BP 150/90 mmHg) with normal pulse volume and character, with all peripheral pulses palpable. The four limb BP did not reveal any discrepancy across different limbs and the fundus did not show any hypertensive changes. The pain was generalized in all limbs, trunk, and back. There were no objective signs of joint swelling/tenderness or inflammation and the examination of the musculoskeletal system was essentially normal. He was conscious, oriented, with an intact memory and intellect, with no cranial nerve involvement. He assumed a fetal posture because of the excruciating pain and resisted examination. The muscles of his upper and lower limbs had strength of 3/5 accompanied with truncal weakness. The deep tendon reflexes were difficult to elicit and the plantar reflexes were flexor. There were no meningeal signs, and his gait could not be examined as he was unable to stand unsupported. There was no evidence of musculoskeletal disease. An echocardiogram and ultrasound of the renal vessels were normal. He continued to have pain and progressive weakness and was given tramadol for pain control. His hypertension was managed using amlodipine. Evaluation for autoimmune disorders including Anti-Nuclear Antibody, Anti-ds DNA, RA factor and C-reactive protein was negative. The blood lead concentration was 0.1 micrograms/dL (<5). He was started on an empirical trial of Vitamin C, and Vitamin D3 was supplemented due to Vitamin D deficiency. A bone marrow aspiration to rule out an occult malignancy was negative.
The patient developed tachypnea and his breathing became labored indicating weakness of the intercostal muscles and the diaphragm. He was started on gabapentin as the pain appeared to be neuropathic, and he showed a slight improvement. Nerve Conduction Velocity (NCV) studies demonstrated a low conduction velocity in the left common peroneal nerve. He developed urinary retention, which was relieved by catheterization. The urine was red, which led to the suspicion of an acute porphyria (Figure 1). A Hoesch test for PBG was strongly positive and a quantitative urine PBG level was 152 μmol/L (normal <10 μmol/L). HMBS mutation analysis identified a novel splice site mutation, IVS4-1G>A, in the HMBS gene confirming the diagnosis of heterozygosity for AIP. Family studies revealed that his asymptomatic mother and two brothers were heterozygous for the novel consensus splice site mutation (Figure 2).
Figure 1.

Reddish-colored urine excreted by the patient during his acute attack
Figure 2.
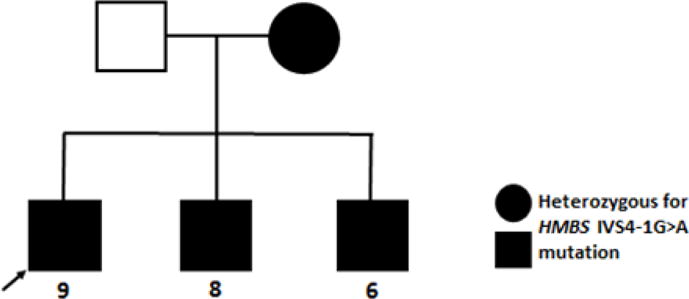
Pedigree of AIP family with HMBS Novel Mutation, IVS4-IG>A
As Hematin, the standard treatment for acute attacks [4], was not commercially available, the patient was started on a high dose dextrose infusion regimen (400 g/day). He received propranolol for hypertension and gabapentin was continued for his neuropathic pain. The patient improved significantly and was discharged on physical therapy. His repeat NCV at the end of 12 weeks demonstrated axonal motor sensory neuropathy. Follow up evaluation 12 weeks after his initial attack showed residual foot and wrist drop. The patient had two subsequent attacks during the nine months after discharge, precipitated by a minor viral illness and poor oral intake, respectively. These attacks were managed by hospitalization and intravenous dextrose (400 gm/day).
REVIEW
Heterozygous AIP typically presents in the second or third to fifth decade of life, most often in women, and very rarely presents before puberty [1, 2, 3, 4]. Previously, Kaplan et al reviewed the literature from 1907–1986 and reported 37 cases of acute porphyria with onset of symptoms in childhood [5]. These patients were predominantly males presenting before age 14 years, who had various manifestations, including abdominal pain, vomiting, fever and tachycardia, with additional mental changes, limb paresis and/or hyporeflexia. Most of these patients presented after treatment of their seizures with porphyrinogenic anti-epilepsy drugs such as sodium valproate and phenytoin. Based on their review, abdominal pain and vomiting were the most predominant presenting symptoms, similar to that in adults. These symptoms were typically preceded by, or concurrent with fever or an upper respiratory infection in children which may be an important precipitating factor in this age group.
A higher incidence of seizures was reported in these children compared to adults with AIP, which often presented before the diagnosis of an acute porphyria. Electrolyte imbalance, particularly hyponatremia due to syndrome of inappropriate antidiuretic hormone secretion, gastrointestinal and renal losses likely contributed to the seizures in these children [5]. Interestingly, irreversible cognitive and behavioral deficits were seen in these pediatric patients, unlike adults, which may be related to the neurotoxic porphyrin precursors and recurrent seizures [5]. Of interest, a survey of 294 Swedish AIP patients indicated that 2.2% of the 268 who responded had seizures, which were mostly due to an unrelated seizure disorder, as AIP patients in Sweden are diagnosed early and effectively treated, thereby limiting the onset of seizures during an acute attack [6]. However, most of these patients were not confirmed diagnostically by elevated levels of urinary PBG. HMBS mutation analysis was not available as the HMBS gene was not isolated until 1986 [6] and the first mutations were identified several years later [8, 9]. Hultdin et al reported 61 Swedish children (<18 years) with confirmed HMBS mutations followed prospectively for an average of 2.5 years. Based on their study, approximately 10% of these children (3 boys and 3 girls) developed clinical evidence of an acute attack before age 15 [10].
By interrogating PubMed, 15 additional heterozygous AIP children were reported since the Kaplan et al review, (Table 1). In contrast to the 37 children reviewed by Kaplan et al (1986), most of these children were confirmed by markedly increased levels of urinary PBG including the case described here (Table 1). These patients had clinical manifestations similar to those reported by Kaplan et al (1986) including weakness/myalgia’s, or intractable seizures [6, 11–19]. Most of these patients also presented after treatment with porphyrogenic anti-seizure medications, particularly with barbiturates and phenytoin. Of the 15 patients, only two had HMBS mutation analyses including the patient reported here (HMBS IVS4-IG>A). The patient reported by Anyaegbu et al (2012) had an elevated urine PBG at 263.2 mM (normal <1.3) and reportedly had an HMBS missense mutation that encoded p.R321H. However, this mutation is the most common benign variant in Caucasians with an allele frequency of ~0.002 and an in vitro expressed activity in the normal range (122% of mean expressed wild-type activity) confirming it as a benign variant (20). In this patient, HMBS sequencing should be repeated to identify the causative mutation.
Table 1.
ChildrenHeterozygous for Acute Porphyrias with reported biochemical findings and concurrent medications (ages 0–14 years)
| Patient Number | Reference | Age in years/Sex | Urinary ALA | Urinary PBG | Erythrocyte HMBS Activity | HMBS Gene Mutation | Medications |
|---|---|---|---|---|---|---|---|
| 1 2 3 |
Bylesjo et al 1996 | 8/F 13/F 14/M |
Increased Increased increased |
Increased Increased Increased |
NR NR NR |
NR NR NR |
phenobarbital, phenytoin, valproate, phenytoin, phenobarbital, valproate, phenytoin |
| 4 | Badcock et al 1990 | 16/M | 230 umol/L | 719 umol/L | Normal | NR | valproate, carbamazepine, phenytoin |
| 5 | Chaix et al 1997 | 9/M | 248umol/L | 285umol/L | decreased | NR | valproate, carbamazepine |
| 6 | Sykes et al 2001 | 3/M | NR | watson-schwartz positive | NR | NR | phenobarbital |
| 7 8 9 |
Varsik et al 2005 | 8/M 8/M 8/M |
24.8 umol/L 41.0 umol/L 12.9 umol/L |
995 umol/L 380 umol/L 12.9 umol/L |
NR NR NR |
NR NR NR |
unspecified Anti-epileptic |
| 10 | Kenny et al 2007 | 8/M | NR | NR | NR | NR | phenobarbital |
| 11 | Bhat et al 2010 | 12/M | NR | 20mg/day | NR | NR | calcium channel blockers |
| 12 | Dibi et al 2010 | 10/M | NR | NR | 92.6mmol/mmol | NR | phenobarbital,valproate |
| 13 | Sunita and Parmar 2011 | 12/M | NR | 49.4 umol/molCr | NR | NR | phenytoin |
| 14 | Anyaegbu et al 2012 | 8/M | 135umol/24h | 177umol/24h | NR | benign variant encoding p.R321H | carbamazepine, topiramate, valproate, clobazam |
| 15 | Zhao et al 2014 | 9/F | 107mg/24h | 74mg/24h | decreased | NR | phenytoin, levetiracetam |
| 16 | (this report) | 9/M | NR | 152umol/L | NR | IVS4-1G>A | Phenytoin, metronidazole |
NR = Not Reported
Interestingly, the vast majority of patients in the Kaplan et al review and our current report were males. This is in contrast to AIP in adults where the majority of symptomatic patients are women. Hormonal factors are likely to play a role in the disease presentation in adult females while this is less likely to be a factor in pre-pubertal children.
In contrast to the symptomatic heterozygous children with autosomal dominant AIP, only a few cases of children with homozygous dominant AIP have been reported [21–24 ]. These children have a severe and progressive neurodegenerative disease secondary to markedly deficient HMBS activity leading to very high levels of urinary ALA and PBG. This disease is markedly distinct from heterozygous AIP, which usually manifests as acute attacks in adults in the third to fourth decades of life. Aminolevulinic acid Dehydratase deficiency porphyria (ADP) is a very rare autosomal recessive porphyria with only a few reported cases worldwide. Patients can present during childhood and adolescence with acute attacks similar to AIP. In contrast to the other acute porphyrias, ALA is substantially increased and PBG levels may be normal or slightly increased in ADP [25]. Acquired causes of ALAD deficiency, such as lead poisoning should also be excluded in patients presenting with symptoms suggestive of an acute attack. Succinylacetone, which accumulates in hereditary tyrosinemia type 1 is structurally similar to ALA, inhibits ALAD, and can cause increased urinary excretion of ALA and clinical manifestations that resemble acute porphyria [1].
Based on the limited number of reported cases of symptomatic AIP children, we interrogated the database of HMBS mutation and biochemically-confirmed AIP patients in the Longitudinal Study of the NIH-supported Porphyrias Consortium (PC) of the Rare Diseases Clinical Research Network (https://www.rarediseasesnetwork.org/cms/porphyrias) (Table 2) [26, 27]. Eleven of the 204 AIP patients (~5%, 10 females, 1 male) in the database reported first symptoms before age 14 by medical history questionnaires (Table 2). Among the 11 patients, there were 10 different HMBS mutations, seven missense, two single base insertions, and one consensus splice-site lesion, all of which had been previously reported in unrelated affected patients. Ten of these patients had a history of recurrent attacks after the initial childhood episode. In contrast to the published reports, the majority (91%) of the childhood onset cases reported in the PC were female. These preliminary results from the Longitudinal Study are limited as medical history, and biochemical tests were not recorded at the time of the attack in childhood and hence these attacks could not be clinically confirmed. These and other studies highlight the importance of identifying HMBS mutation carriers in childhood and following them prospectively to understand the natural history and clinical presentation in children. Long-term follow up of these reported children with AIP is particularly important to see if there is an increased risk of recurrent attacks or improvement of acute attack symptoms or seizures with age.
Table 2.
Review of Biochemically and HMBS Mutation-Confirmed AIP Cases Reporting Symptoms Before Puberty in the Longitudinal Study of the Porphyrias [13]
| Patient Number | HMBS Mutation | Urine PBG at enrollment* (lab range) | Sex | AGE AT | Recurrent Attacks | ||
|---|---|---|---|---|---|---|---|
| Database Enrollment | Symptom Onset | Menarche | |||||
| 1 | c.863_86 4insT | 111.5 mg (0–4) | F | 33 | 3 | 12.5 | Yes |
| 2 | IVS10-2A>G | 29 mg/24 hr (<2.4) | F | 21 | 3 | 10 | Yes |
| 3 | p.R201W | 6 mg/g creat (<2.0) | F | 19 | 5 | 9 | Yes |
| 4 | p.R167Q | 13.3 mg/24 hr (0–0.5) | F | 72 | 8 | 16 | Yes |
| 5 | p.G111R | 3.3 mmol/mol creat (0.09–2.97) | F | 72 | 9 | 11 | Yes |
| 6 | p.R116Q | 16.9 mg (0–4) | F | 41 | 10 | 11 | Yes |
| 7 | p.V267M | 5.2 mg (0–4) | F | 36 | 10 | 11 | No |
| 8 | p.R173W | 8.3 mg (0–4) | M | 53 | 10 | N/A | Yes |
| 9 | p.A33IV | 45 umol/L (0.0–8.8) | F | 59 | 11 | 11 | Yes |
| 10 | c.633_63 4insC | 55.8 mg/g creat (<2.0) | F | 39 | 11 | 11 | Yes |
| 11 | p.A33IV | Not available | F | 39 | 13 | 17 | Yes |
HMBS: Hydroxylmethylbilane Synthase enzymatic activity (also known as Porphobilinogen Deaminase PBGD)
Patients were asymptomatic at the time of enrollment in longitudinal study
PBG: Urine porphobilinogen concentration
Clearly, the diagnosis and management of AIP in children has been and continues to be challenging. Our patient, who was heterozygous for the novel HMBS consensus splice-site mutation, IVS4-1G>A, had multiple precipitating factors including a febrile illness, poor caloric intake, and the use of the porphyrinognenic drug, phenytoin, and the possibly porphyrinogenic antibiotics like metronidazole, all of which likely contributed to his initial symptoms and prolonged clinical course. As his clinical symptoms were non-specific, he experienced a series of evaluations for other causes which were all negative. The presence of red urine and axonal motor neuropathy on the nerve conduction velocity suggested an acute porphyria and subsequent biochemical and HMBS mutation analyses confirmed the diagnosis of AIP.
This case emphasizes the importance of determining the urinary PBG levels during an attack and confirming the diagnosis of AIP – or the other two autosomal dominant hepatic porphyrias, Hereditary Copropophyria (deficient coproporphyrinogen oxidase) and Variegate Porphyria (deficient protoporphyrinogen oxidase) – by mutation analysis of their specific genes. Early diagnosis of an acute attack will lead to early therapeutic intervention by eliminating all triggering factors and initiating hematin infusions. Symptomatic individuals with AIP also carry the potential to develop complications like hypertension, hepatocellular carcinoma and renal insufficiency and should be monitored annually [2, 4]
In conclusion, acute porphyrias should be considered in the differential diagnosis of cases with unexplained neurological weakness and myalgias, as well as in children with hypertension in association with behavioral abnormalities including irritability and lethargy. It should be noted that acute abdominal pain may resolve early with conservative management, and may go unnoticed if the patient presents later in the clinical course. In addition, the possibility of AIP should be explored in selected cases of unexplained seizures, as the commonly used antiepileptic’s (phenobarbital, valproate, phenytoin) can precipitate acute attacks in children. Early diagnosis and hematin therapy for acute attacks is essential to prevent long-term neurologic deficits and chronic complications.
Highlights.
Acute Intermittent Porphyria (AIP) rarely presents in childhood
First report of a biochemically and genetically confirmed Pediatric patient with AIP
Seizures may precede the diagnosis in pediatric patients
Clinical presentation may include neuropathy, hypertension, behavioral changes
Long term natural history studies are needed in mutation confirmed pediatric patients
Acknowledgments
Funding: This work was supported in part by The Porphyrias Consortium (U54DK083909), which is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through collaboration between NCATS and the NIDDK. MB is supported in part by the NIH Career Development Award (K23DK095946).
Abbreviations
- ALAS1
Aminolevulinic acid synthase 1
- HMBS
Hydroxymethylbilane synthase
- ADP
Aminolevulinic acid Dehydratase deficiency porphyria
- PBG
Porphobilinogen
- ALA
Aminolevulinic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson K. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Beadet ALVD, Sly WS, editors. The metabolic and molecular bases of inherited disease. New York: McGraw Hill; 2001. pp. 2991–3062. [Google Scholar]
- 2.Puy H, Gouya L, Deybach J-C. Porphyrias. Lancet. 2010;375:924–37. doi: 10.1016/S0140-6736(09)61925-5. [DOI] [PubMed] [Google Scholar]
- 3.Badminton MN, Whatley SD, Deacon AC, Elder GH. The porphyrias and other disorders of porphyrin metabolism. In: Burtis CA, Ashwood ER, Bruns DE, editors. Tietz Textbook of Clinical Chemistry and Molecular Diagnostics. 5. St Louis, MO: Elsevier Saunders; 2012. pp. 1031–55. [Google Scholar]
- 4.Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;142:439–50. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- 5.Kaplan PW, Lewis DV. Juvenile acute intermittent porphyria with hypercholesterolemia and epilepsy: a case report and review of the literature. J Child Neurol. 1986;1:38–45. doi: 10.1177/088307388600100106. [DOI] [PubMed] [Google Scholar]
- 6.Bylesjö I, Forsgren L, Lithner F, Boman K. Epidemiology and clinical characteristics of seizures in patients with acute intermittent porphyria. Epilepsia. 1996;37:230–5. doi: 10.1111/j.1528-1157.1996.tb00018.x. [DOI] [PubMed] [Google Scholar]
- 7.Raich N, Romeo PH, Dubart A, Beaupain D, Cohen-Solal M, Goossens M. Molecular cloning and complete primary sequence of human erythrocyte porphobilinogen deaminase. Nucleic Acids Res. 1986 Aug 11;14(15):5955–68. doi: 10.1093/nar/14.15.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grandchamp B, Picat C, Mignotte V, Wilson J, Te Velde K, Sandkuyl L, Romeo P, Goossens M, Nordmann Y. Tissue specific splicing mutation in acute intermittent porphyria. Proc Natl Acad Sci. 1989;86:661–664. doi: 10.1073/pnas.86.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grandchamp B, Picat C, Kaupinen R, Mignotte V, Peltonen L, Mustajoki P, Romeo P, Goossens M, Nordmann Y. Molecular Analysis of acute intermittent porphyria in a finnish family with normal erythrocyte porphobilinogen deaminase. Eur J Clin Invest. 1989b;19:415–418. doi: 10.1111/j.1365-2362.1989.tb00252.x. [DOI] [PubMed] [Google Scholar]
- 10.Hultdin J, Schmauch A, Wikberg A, Dahlquist G, Andersson C. Acute intermittent porphyria in childhood: a population-based study. Acta Paediatr. 2003 May;92(5):562–8. [PubMed] [Google Scholar]
- 11.Badcock NR, Zoanetti GD, O’Reilly DA, Robertson EF. Variant acute intermittent porphyria in a child. Clin Chem. 1990;36:812–814. [PubMed] [Google Scholar]
- 12.Chaix Y, Gencourt C, Grouteau E, Carrière JP. Acute intermittent porphyria associated with epilepsy in a child: diagnostic and therapeutic difficulties. Arch Pediatr. 1997;4:971–4. doi: 10.1016/s0929-693x(97)86093-9. [DOI] [PubMed] [Google Scholar]
- 13.Kenny JM, Chipolombwe MJ, Molyneux E. A case of repeated coma in an 8-year-old boy. Trop Doct. 2007;37:264–5. doi: 10.1258/004947507782332919. [DOI] [PubMed] [Google Scholar]
- 14.Sykes RM. Acute intermittent porphyria, seizures, and antiepileptic drugs: a report on a 3-year-old Nigerian boy. Seizure. 2001;10:64–66. doi: 10.1053/seiz.2000.0473. [DOI] [PubMed] [Google Scholar]
- 15.Sunita A, Shweta P. Peripheral neuropathy with severe intractable hyponatremia as a presentation of acute intermittent porphyria. The West London Medical Journal. 2011;3:20–24. [Google Scholar]
- 16.Varsik P, Buranova D, Kollar B, Traubner P, Bozek P, Mikulecky M. Familial occurrence of myoclonic epilepsy syndrome and acute intermittent porphyria. Neuro Endocrinol Lett. 2005;26:7–12. [PubMed] [Google Scholar]
- 17.Bhat JI, Qureeshi UA, Bhat MA. Acute Intermittent Porphyria with Transient Cortical Blindness. Indian Pediatr. 2010;47:977–978. doi: 10.1007/s13312-010-0152-9. [DOI] [PubMed] [Google Scholar]
- 18.Dibi A, Aitouamar H, Bentahila A. Recurrent flaccid paralysis indicative of acute intermittent porphyria in a child. Arch Pediatr. 2010;17:1670–2. doi: 10.1016/j.arcped.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Anyaegbu E, Goodman M, Ahn SY, Thangarajh M, Wong M, Shinawi M. Acute intermittent porphyria: a diagnostic challenge. J Child Neurol. 2012;27:917–21. doi: 10.1177/0883073811427603. [DOI] [PubMed] [Google Scholar]
- 20.Chen B, Solis-Villa C, Hakenberg J. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum Mutat. 2016 doi: 10.1002/humu.23067. Epub Aug 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao B, Wei Q, Wang Y, Chen Y, Shang H. Posterior reversible encephalopathy syndrome in acute intermittent porphyria. Pediatr Neurol. 2014;5:457–60. doi: 10.1016/j.pediatrneurol.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 22.Beukeveld GJ, Wolthers BG, Nordmann Y, Deybach JC, Grandchamp B, Wadman SK. A retrospective study of a patient with homozygous form of acute intermittent porphyria. J Inherit Metab Dis. 1990;13:673–83. doi: 10.1007/BF01799566. [DOI] [PubMed] [Google Scholar]
- 23.Llewellyn DH, Smyth SJ, Elder GH, Hutchesson AC, Rattenbury JM, Smith MF. Homozygous acute intermittent porphyria: compound heterozygosity for adjacent base transitions in the same codon of the porphobilinogen deaminase gene. Human Genetics. 1992;89:97–98. doi: 10.1007/BF00207051. [DOI] [PubMed] [Google Scholar]
- 24.Hessels J, Voortman G, van der Wagen A, van der Elzen C, Scheffer H, Zuijderhoudt FM. Homozygous acute intermittent porphyria in a 7-year-old boy with massive excretions of porphyrins and porphyrin precursors. J Inherit Metab Dis. 2004;27(1):19–27. doi: 10.1023/B:BOLI.0000016613.75677.05. Review. [DOI] [PubMed] [Google Scholar]
- 25.Solis C, Martinez-Bermejo A, Naidich TP, et al. Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias. Arch Neurol. 2004;61:1764–70. doi: 10.1001/archneur.61.11.1764. [DOI] [PubMed] [Google Scholar]
- 26.Sassa S. ALAD porphyria. Semin Liver Dis. 1998;18(1):95–101. doi: 10.1055/s-2007-1007145. Review. [DOI] [PubMed] [Google Scholar]
- 27.Porphyrias Consortium of the National Institutes of Health Rare Disease Clinical Research Network. Available at: http://www.rarediseasesnetwork.org/cms/porphyrias Accessed April 11, 2016.
- 28.ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Longitudinal Study of the Porphyrias. [cited 2016 Apr 11]. Available from: https://clinicaltrials.gov/ct2/show/NCT01561157 NLM Identifier: NCT01561157. [Google Scholar]