

Abstract
The polymyxin antibiotics [colistin and polymyxin B (PMB)] are increasingly used as a last-line option for the treatment of infections caused by extensively drug-resistant Gram-negative bacteria. Despite having similar structures and antibacterial activity in vitro, the two clinically available polymyxins have very different pharmacological properties, as colistin (polymyxin E) is intravenously administered to patients in the form of an inactive prodrug colistin methanesulphonate (sodium). This review will discuss recent progress in the pharmacokinetics/pharmacodynamics and toxicity of colistin and PMB, the factors that affect their pharmacological profiles, and the challenges for the effective use of both polymyxins. Strategies are proposed for optimising their clinical utility based upon the recent pharmacological studies in vitro, in animals and patients. In the ‘bad bugs, no drugs’ era, polymyxins are a critically important component of the antibiotic armamentarium against difficult-to-treat Gram-negative ‘superbugs’. Rational approaches to the use of polymyxins must be pursued to increase their effectiveness and to minimise resistance and toxicity.
Keywords: Polymyxins, Pharmacokinetics, Pharmacodynamics, Nephrotoxicity
1. Introduction
Colistin (polymyxin E) and polymyxin B (PMB) are lipopeptide antibiotics with activity against many Gram-negative bacteria [1,2]. The polymyxins were approved for clinical use in the late 1950s but fell out of favour during the mid-1970s owing to concerns over their potential to cause nephrotoxicity and neurotoxicity [3]. Over the last two decades, clinical interest in polymyxins has increased due to the emergence of extensively drug-resistant Gram-negative bacteria coupled with the dry antibiotic development pipeline [1]. Colistin and PMB are currently considered a last-line defence against the problematic Gram-negative ‘superbugs’, notably carbapenem-resistant Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii, which are classified under ‘Urgent’ or ‘Serious’ threat level by the US Centers for Disease Control and Prevention (CDC) [4]. It is used against these pathogens that will be the focus of this mini-review.
Colistin and PMB possess very similar chemical structures, differing only by one amino acid at position 6 in the peptide ring, with a D-leucine and D-phenylalanine, respectively [5]. Not surprisingly, they have very similar antimicrobial spectra and resistance mechanisms [6]. A major difference between the polymyxins is the form in which they are administered parenterally. Colistin is administered in the form of an inactive prodrug, colistin methanesulphonate (CMS) (a polyanion at physiological pH), while PMB (a polycation at physiological pH) is administered directly as its active form [1]. The different chemical forms administered have significant impacts on their pharmacokinetics and toxicity [7]. For optimal use of CMS/colistin and PMB, it is important to understand their pharmacological differences. In this mini-review, we will discuss the latest progress in the pharmacokinetics/pharmacodynamics and toxicity of colistin and PMB as well as the challenges for optimal use of both polymyxins.
2. Different labelling of polymyxin products
Undoubtedly, a major contributing factor to the confusion surrounding the effective use of CMS is differences in the dosing terminology [2]. In many parts of the world, such as Europe and India, International Units (IU) are used, whereas in North and South America, Southeast Asia and Oceania colistin base activity (CBA) is used [1,2]. One million IU (MIU) of CMS is equal to ca. 80 mg of CMS or 34 mg of CBA; a more detailed discussion on differences in labelling and dosage recommendations can be found in our previous reviews [1,2]. Understanding the labelling differences is critical for the optimal use of CMS in patients. For PMB, which is available in North and South America, Southeast Asia and Japan, all products are labelled using IU (1 mg = 10 000 IU).
3. Minimum inhibitory concentrations (MICs) and mode of action
As CMS is an inactive prodrug of colistin, colistin sulphate should be used in MIC measurements for colistin [1]. To date, SENTRY Antimicrobial Surveillance Program (2006–2009) is the largest surveillance programme examining the MICs of the polymyxins. The compiled data from this programme showed that PMB and colistin have similar in vitro activities (MIC90, ≤0.5–1 mg/L) against P. aeruginosa, A. baumannii and Klebsiella pneumoniae, with very low resistance rates globally (<0.1–1.5%) [8]. However, questions have been raised regarding the susceptibility testing methods used for polymyxins, including their potential adsorption to plastic devices used in the MIC measurement and poor diffusion of polymyxins in agar [9]. In this regard, polysorbate 80 (P-80) was initially proposed to improve the broth microdilution MIC results for colistin and PMB as it can prevent the binding of polymyxins to plastic panels. However, its use was contraindicated by the Clinical and Laboratory Standards Institute (CLSI) owing to potential synergism between P-80 and the polymyxins [9,10]. In the most recent CLSI protocol, P-80 is not recommended in the measurement of colistin and PMB MICs. Presently, broth microdilution is regarded as the best method for polymyxin susceptibility testing. Susceptibility breakpoints for colistin and PMB set by the CLSI for P. aeruginosa, Acinetobacter spp. and other non-Enterobacteriaceae are identical, where an MIC of ≤2 mg/L is regarded as susceptible [11]. The susceptibility breakpoints of colistin by the European Committee on Antimicrobial Susceptibility Testing (EUCAST) are ≤4 mg/L for Pseudomonas spp. and ≤2 mg/L for Acinetobacter spp. and Enterobacteriaceae [12]. However, as will be discussed in Section 5 below on pharmacodynamics, data from recent pharmacokinetic/pharmacodynamic (PK/PD) studies suggest the breakpoints for the above Gram-negative pathogens could be even lower. Consequently, a joint CLSI and EUCAST Working Group is currently re-evaluating the existing breakpoints [1,9].
The precise mechanism of action of the polymyxins is currently unclear. However, it is believed that activity is related, in part, to disruption of the bacterial outer and inner membranes via a ‘self-promoted uptake’ mechanism [13]. The initial step involves binding of the positively charged polymyxins to negatively charged lipopolysaccharide (LPS) on the outer membrane of Gram-negative bacteria both via electrostatic and hydrophobic interactions (Fig. 1) [5]. Bacteria can become resistant to polymyxins by modifications of the negatively charged phosphate groups of lipid A [14] or by loss of LPS [15]. For more details, we direct the reader to the review in this Theme Issue on the mechanism of polymyxin resistance.
Fig. 1.
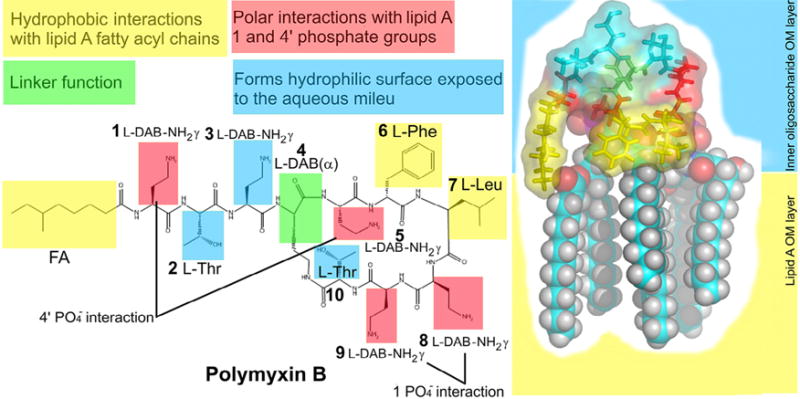
Schematic diagram showing key contacts involved in the complex formation between polymyxin B and the lipid A component of lipopolysaccharide. FA, N-terminal fatty acyl chain; OM, outer membrane. Figure reproduced from Velkov et al. [5] with permission. Published 2010 by the American Chemical Society.
4. Pharmacokinetics of polymyxins
4.1. Colistin methanesulphonate/colistin
The positively charged colistin exhibits a markedly different PK profile to that of the sulfomethylated derivative [1]. CMS is eliminated predominantly by the kidneys, whereas colistin is mainly cleared by a route other than renal excretion [2]. Following parenteral administration of CMS, colistin is generally formed slowly, with the plasma concentration increasing slowly. Plachouras et al. [16] showed that it can take >36 h to reach a colistin steady-state plasma concentration of 2 mg/L with intravenous (i.v.) administration of 3 MIU CMS every 8 h (q8h) in patients with good renal function. This finding highlights that the low initial exposure to formed colistin is a significant PK/PD challenge for optimising CMS use in patients. This dilemma can be partially counteracted with the use of a loading dose. In studies that evaluated CMS loading doses of 6 MIU and 9 MIU, the average colistin plasma concentrations reached 1.34 mg/L and 2.65 mg/L, respectively, at 8 h after the loading dose, with the likelihood of earlier eradication of the infecting bacteria [17,18]. In critically ill patients, kidney function and renal replacement therapy (RRT) have a dramatic impact on the pharmacokinetics of CMS and formed colistin [19,20]. One of the largest population PK studies reported thus far in critically ill patients involved 105 patients with varying degrees of renal function [creatinine clearance (CLCr) of 3–169 mL/min/1.73 m2], including 12 patients on intermittent haemodialysis and 4 on continuous RRT (CRRT) [19]. Even though there was only a ca. 5.5-fold range in the daily doses (2.5–13.7 MIU), substantial interpatient variation (0.48–9.38 mg/L, ca. 19.5-fold) in the average steady-state plasma colistin concentration (Css,avg) was observed in the 105 patients. Significant interpatient variation was observed even among patients with similar CLCr and those receiving the same daily dose of CMS (Fig. 2). In patients on RRT, both CMS and formed colistin were cleared [19,20]. Clearly, given that the plasma concentration of formed colistin is highly influenced by renal function, it is essential that the dosage regimen of CMS is adjusted in patients with varying renal function to ensure that appropriate colistin exposure is obtained. In patients with a CLCr of >80 mL/min, only 65–75% of patients receiving the approved updated dose recommended by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) achieved a Css,avg of formed colistin ≥1 mg/L [22]. As the MIC90 for colistin is ≤0.5–1 mg/L against P. aeruginosa, A. baumannii and K. pneumoniae [8], it would be clinically useful to administer the maximal CMS dose in patients with CLCr > 80 mL/min, ideally in combination with another antibiotic that may provide synergistic bacterial killing [19,23]. As colistin is ca. 50% unbound in human plasma [23] (and unpublished data), a colistin Css,avg of ca. 2 mg/L is necessary for effective treatment of bacteria with an MIC of 1 mg/L. For patients on RRT, in order to achieve a colistin Css,avg of 2 mg/L, the current recommendation suggests a CMS loading dose of 9 MIU followed at 24 h by 1 MIU every 12 h (q12h) for patients on intermittent haemodialysis, and 4.3 MIU q8h or 6.3 MIU q12h for patients on CRRT [19]. Furthermore, haemodialysis patients should aim to have their dialysis performed towards the end of the CMS dosing interval to avoid excessive removal of CMS from the body. After dialysis, a CMS dose of 1.7 MIU is required to replenish the removed CMS.
Fig. 2.
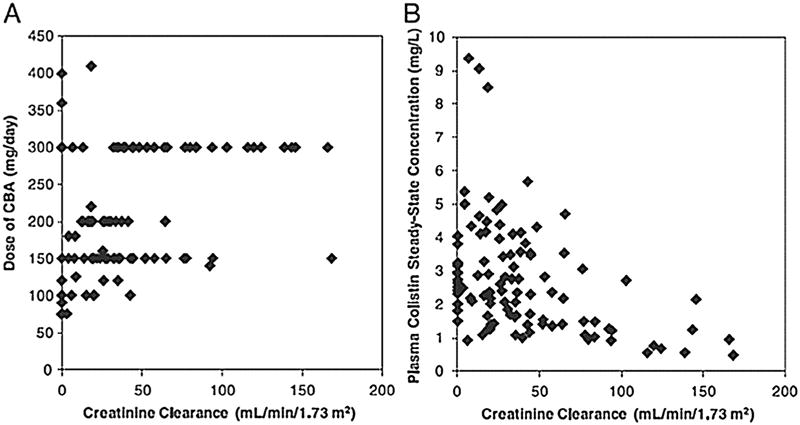
Relationship of physician-selected daily dose of colistin base activity (CBA) (A) and the resultant average steady-state plasma colistin concentration (B) versus creatinine clearance (CLCr) in 105 critically ill patients. CLCr was calculated using the Jelliffe equation [21]. Figure reproduced from Garonzik et al. [19] with permission. Published 2011 by the American Society for Microbiology.
Currently, little is known about the pharmacokinetics of CMS and formed colistin in extravascular sites. In critically ill patients with and without central nervous system (CNS) infection, the distribution of colistin into the cerebral spinal fluid (CSF) appears to be very low following i.v. CMS administration. In a study by Ziaka et al. [24], the CSF concentrations of formed colistin (at 1, 4 and 8 h) following i.v. administration of 3 MIU CMS q8h were only ca. 7% of the total serum colistin concentrations in patients without CNS infection and ca. 11% in patients with external ventricular drain-associated ventriculitis (EVDV). When a combination of i.v. (3 MIU CMS q8h) and intraventricular (0.125 MIU CMS once daily) CMS was administered to patients with EVDV, concentrations of formed colistin in the CSF were ca. 1.45, 0.84 and 0.62 mg/L, respectively, at 1, 4 and 8 h and were >40% of the total colistin serum concentration at each time point [24]. It is evident that the combination of i.v. and intraventricular CMS may be useful for the treatment of CNS infection caused by Gram-negative bacteria; however, further clinical studies are required.
A recent study in cystic fibrosis (CF) patients showed that the concentration of formed colistin in sputum following i.v. administration of CMS is minimal. When six patients with CF were administered an i.v. CMS dose of 5 MIU at 3 days post-nebulisation of 4 MIU of CMS, the formed colistin concentrations in the sputum over 12 h were similar to their carryover concentrations in the pre-dose sputum (0.12–0.72 mg/L) [25]. Higher concentrations (>10-fold) of formed colistin in the sputum were achieved via inhalation (4 MIU/day of CMS). After a single inhalation dose, an average maximum colistin concentration of ca. 6.0 mg/L was achieved in the sputum at ca. 3 h for 2 MIU of CMS and ca. 12.8 mg/L at ca. 4.6 h for 4 MIU of CMS [25]. However, plasma concentrations of CMS and formed colistin were very low following inhalation. Following a single nebulisation dose of CMS at 2 MIU or 4 MIU, the maximum plasma CMS concentrations were 0.22 ± 0.055 mg/L at ca. 1.3 h and 0.33 ± 0.092 mg/L at ca. 1.9 h, respectively, with <3% of the nebulised CMS dose recovered in the urine by 24 h. In a study comparing the intrapulmonary and systemic pharmacokinetics of formed colistin in critically ill patients following administration of 2 MIU of CMS via inhalation, the steady-state colistin concentrations in the epithelial lining fluid were much higher than the steady-state plasma colistin concentrations (9.53–1137 mg/L vs. 0.15–0.73 mg/L) [26]. These findings highlight the potential to administer CMS by inhalation for the treatment of Gram-negative bacterial pneumonia, maximising the exposure of formed colistin in the lungs while minimising plasma concentrations and associated systemic toxicity. Clearly, further PK/PD studies are warranted for optimising the use of inhaled CMS.
4.2. Polymyxin B
Compared with CMS, only a very small number of studies have examined the pharmacokinetics of PMB following i.v. administration. One study involving eight critically ill patients showed that PMB is mainly eliminated by non-renal pathway(s), with <1% recovered in the unchanged form in urine [27], which is very similar to colistin in rats [28]. The largest population PK study to date involved 24 critically ill patients with a wide range of kidney function (CLCr of 10–143 mL/min), including two patients on CRRT [29]. With i.v. doses ranging from 0.45 mg/kg/day to 3.38 mg/kg/day (i.e. ca. 7.5-fold), the PMB Css,avg ranged from 0.68 mg/L to 4.88 mg/L (ca. 7.2-fold) (Fig. 3) and the median urinary recovery (4.04%) was very low. The PMB clearance scaled by total body weight from this study showed minimal interpatient variability in the PMB Css,avg (range, 0.02–0.06 L/h/kg; ca. 3-fold), a finding in marked contrast to the influence of renal function on the Css,avg of plasma colistin following administration of CMS as discussed above. Thus, renal function does not markedly affect PMB plasma concentrations and should not be used for dose adjustment. In the two patients on CRRT, 12.2% and 5.62% of the dose was recovered as unchanged PMB in the dialysates during the 12-h dosing interval [29]. Similar to colistin, PMB is cleared during dialysis; however, dosage adjustments are currently not recommended for patients on CRRT owing to limited clinical data. A National Institutes of Health (NIH)-funded clinical study is investigating the PK, PD and toxicodynamic (TD) relationships of i.v. PMB in critically ill patients, which aims to develop scientifically-based dosing recommendations for this important polymyxin antibiotic (NCT02682355, http://www.clinicaltrials.gov). In addition, little is known about the distribution of PMB into extravascular sites following i.v. administration, and studies in this area will be essential to determine the usefulness of i.v. PMB for the treatment of infections such as pneumonia and meningitis.
Fig. 3.
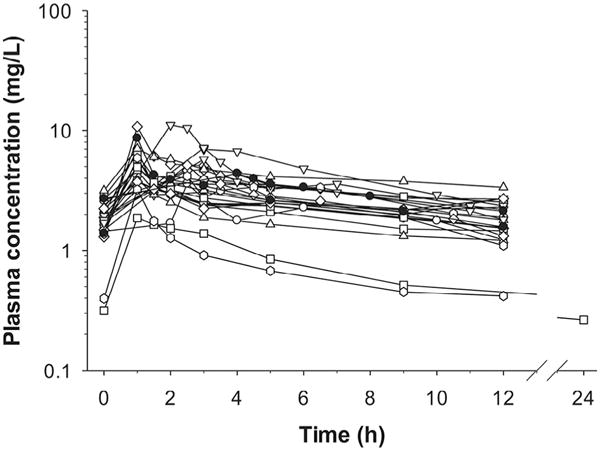
Plasma concentration–time profiles of polymyxin B in 24 critically ill patients. Concentrations from patients undergoing continuous venovenous haemodialysis are shown by filled symbols. Figure reproduced from Sandri et al. [29] with permission. Published 2013 by Oxford University Press.
In summary, the pharmacokinetics of CMS/colistin is influenced by renal function, with dosage regimens requiring adjustment in different types of patients. However, such an adjustment is not required for PMB, which is mainly cleared by non-renal pathway(s). As it is difficult to achieve a Css,avg of even 1 mg/L in patients with good renal function following i.v. administration of CMS [19], PMB may be a better option for treatment of bloodstream infections, with less interpatient variability and higher Css,avg [7,29]. Since CMS is mainly eliminated by the kidneys with high levels of colistin produced in the urinary tract, it may be a better option than PMB for the treatment of urinary tract infections. Inhaled CMS has been successfully employed for the treatment of lung infections caused by P. aeruginosa in patients with CF over the last three decades [30]. Given that inhaled PMB has been associated with a greater incidence of local airway irritation compared with CMS [31], CMS may be a better choice for inhalation. Nevertheless, prospective randomised controlled clinical studies are warranted to compare the efficacy of both polymyxins for the treatment of different types of infections.
5. Pharmacodynamics of polymyxins
Most studies examining the pharmacodynamics of the polymyxins have been conducted using colistin [23,32–34]. In in vitro studies, colistin shows rapid concentration-dependent killing against A. baumannii, K. pneumoniae and P. aeruginosa, with a minimal post-antibiotic effect at clinically achievable concentrations [32–34]. However, despite rapid initial killing, re-growth often occurs quickly (as early as within 2 h of the initial exposure). PMB displays very similar pharmacodynamics to that of colistin, with similar rapid killing against A. baumannii, K. pneumoniae and P. aeruginosa in vitro, followed by rapid re-growth [35–37]. In polymyxin-heteroresistant strains, amplification of polymyxin-resistant subpopulations has been shown to play an important role in the rapid emergence of resistance [38–40]. An inoculum effect has been reported both with colistin and PMB in vitro [37,40].
Using P. aeruginosa and A. baumannii in neutropenic mouse thigh and lung infection models, the PK/PD index that best describes the antimicrobial activity of colistin is the ratio of the area under the unbound (free) concentration–time curve to the MIC (fAUC/MIC) (Fig. 4) [23]; for P. aeruginosa, this has also been demonstrated in vitro [34]. Owing to the potential binding of polymyxins to the plasticware or ultrafiltration membranes, our group identified that ultrafiltration can be problematic [28], and ultracentrifugation and rapid equilibrium dialysis methods are superior for measuring plasma binding of polymyxins [23]. Our recent PK/PD study using ultracentrifugation and rapid equilibrium dialysis methods in neutropenic mice showed that the unbound fraction of colistin of 0.084 is ca. 6-fold lower than in humans (ca. 0.5) [23] (and unpublished data). For three strains of P. aeruginosa [ATCC 27853, PAO1 and a multidrug-resistant (MDR) clinical isolate] and three strains of A. baumannii (ATCC 19606 and two MDR clinical isolates), an fAUC/MIC value of 7.4–13.7 and 7.4–17.6, respectively, was required for a 2 log10 reduction in bacterial load in the thigh of neutropenic mice. In the neutropenic mouse lung infection model, subcutaneous colistin was substantially less effective at killing P. aeruginosa and A. baumannii compared with in the thigh infection model [23]. With the highest tolerable dose (40 mg/kg administered 6- or 8-hourly with cumulative daily doses of 120–160 mg/kg), 2 log10 killing in the lungs was not achievable for all six of the tested strains. The lower antibacterial activity in the lungs relative to the thigh is most likely due to limited drug exposure in the lungs following parenteral administration. Currently available data from animal and clinical studies suggest that colistin (and CMS) may have limited efficacy against respiratory tract infections [23,25].
Fig. 4.
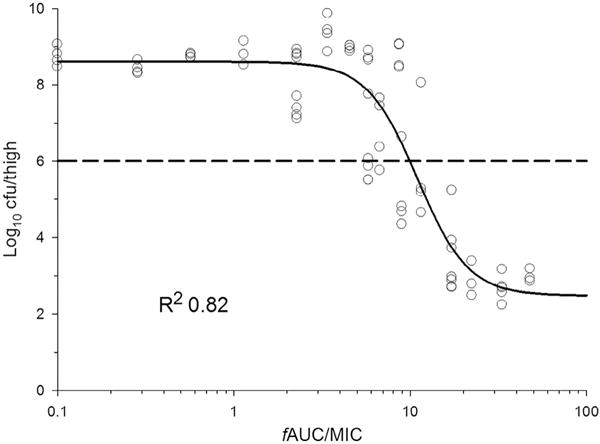
Relationship between bacterial load in the thighs of neutropenic mice at 24 h and the ratio of the area under the unbound (free) concentration–time curve to the MIC (fAUC/MIC) of colistin for Pseudomonas aeruginosa ATCC 27853. Figure adapted from Cheah et al. [23] with permission. Published 2015 by Oxford University Press.
Limited studies to date have examined the PK/PD index driving the activity of PMB. Given the similarity in the structure, it is very likely that fAUC/MIC is the most predictive PK/PD parameter for parenteral PMB [37]. In patients with good renal function, however, administration of PMB is very likely to generate higher fAUC/MIC values than CMS because: (i) CMS distribution is influenced by kidney function while PMB is not; and (ii) CMS conversion to colistin in vivo is slow and incomplete. To optimise the clinical use of PMB, more PD studies are needed.
6. Toxicodynamics of polymyxins
In the early years of their use, polymyxin-associated neurotoxicity occurred in patients with an incidence as high as 27% following parenteral administration [3,41]. However, recent retrospective clinical studies have not shown neurotoxicity to be a major concern [42,43]. Nephrotoxicity is by far the most common and dose-limiting side effect associated with parenteral polymyxins, with incidence rates in patients as high as 60% [44,45]. However, the rate of nephrotoxicity in patients receiving i.v. polymyxins is somewhat variable and depends on the definition of nephrotoxicity employed [e.g. RIFLE (risk, injury, failure, loss, and end-stage kidney disease) and AKIN (Acute Kidney Injury Network) scoring systems] [46].
Nephrotoxicity has been observed both with colistin and PMB following parenteral administration [46–49]. Recent TD analyses of colistin showed that patients with colistin Css,avg > 2.5 mg/L and patients with CLCr > 80 mL/min are more likely to develop nephrotoxicity [47,48]. The minimum colistin plasma concentration was also identified as an independent risk factor for nephrotoxicity, which occurred in the majority of patients when the minimum colistin plasma concentration was ≥2.2 mg/L (odds ratio = 4.6 on Day 7) [47]. For PMB, a daily dose of ≥150 mg (hazard ratio = 1.92) has been identified as the risk factor of nephrotoxicity [49]. A retrospective study showed the earliest onset of nephrotoxicity reported for i.v. CMS or PMB occurred 2 days after initiation of therapy, with the majority of cases occurring after 15 days of therapy [46]. Fortunately, polymyxin-associated nephrotoxicity was, however, reversible in most patients [47,50].
With regard to the mechanism of polymyxin-induced nephrotoxicity, cell culture and animal studies have demonstrated that colistin and PMB accumulate in renal tubular cells possibly through active uptake mechanisms mediated by megalin and PEPT2 transporters [51,52]. The resultant extremely high intracellular concentration of polymyxins in renal tubular cells causes dramatic changes in the morphology of mitochondria, loss of cytoplasmic membrane potential, apoptosis and cell cycle arrest [53,54]. The precise mechanisms of the uptake of polymyxins by renal tubular cells and subsequent cell death remain unanswered. However, elucidating these mechanisms is crucial for optimising their use in patients, development of novel approaches to attenuate polymyxin-induced nephrotoxicity, and the discovery of safer new-generation polymyxins.
7. Conclusions
Significant progress in understanding the pharmacology of polymyxins has been made over the past 15 years, although many gaps still remain. Scientifically-based dosing recommendations have now been developed for i.v. administration of CMS in critically ill patients and more recent studies are generating valuable insights for PMB. It is evident now that only the dose of CMS/colistin, not PMB, should be adjusted according to the patient’s renal function. As CRRT can efficiently eliminate both colistin and PMB, further clinical PK/PD/TD studies are warranted in order to optimise their use in this type of patient. Other high-priority research areas include evaluation of the efficacy of i.v. CMS/colistin and PMB for the treatment of respiratory tract infections and clinical PK/PD/TD studies of intrathecal and intraventricular administration of both polymyxins for the treatment of meningitis. While we await the development of novel antibiotics for the treatment of infections caused by Gram-negative ‘superbugs’, every effort must be made to optimise the clinical use of the polymyxins to maximise their efficacy while minimising the emergence of resistance and toxicity.
Acknowledgments
Funding: JL, TV and AF are supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH-NIAID) [R01 AI111965]. BTT, JL, RLN and AF are supported by the NIH-NIAID [R01 AI111990]. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH-NIAID. JL is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow, and TV is an Australian NHMRC Career Development Research Fellow.
Footnotes
Competing interests: None declared.
Ethical approval: Not required.
References
- 1.Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, et al. Framework for optimisation of the clinical use of colistin and polymyxin B: the Prato polymyxin consensus. Lancet Infect Dis. 2015;15:225–34. doi: 10.1016/S1473-3099(14)70850-3. [DOI] [PubMed] [Google Scholar]
- 2.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, et al. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis. 2006;6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 3.Koch-Weser J, Sidel VW, Federman EB, Kanarek P, Finer DC, Eaton AE. Adverse effects of sodium colistimethate. Manifestations and specific reaction rates during 317 courses of therapy. Ann Intern Med. 1970;72:857–68. doi: 10.7326/0003-4819-72-6-857. [DOI] [PubMed] [Google Scholar]
- 4.US Centers for Disease Control and Prevention (CDC) Antibiotic/antimicrobial resistance: antibiotic resistance threats in the United States. Atlanta, GA: National Center for Emerging and Zoonotic Infectious Diseases, CDC; 2013. [Google Scholar]
- 5.Velkov T, Thompson PE, Nation RL, Li J. Structure–activity relationships of polymyxin antibiotics. J Med Chem. 2010;53:1898–916. doi: 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baron S, Hadjadj L, Rolain JM, Olaitan AO. Molecular mechanisms of polymyxin resistance: knowns and unknowns. Int J Antimicrob Agents. 2016 doi: 10.1016/j.ijantimicag.2016.06.023. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 7.Nation RL, Velkov T, Li J. Colistin and polymyxin B: peas in a pod, or chalk and cheese? Clin Infect Dis. 2014;59:88–94. doi: 10.1093/cid/ciu213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gales AC, Jones RN, Sader HS. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006–09) J Antimicrob Chemother. 2011;66:2070–4. doi: 10.1093/jac/dkr239. [DOI] [PubMed] [Google Scholar]
- 9.Humphries RM. Susceptibility testing of the polymyxins: where are we now? Pharmacotherapy. 2015;35:22–7. doi: 10.1002/phar.1505. [DOI] [PubMed] [Google Scholar]
- 10.European Committee on Antimicrobial Susceptibility Testing. Recommendations for MIC determination of colistin (polymyxin E) As recommended by the joint CLSI–EUCAST Polymyxin Breakpoints Working Group. EUCAST; 2016. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/General_documents/Recommendations_for_MIC_determination_of_colistin_March_2016.pdf; [accessed 10.10.16] [Google Scholar]
- 11.Clinical and Laboratory Standards Institutes. Performance standards for antimicrobial susceptibility testing; twenty-fourth informational supplement. Wayne, PA: CLSI; 2014. Document M100-S24. [Google Scholar]
- 12.European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters Version 6.0. EUCAST; 2016. http://www.eucast.org; 2016 [accessed 10.10.16] [Google Scholar]
- 13.Trimble MJ, Mlynárčik P, Kolář M, Hancock RE. Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harb Perspect Med. 2016;6 doi: 10.1101/cshperspect.a025288. a025288[pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olaitan AO, Morand S, Rolain JM. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, et al. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother. 2010;54:4971–7. doi: 10.1128/AAC.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plachouras D, Karvanen M, Friberg LE, Papadomichelakis E, Antoniadou A, Tsangaris I, et al. Population pharmacokinetic analysis of colistin methanesulfonate and colistin after intravenous administration in critically ill patients with infections caused by Gram-negative bacteria. Antimicrob Agents Chemother. 2009;53:3430–6. doi: 10.1128/AAC.01361-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohamed AF, Karaiskos I, Plachouras D, Karvanen M, Pontikis K, Jansson B, et al. Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother. 2012;56:4241–9. doi: 10.1128/AAC.06426-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karaiskos I, Friberg LE, Pontikis K, Ioannidis K, Tsagkari V, Galani L, et al. Colistin population pharmacokinetics after application of a loading dose of 9 MU colistin methanesulfonate in critically ill patients. Antimicrob Agents Chemother. 2015;59:7240–8. doi: 10.1128/AAC.00554-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garonzik SM, Li J, Thamlikitkul V, Paterson DL, Shoham S, Jacob J, et al. Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother. 2011;55:3284–94. doi: 10.1128/AAC.01733-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karvanen M, Plachouras D, Friberg LE, Paramythiotou E, Papadomichelakis E, Karaiskos I, et al. Colistin methanesulfonate and colistin pharmacokinetics in critically ill patients receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother. 2013;57:668–71. doi: 10.1128/AAC.00985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jelliffe R. Estimation of creatinine clearance in patients with unstable renal function, without a urine specimen. Am J Nephrol. 2002;22:320–4. doi: 10.1159/000065221. [DOI] [PubMed] [Google Scholar]
- 22.Nation RL, Garonzik SM, Li J, Thamlikitkul V, Giamarellos-Bourboulis EJ, Paterson DL, et al. Updated US and European dose recommendations for intravenous colistin: how do they perform? Clin Infect Dis. 2016;62:552–8. doi: 10.1093/cid/civ964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheah SE, Wang J, Nguyen VT, Turnidge JD, Li J, Nation RL. New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and Acinetobacter baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother. 2015;70:3291–7. doi: 10.1093/jac/dkv267. [DOI] [PubMed] [Google Scholar]
- 24.Ziaka M, Markantonis SL, Fousteri M, Zygoulis P, Panidis D, Karvouniaris M, et al. Combined intravenous and intraventricular administration of colistin methanesulfonate in critically ill patients with central nervous system infection. Antimicrob Agents Chemother. 2013;57:1938–40. doi: 10.1128/AAC.01461-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yapa SWS, Li J, Patel K, Wilson JW, Dooley MJ, George J, et al. Pulmonary and systemic pharmacokinetics of inhaled and intravenous colistin methanesulfonate in cystic fibrosis patients: targeting advantage of inhalational administration. Antimicrob Agents Chemother. 2014;58:2570–9. doi: 10.1128/AAC.01705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boisson M, Jacobs M, Grégoire N, Gobin P, Marchand S, Couet W, et al. Comparison of intrapulmonary and systemic pharmacokinetics of colistin methanesulfonate (CMS) and colistin after aerosol delivery and intravenous administration of CMS in critically ill patients. Antimicrob Agents Chemother. 2014;58:7331–9. doi: 10.1128/AAC.03510-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zavascki AP, Goldani LZ, Li J, Nation RL. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother. 2007;60:1206–15. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Milne RW, Nation RL, Turnidge JD, Smeaton TC, Coulthard K. Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob Agents Chemother. 2003;47:1766–70. doi: 10.1128/AAC.47.5.1766-1770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandri AM, Landersdorfer CB, Jacob J, Boniatti MM, Dalarosa MG, Falci DR, et al. Population pharmacokinetics of intravenous polymyxin B in critically ill patients: implications for selection of dosage regimens. Clin Infect Dis. 2013;57:524–31. doi: 10.1093/cid/cit334. [DOI] [PubMed] [Google Scholar]
- 30.Banerjee D, Stableforth D. The treatment of respiratory Pseudomonas infection in cystic fibrosis: what drug and which way? Drugs. 2000;60:1053–64. doi: 10.2165/00003495-200060050-00006. [DOI] [PubMed] [Google Scholar]
- 31.Marschke G, Sarauw A. Polymyxin inhalation therapeutic hazard. Ann Intern Med. 1971;74:144–5. doi: 10.7326/0003-4819-74-1-144. [DOI] [PubMed] [Google Scholar]
- 32.Owen RJ, Li J, Nation RL, Spelman D. In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J Antimicrob Chemother. 2007;59:473–7. doi: 10.1093/jac/dkl512. [DOI] [PubMed] [Google Scholar]
- 33.Poudyal A, Howden BP, Bell JM, Gao W, Owen RJ, Turnidge JD, et al. In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother. 2008;62:1311–18. doi: 10.1093/jac/dkn425. [DOI] [PubMed] [Google Scholar]
- 34.Bergen PJ, Bulitta JB, Forrest A, Tsuji BT, Li J, Nation RL. Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob Agents Chemother. 2010;54:3783–9. doi: 10.1128/AAC.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran TB, Cheah SE, Yu HH, Bergen PJ, Nation RL, Creek DJ, et al. Anthelmintic closantel enhances bacterial killing of polymyxin B against multidrug-resistant Acinetobacter baumannii. J Antibiot (Tokyo) 2016;69:415–21. doi: 10.1038/ja.2015.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abdul Rahim N, Cheah SE, Johnson MD, Yu H, Sidjabat HE, Boyce J, et al. Synergistic killing of NDM-producing MDR Klebsiella pneumoniae by two ‘old’ antibiotics—polymyxin B and chloramphenicol. J Antimicrob Chemother. 2015;70:2589–97. doi: 10.1093/jac/dkv135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tam VH, Schilling AN, Vo G, Kabbara S, Kwa AL, Wiederhold NP, et al. Pharmacodynamics of polymyxin B against Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2005;49:3624–30. doi: 10.1128/AAC.49.9.3624-3630.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Rayner CR, Nation RL, Owen RJ, Spelman D, Tan KE, et al. Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother. 2006;50:2946–50. doi: 10.1128/AAC.00103-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meletis G, Tzampaz E, Sianou E, Tzavaras I, Sofianou D. Colistin heteroresistance in carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother. 2011;66:946–7. doi: 10.1093/jac/dkr007. [DOI] [PubMed] [Google Scholar]
- 40.Bergen PJ, Forrest A, Bulitta JB, Tsuji BT, Sidjabat HE, Paterson DL, et al. Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant Pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother. 2011;55:5134–42. doi: 10.1128/AAC.05028-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fekety FR, Jr, Norman PS, Cluff LE. The treatment of Gram-negative bacillary infections with colistin. The toxicity and efficacy of large doses in forty-eight patients. Ann Intern Med. 1962;57:214–29. doi: 10.7326/0003-4819-57-2-214. [DOI] [PubMed] [Google Scholar]
- 42.Falagas ME, Rizos M, Bliziotis IA, Rellos K, Kasiakou SK, Michalopoulos A. Toxicity after prolonged (more than four weeks) administration of intravenous colistin. BMC Infect Dis. 2005;5:1. doi: 10.1186/1471-2334-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliveira MS, Prado GV, Costa SF, Grinbaum RS, Levin AS. Polymyxin B and colistimethate are comparable as to efficacy and renal toxicity. Diagn Microbiol Infect Dis. 2009;65:431–4. doi: 10.1016/j.diagmicrobio.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 44.Kubin CJ, Ellman TM, Phadke V, Haynes LJ, Calfee DP, Yin MT. Incidence and predictors of acute kidney injury associated with intravenous polymyxin B therapy. J Infect. 2012;65:80–7. doi: 10.1016/j.jinf.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 45.Akajagbor DS, Wilson SL, Shere-Wolfe KD, Dakum P, Charurat ME, Gilliam BL. Higher incidence of acute kidney injury with intravenous colistimethate sodium compared with polymyxin B in critically ill patients at a tertiary care medical center. Clin Infect Dis. 2013;57:1300–3. doi: 10.1093/cid/cit453. [DOI] [PubMed] [Google Scholar]
- 46.Phe K, Lee Y, McDaneld PM, Prasad N, Yin T, Figueroa DA, et al. In vitro assessment and multicenter cohort study of comparative nephrotoxicity rates associated with colistimethate versus polymyxin B therapy. Antimicrob Agents Chemother. 2014;58:2740–6. doi: 10.1128/AAC.02476-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorlí L, Luque S, Grau S, Berenguer N, Segura C, Montero MM, et al. Trough colistin plasma level is an independent risk factor for nephrotoxicity: a prospective observational cohort study. BMC Infect Dis. 2013;13:380. doi: 10.1186/1471-2334-13-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forrest A, Silveira FP, Thamlikitkul V, Garonzik SM, Mandragos K, Shoham S, et al. Toxicodynamics for colistin-associated changes in creatinine clearance. 54th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC); 5–9 September 2014; Washington, DC: ASM Press; 2014. [Google Scholar]
- 49.Rigatto MH, Behle TF, Falci DR, Freitas T, Lopes NT, Nunes M, et al. Risk factors for acute kidney injury (AKI) in patients treated with polymyxin B and influence of AKI on mortality: a multicentre prospective cohort study. J Antimicrob Chemother. 2015;70:1552–7. doi: 10.1093/jac/dku561. [DOI] [PubMed] [Google Scholar]
- 50.Dubrovskaya Y, Prasad N, Lee Y, Esaian D, Figueroa DA, Tam VH. Risk factors for nephrotoxicity onset associated with polymyxin B therapy. J Antimicrob Chemother. 2015;70:1903–7. doi: 10.1093/jac/dkv014. [DOI] [PubMed] [Google Scholar]
- 51.Suzuki T, Yamaguchi H, Ogura J, Kobayashi M, Yamada T, Iseki K. Megalin contributes to kidney accumulation and nephrotoxicity of colistin. Antimicrob Agents Chemother. 2013;57:6319–24. doi: 10.1128/AAC.00254-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu X, Chan T, Xu C, Zhu L, Zhou QT, Roberts KD, et al. Human oligopeptide transporter 2 (PEPT2) mediates cellular uptake of polymyxins. J Antimicrob Chemother. 2016;71:403–12. doi: 10.1093/jac/dkv340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Azad MA, Finnin BA, Poudyal A, Davis K, Li J, Hill PA, et al. Polymyxin B induces apoptosis in kidney proximal tubular cells. Antimicrob Agents Chemother. 2013;57:4329–35. doi: 10.1128/AAC.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eadon MT, Hack BK, Alexander JJ, Xu C, Dolan ME, Cunningham PN. Cell cycle arrest in a model of colistin nephrotoxicity. Physiol Genomics. 2013;45:877–88. doi: 10.1152/physiolgenomics.00076.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]