

Abstract
A broad range of redox-regulated proteins undergo reversible disulfide bond formation on oxidation-prone cysteine residues. Heightened reactivity of the thiol groups in these cysteines also increases susceptibility to modification by organic electrophiles, a property that can be exploited in the study of redox networks. Here, we explored whether divinyl sulfone (DVSF), a thiol-reactive bifunctional electrophile, cross-links oxidant-sensitive proteins to their putative redox partners in cells. To test this idea, previously identified oxidant targets involved in oxidant defense (namely, peroxiredoxins, methionine sulfoxide reductases, sulfiredoxin, and glutathione peroxidases), metabolism, and proteostasis were monitored for cross-link formation following treatment of Saccharomyces cerevisiae with DVSF. Several proteins screened, including multiple oxidant defense proteins, underwent intermolecular and/or intramolecular cross-linking in response to DVSF. Specific redox-active cysteines within a subset of DVSF targets were found to influence cross-linking; in addition, DVSF-mediated cross-linking of its targets was impaired in cells first exposed to oxidants. Since cross-linking appeared to involve redox-active cysteines in these proteins, we examined whether potential redox partners became cross-linked to them upon DVSF treatment. Specifically, we found that several substrates of thioredoxins were cross-linked to the cytosolic thioredoxin Trx2 in cells treated with DVSF. However, other DVSF targets, like the peroxiredoxin Ahp1, principally formed intra-protein cross-links upon DVSF treatment. Moreover, additional protein targets, including several known to undergo S-glutathionylation, were conjugated via DVSF to glutathione. Our results indicate that DVSF is of potential use as a chemical tool for irreversibly trapping and discovering thiol-based redox partnerships within cells.
Keywords: thiol, disulfide, electrophile, cross-linker, thioredoxin, peroxiredoxin, methionine sulfoxide reductase, sulfiredoxin, glutathione peroxidase, glutathionylation
Graphical abstract

Introduction
Elevated levels of oxidants play a key role in pathogen defense, development, and degeneration associated with aging and numerous age-related diseases [1-3]. The reactive oxygen species (ROS) formed during these processes oxidize a variety of molecular targets, including DNA, lipids, and proteins, thereby leading to alterations in cellular homeostasis and, when damage is pronounced, cellular dysfunction and death [4, 5]. Protein targets of ROS are commonly modified on specific cysteine residues that contribute to the protein’s function [6-8]. A cysteine residue’s susceptibility to oxidation is influenced by many factors, including its accessibility and an appropriate microenvironment that lowers its thiol pKa to form a thiolate [8, 9]. Following oxidation of a cysteine thiolate to a sulfenic acid, the sulfenic acid can react with another thiol in a protein or in glutathione to form a disulfide bond or may, in some instances, condense with the peptide backbone to form a sulfenamide [10]; such modifications are reversible by thiol reductases (e.g., thioredoxins, glutaredoxins) [11]. However, more extensive oxidation of cysteine thiols to sulfinic acids is considered largely irreversible [10]. Cysteine sulfinic acids are thought to undergo reduction only in a subset of peroxiredoxins that are recognized and repaired by sulfiredoxins [12].
Because cysteine oxidation often serves as a regulatory post-translational modification, considerable attention has been focused on identifying oxidation-prone proteins within the proteome [13, 14]. Proteome-wide screens have revealed a number of proteins undergoing sulfenylation, S-glutathionylation, and disulfide formation [15-22], as well as proteins that are redox partners of thioredoxins [23-28]. The proteins identified from these studies participate in a variety of cellular processes, including oxidant defense, metabolism, gene expression, maintenance of cell structure, and proteostasis. Although distinct networks of proteins that partner in oxidant defense and other processes have emerged from this work, the redox partnerships in certain cases have not been fully elucidated, indicating a need for alternative methodological approaches to establish such relationships.
Many of the same protein targets of ROS are prone to alkylation by thiol-targeted electrophiles, due to the heightened reactivity and accessibility of their oxidation-prone cysteines [29, 30]. Indeed, there is overlap between the proteins identified as oxidant and electrophile targets in many species [31]. For example, oxidant-defense proteins that employ Cys-based mechanisms for peroxide detoxification and disulfide reduction exhibit heightened reactivity toward numerous thiol-reactive electrophiles [32-38]. Moreover, work from our laboratory and others has revealed that several proteins which participate in a prominent oxidant defense relay involving peroxiredoxins, thioredoxins, and thioredoxin reductases undergo cross-linking to one another at their redox centers following exposure to homobifunctional electrophiles [39-42]. Cross-linking of these partners in Saccharomyces cerevisiae was particularly pronounced with the bifunctional electrophile divinyl sulfone (DVSF) [39].
Based on our earlier findings, we explored whether DVSF, due to its small size and irreversible reactivity with thiols, traps redox-active cysteine pairs that normally undergo reversible disulfide formation in oxidant-sensitive proteins. To this end, we tested whether a panel of oxidation-prone proteins underwent cross-linking in S. cerevisiae upon treatment with DVSF. Several of the DVSF target proteins that we identified formed cross-links with thioredoxins (their proposed intracellular reductant), whereas others formed intra-protein cross-links or were conjugated through DVSF to glutathione (GSH). Such a chemical approach may be useful in characterizing novel redox partnerships in and between proteins across a broad range of species.
Materials and Methods
Cloning and Mutagenesis of Candidate Genes
S. cerevisiae genes encoding oxidant-sensitive proteins were amplified from genomic DNA using standard PCR procedures and primers indicated in Supplementary Table 1. FLAG tags were incorporated at the N-terminus for most proteins. C-terminal FLAG tags were incorporated into proteins localized to mitochondria (i.e., Prx1 and Mxr2) or when poor expression was observed with the N-terminal tag (i.e., Mxr1). PCR products were subjected to restriction digestion with the appropriate restriction enzymes and ligated into the yeast expression vector p416-GPD or the bacterial protein expression vector pET45b (Novagen) using standard molecular techniques. Cloning was verified by restriction analysis and DNA sequencing of the full gene. Site-directed mutagenesis of genes encoding DVSF targets was performed by PCR with mutagenic primers and DpnI digestion of the template vector as described in the QuikChange PCR Method (Agilent). Primers used for site-directed mutagenesis are included in Supplementary Table 2. All mutations were confirmed by DNA sequencing.
S. cerevisiae Strains and Culture
Yeast strains used in this study are described in Supplementary Table 3. A strain lacking cytosolic thioredoxins (trx1Δ trx2Δ or trxΔ) was generated by disrupting the TRX2 locus with a HIS3 cassette in the trx1Δ strain from the yeast deletion library (Open Biosystems). All deletion strains were genotyped by PCR and analyzed for peroxide sensitivity. BY4741 and gsh1Δ strains were maintained in YPD culture medium, which has adequate GSH for survival of the gsh1Δ strain [43]. Strains expressing FLAG-tagged proteins (encoded by the corresponding p416-GPD clones) were maintained in synthetic complete medium lacking uracil (SC-Ura) containing 2% (w/v) glucose (Sunrise Science).
Cell Treatment, Protein Lysis, and Western Blotting to Detect DVSF-Mediated Cross-Linking
Cells grown to mid-log phase were treated with DVSF (Sigma) for 1 h at 30°C. In some experiments, cells were pre-incubated with H2O2 (Sigma) or tert-butylhydroperoxide (TBHP, Sigma). Cells were lysed by vortexing with acid-washed glass beads in a buffer containing 20 mM Tris-HCl (pH 8.0), 0.5 mM Na2EDTA, 10% glycerol, 50 mM NaCl, and a protease inhibitor cocktail as described previously [39]. Protein concentrations in lysates were determined using a Bradford assay (Sigma) with bovine serum albumin (Pierce) as a standard. Lysates were resolved on SDS-PAGE under reducing conditions and transferred to polyvinyl difluoride membrane. Membranes were blocked with TBS-T buffer (100 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.1% (v/v) Tween 20) containing 5% (w/v) non-fat dry milk. Membranes were incubated with primary antibodies against the FLAG tag (M2, mouse monoclonal, Sigma), Pgk1 (mouse monoclonal, Invitrogen), glutathione (mouse monoclonal, Santa Cruz), protein A (which detects a tandem affinity purification (TAP) tag; rabbit polyclonal, Sigma), and green fluorescent protein (GFP, rabbit polyclonal, Invitrogen) at 4°C overnight. Subsequently, membranes were washed three times for 10 min with TBS-T, exposed to appropriate horseradish peroxidase-linked secondary antibodies (Cell Signaling Technologies) for 45 min, washed with TBS-T four times for 15 min, and visualized with chemiluminescent detection.
Immunoprecipitation of FLAG-Tagged Proteins
The immunoprecipitation procedure was adapted from a procedure described previously [39]. Briefly, protein lysates from DVSF-treated yeast cultures (100 μg) were diluted in lysis buffer to 600 μL and subjected to immunoprecipitation with 8 μL EZView anti-FLAG beads (Sigma) on a tube rotator for 4 h at 4°C. Beads were pelleted and washed six times with lysis buffer (20 mM Tris-HCl (pH 8.0), 0.5 mM Na2EDTA, 10% glycerol, 50 mM NaCl) supplemented with 0.1% (w/v) SDS and 0.5% (w/v) sodium deoxycholate. Elution of immunoprecipitated proteins was carried out at room temperature for 30 min with 0.5 mg/mL 3X FLAG peptide dissolved in a buffer containing 50 mM Tris-HCl (pH 7.5) and 150 mM NaCl. The supernatant containing the eluant was recovered and subjected to analysis by Western blot with the indicated antibodies.
Purification of Ahp1 Interface and Active Site Mutants
pET45b-Ahp1 expression constructs were transformed into Rosetta DE3 cells (Novagen). Cultures of transformants were grown at 37°C to mid-log phase in 400 mL LB broth containing 100 μg/mL ampicillin, prior to induction for 6 h at 37°C with 1 mM isopropyl β-D-thiogalactopyranoside. Cells were pelleted, and His-tagged proteins were purified using a Fast Start Ni-NTA kit (Qiagen) according to manufacturer’s instructions. Eluted proteins were desalted on a PD25 Sephadex gel filtration column (GE Healthcare) equilibrated with 50 mM Tris-HCl (pH 7.5), 2 mM DTT, 10% glycerol, and protease inhibitor cocktail (G Biosciences) and, upon confirming purity by SDS-PAGE, frozen at −80°C. Proteins were quantified at A280 using an estimated extinction coefficient of 37,930 M−1•cm−1 (http://web.expasy.org/protparam/).
Analysis of Ahp1 Variant Proteins
To evaluate oligomeric states, purified Ahp1 dimer interface mutants (20 μM) were resolved on 4-16% Bis-Tris native PAGE gels (Invitrogen) run at 150 V. Following electrophoresis, proteins were detected by staining with Coomassie blue. For cross-linking experiments with purified Ahp1, recombinant proteins were reduced with 20 mM DTT at 37°C for 1 h prior to desalting on a BioSpin6 column (Biorad) equilibrated with 50 mM HEPES-OH (pH 8.0). Protein cross-linking reactions (20 μL) were carried out with 10 μM purified protein, 50 mM HEPES-OH (pH 8.0), 150 mM NaCl, 2.5 mM Na2EDTA, and varying doses of DVSF (or DMSO, vehicle control) for 3 h at 37°C. Reactions were quenched by boiling in reducing SDS-PAGE sample buffer prior to resolution by SDS-PAGE. Proteins were visualized with Coomassie blue. To monitor S-glutathionylation in Ahp1 bearing individual cysteine to alanine substitutions, purified recombinant proteins were reduced with 20 mM DTT at 37°C for 1 h prior to desalting on a BioSpin6 column equilibrated with 50 mM Tris-HCl (pH 8.0). Protein cross-linking reactions (20 μL) were carried out with 10 μM purified protein, 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 2.5 mM Na2EDTA, and varying doses of oxidized GSH (GSSG) for 20 min at 37°C. Reactions were terminated by incubation with 10 mM N-ethylmaleimide for 10 min at 37°C. Proteins were denatured by boiling in non-reducing SDS-PAGE sample buffer prior to resolution by SDS-PAGE. S-glutathionylation was detected by Western blot; protein loading and intersubunit disulfide formation were visualized with Coomassie blue.
Toxicity Assays with tert-Butyl Hydroperoxide
Cultures of BY4741 or ahp1Δ transformed with empty vector (p416-GPD) or Ahp1 expression constructs were grown overnight in uracil-dropout medium to saturation at 30°C. Yeast cultures were diluted to an OD600 of 0.5, from which 10-fold serial dilutions were prepared. Dilutions were plated onto YPD medium lacking or containing 2 mM TBHP. Plates were incubated at 30°C for 48 h prior to growth analysis to measure peroxide sensitivity.
Results
Cross-Linking of Oxidation-Prone Proteins in Cells Treated with DVSF
Previously, we reported that DVSF cross-links the cytosolic thioredoxin Trx2 to two of its major redox partners, thioredoxin reductase (Trr1) and an abundant 2-Cys peroxiredoxin (Tsa1) in S. cerevisiae [39]. Based on these results, we hypothesized that other oxidation-prone proteins undergo DVSF-mediated cross-linking to potential redox partners. To test this hypothesis, we screened a panel of 19 oxidant-sensitive FLAG-tagged proteins that participate in a number of different cellular processes (oxidant defense (peroxiredoxins, methionine sulfoxide reductases, sulfiredoxin, and glutathione peroxidases), metabolism, and proteostasis) [17, 19, 21]. We monitored whether each of these proteins underwent a molecular weight shift (indicative of cross-linking) when isolated from cells treated with a cytotoxic dose of DVSF [44, 45]. Under these conditions, each cytosolic peroxiredoxin (Tsa1, Tsa2, and Ahp1), each methionine sulfoxide reductase (Mxr1, Mxr2, and Ykg9), sulfiredoxin (Srx1), a glutathione peroxidase (Gpx1), isopropylmalate isomerase (Leu1), and ribonucleotide reductase (Rnr1) formed intermolecular cross-links, as detected by the migration of proteins at a higher molecular weight when compared with the unmodified, monomeric form (Fig. 1). In addition, a fraction of the Mxr1, Ykg9, Gpx2, and Gpx3 expressed electrophoresed more quickly in lysates from cells treated with DVSF (Fig. 1), suggesting that an intramolecular cross-link forms in these proteins similar to what has been observed with intramolecular disulfide formation [46, 47]. Several proteins, including a mitochondrial peroxiredoxin (Prx1), a nuclear peroxiredoxin (Dot5), a glyceraldehyde-3-phosphate dehydrogenase (Tdh3), a ribosomal small subunit protein (Rps5), two molecular chaperones (Sse1 and Ssa1), and an ubiquitin-conjugating enzyme (Ubc1), did not undergo appreciable molecular weight changes upon DVSF treatment, indicating that they may not be readily cross-linked to other proteins or internally cross-linked under these conditions. Therefore, we focused most of our subsequent work on proteins that underwent pronounced molecular weight shifts upon treatment with DVSF.
Figure 1. Cross-Linking of Multiple Oxidant-Sensitive Proteins in Cells Treated with DVSF.

(A) Scheme of DVSF-mediated thiol cross-linking. (B) Log-phase cells expressing FLAG-tagged forms of oxidant-sensitive proteins were treated with vehicle (DMSO) or 1 mM DVSF for 1 h at 30°C. Protein lysates (10-20 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. An asterisk (*) represents a background band that appears with the FLAG-antibody in certain blots. A double asterisk (**) represents a species of the protein that electrophoreses more quickly, suggesting intramolecular cross-linking. Results are representative of three independent experiments.
DVSF-Mediated Cross-Linking through Modification of Redox-Active Thiols
To ascertain whether redox-active cysteines are responsible for cross-linking, we introduced individual alanine substitutions at all cysteine residues in the primary sequence for several of the proteins we identified as DVSF targets. In Ahp1, substitution of either the resolving or the peroxidatic cysteine (Cys31 and Cys62, respectively) to alanine prevented cross-linking by DVSF (Fig. 2), implying that the principal redox-active cysteine residues in the active site were modified by DVSF [48]. Likewise, alanine substitutions of the primary catalytic residues in Srx1 (Cys84) and Gpx3 (Cys36 and Cys82) decreased cross-linking with DVSF (Fig. 2) [46, 49, 50]. In the case of Ykg9, an internal redox relay involving Cys91, Cys101, and Cys125 has been proposed, with Cys125 playing a key role in catalysis [51, 52]. While the cross-linking patterns with Ykg9 mutants were more complex, internal cross-linking of Ykg9 by DVSF was decreased when Cys125 was changed to Ala (Supp. Fig. 1). To provide further support that the redox-active cysteines of DVSF targets are involved in cross-linking, we tested whether a subset of these proteins undergo cross-linking in cells first treated with oxidants (either H2O2 or TBHP, an organic peroxide and preferential substrate for Ahp1 [48]). Pre-incubation of cells with peroxides decreased DVSF-mediated cross-linking of Tsa1, Srx1, and Ahp1 (Fig. 3), suggesting that they are incapable of reacting with DVSF when in their oxidized forms. Collectively, these results suggest that DVSF modifies key oxidation-prone cysteine residues in its target proteins to promote cross-linking.
Figure 2. Involvement of Redox-Active Cys Residues in Cross-Linking of DVSF Targets.

Log-phase cells expressing wild-type (wt) proteins or ones with individual Cys-to-Ala substitutions were treated with DMSO (vehicle) or 1 mM DVSF for 1 h at 30°C. Protein lysates (10-20 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. An asterisk (*) represents a background band that appears with the FLAG-antibody in certain blots. A double asterisk (**) indicates potential intramolecular cross-linking in the protein. Results are representative of three independent experiments.
Figure 3. Decreased Cross-Linking of DVSF Targets in Cells Pretreated with H2O2.
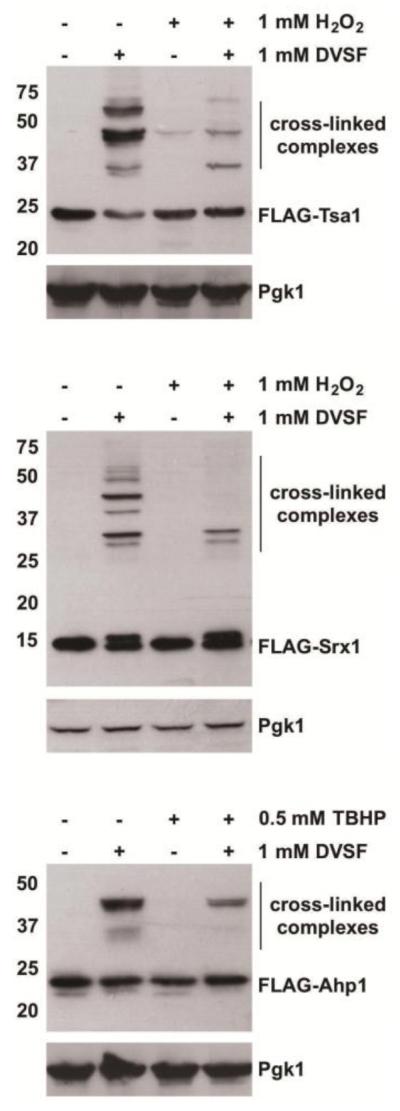
Log-phase cells expressing DVSF targets were incubated for 10 min with either vehicle (H2O or DMSO) or oxidant (1 mM H2O2 or 0.5 mM TBHP, respectively) prior to treatment for 1 h at 30°C with DMSO (vehicle) or 1 mM DVSF. Protein lysates (10 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. An asterisk (*) represents a background band that appears with the FLAG-antibody in certain blots. Results are representative of three independent experiments.
Stabilizing Interactions between Proteins Involved in Redox Networks with DVSF
Previously, we found that Trx2 formed cross-linked complexes with known redox partners, Tsa1 and Trr1, following treatment of cells with DVSF [39]. Since both Tsa1 and Trr1 are abundant proteins [53], we sought to determine whether other low-abundance Trx2 interaction partners–in this case, other Trx2 substrates–undergo cross-linking to Trx2 when exposed to DVSF (Fig. 4A). Several of the proteins that formed intermolecular cross-links upon treatment with DVSF in our initial screen were expressed in a yeast strain lacking the two cytosolic thioredoxins (trx1Δ trx2Δ, labeled as trxΔ). When these strains were treated with DVSF, we observed that Tsa1, Tsa2, and Ykg9 formed fewer cross-linked species compared with the wild-type control (BY4741) (Fig. 4B), suggesting that thioredoxins potentially form one or more cross-linked complexes with these substrates in cells treated with DVSF. Likewise, Srx1, which interacts with both cytosolic peroxiredoxins (Tsa1 and Tsa2) and thioredoxins (Trx1 and Trx2), exhibited differential DVSF cross-linking patterns with protein targets in both trx1Δ trx2Δ and tsa1Δ tsa2Δ (labeled as tsaΔ) backgrounds, implying that these proteins become cross-linked to Srx1 upon DVSF treatment. To further support our findings, the FLAG-tagged proteins were co-expressed in a strain containing a TAP-tagged Trx2 (Trx2-TAP) for co-immunoprecipitation experiments. Following treatment of these strains with DVSF, Trx2-TAP co-immunoprecipitated in cross-linked complexes with FLAG-tagged Tsa1, Tsa2, Ykg9, and Srx1 (Fig. 4C). Moreover, complexes of cross-linked Tsa1 which resolved at a similar molecular weight as some Srx1 complexes were isolated upon immunoprecipitation of Srx1 (Supp. Fig. 2). While we were able to isolate cross-linked complexes of Trx2 with particular substrates, not all DVSF targets studied (e.g., Ahp1) underwent extensive cross-linking with Trx2, despite being known substrates for thioredoxins (Supp. Fig. 3). The latter result prompted us to examine other potential redox partners, including cysteine redox pairs within an individual protein’s structure and small molecule thiols like GSH, that might become cross-linked to these proteins in lieu of thioredoxin upon DVSF treatment.
Figure 4. Cross-Linking of Selected DVSF Targets to Thioredoxin.

(A) Scheme depicting cross-linking between Trx2 and its interaction partners. (B) Indicated proteins were expressed in wild-type (BY4741), trx1Δ trx2Δ (labeled as trxΔ) or, in the case of Srx1, tsa1Δ tsa2Δ (labeled as tsaΔ) cells and treated with DMSO (vehicle) or 1 mM DVSF for 1 h at 30°C. Protein lysates (10-20 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. Dots (•) represent potential intermolecular cross-links to Trx1 or Trx2, whereas an arrow head (<) indicates potential cross-links between Srx1 and Tsa1 or Tsa2. (C) FLAG-tagged proteins were co-expressed in yeast containing a TAP-tagged Trx2 (Trx2-TAP) and treated with DMSO (vehicle) or 1 mM DVSF for 1 h at 30°C. FLAG-tagged proteins were immunoprecipitated from cell lysates (100 μg) and detected, along with Trx2-TAP isolated in cross-linked complexes, via Western blot. Results are representative of three independent experiments.
Studying Redox Centers within Individual Proteins with DVSF
Since the two cysteine residues that form an intersubunit disulfide bond in Ahp1 (Cys31 and Cys62) are also required for cross-linking by DVSF (Fig. 2), we sought to disrupt this natural redox pairing in the protein by decreasing dimer formation. Two conserved phenylalanine residues (Phe58 and Phe95) reside at the interface of the Ahp1 dimer (Fig. 5A, Supp. Fig. 4) [48]. We hypothesized that these residues allow for hydrophobic and aromatic stacking interactions to promote dimer assembly. Therefore, Phe58 and Phe95 were mutated to alanine or leucine to investigate a potential link between Ahp1 oligomerization and cellular protection against organic peroxides. Upon substitution of both Phe58 and Phe95 to either alanine or leucine, purified recombinant Ahp1 exhibited decreased subunit dimerization by native PAGE, whereas dimer disruption was less pronounced with individual mutations at these sites (Fig. 5B, Supp. Fig. 5A). Alterations of these aromatic residues, especially when introduced in tandem, compromised Ahp1’s ability to protect against TBHP when expressed in an ahp1-deficient (ahp1Δ) background (Fig. 5C, Supp. Fig. 5B), suggesting that Phe58 and Phe95 collectively play a structural role in dimer formation and are critical for the protein’s function in cells. Using the Ahp1 interface mutants, we tested whether the protein undergoes inter-subunit cross-linking with DVSF (Fig. 5D). Combined substitution of Phe58 and Phe95 to alanine or leucine at the dimer interface decreased inter-subunit cross-linking of purified Ahp1 by DVSF in vitro (Fig. 5E, Supp. Fig. 5C). Likewise, these mutant Ahp1 proteins were cross-linked less readily by DVSF in cells (Fig. 5F, Supp. Fig. 5D). Collectively, these results suggest a correlation between the oligomeric state of Ahp1 and susceptibility to DVSF-mediated intersubunit cross-linking, but they do not rule out the possibility that shifting Ahp1 toward a monomeric state influences the reactive nature of its active site cysteines and thereby limits peroxide clearance and electrophilic modification. On a more general level, however, our studies with the Ahp1 dimer indicate that DVSF has utility in probing pairs of redox-active Cys residues within individual proteins in cells.
Figure 5. Disruption of the Redox Center at the Dimer Interface of Ahp1 Compromises Cellular Defense against Organic Peroxides and Impairs Cross-Linking by DVSF.

(A) Structure of Ahp1 depicting catalytic Cys residues (C62 (peroxidatic Cys) and C31 (resolving Cys)) and conserved Phe residues at the dimer interface. Protein structures were generated with Chimera (https://www.cgl.ucsf.edu/chimera/) using PDB 4DSR. (B) Purified recombinant Ahp1 (wild-type (wt) or dimer interface variants, 20 μM) were resolved on native-PAGE and detected with Coomassie blue to determine oligomeric state. (C) Wild-type (wt) or mutant forms of Ahp1 were expressed in ahp1Δ yeast. Serial dilutions of these cultures and corresponding controls were grown on YPD medium containing 2 mM TBHP for 48 h at 30°C to determine the effect of dimer interface disruption on oxidant defense. (D) Scheme depicting proposed inter-subunit cross-linking in Ahp1 by DVSF. (E) Purified Ahp1 proteins (10 μM) were treated with increasing concentrations of DVSF for 3 h at 37°C, prior to electrophoresis on SDS-PAGE and detection by staining with Coomassie blue. (F) Log-phase yeast cells expressing Ahp1 variants were treated with 1 mM DVSF for 1 h at 30°C. Protein lysates (10-20 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. Results for all experiments are representative of three independent trials.
Exploring Putative Glutathionylation Targets with DVSF
While some proteins form intermolecular disulfide bonds with other thiol oxidoreductases (e.g., thioredoxins), many proteins also undergo reversible redox chemistry with small molecule thiols like GSH [54]. Therefore, we explored whether DVSF could be used to detect GSH conjugates of several proteins that are prone to S-glutathionylation during oxidative stress (Fig. 6A). We found that protein-GSH conjugates increased in a dose-dependent fashion following DVSF treatment (Fig. 6B). In yeast lacking the ability to synthesize GSH de novo (gsh1Δ) [43], GSH underwent adduction to fewer proteins in the presence of DVSF (Fig. 6C). Moreover, several yeast proteins with mammalian orthologs that are susceptible to regulation by glutathionylation, including Tdh3, Tsa1, Ssa1, Ahp1, Gpx1, and Srx1, were conjugated to GSH in cells treated with DVSF (Fig. 6D and E, Supp. Fig. 6) [15, 16, 55-57]. In addition, several putative targets for glutathionylation, including Rnr1 and Leu1, were cross-linked to GSH upon DVSF treatment (Supp. Fig. 6). In contrast, we were unable detect GSH conjugates of the glutathione peroxidases Gpx2 and Gpx3 (both of which undergo strong intramolecular cross-linking) or of the proteostasis factors Sse1, Ubc1, or Rps5 in DVSF-treated cells, indicating that certain proteins in our panel are more susceptible to this modification than others (Supp. Fig. 7).
Figure 6. Cross-Linking of GSH to Redox-Active Proteins in Cells Treated with DVSF.

(A) Scheme depicting GSH cross-linking to putative DVSF targets. (B) Log-phase yeast cells (BY4741) were treated with increasing doses of DVSF for 1 h at 30°C. Protein lysates (10 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. (C) Log-phase yeast cells (BY4741 or gsh1Δ) were treated with 1 mM DVSF for 1 h at 30°C. Protein lysates (10 μg) were resolved by SDS-PAGE, transferred to PVDF membrane, and detected with an antibody against the FLAG-tag or Pgk1 (loading control) via Western blot. (D and E) Cells expressing FLAG-tagged proteins were treated with 1 mM DVSF for 1 h at 30°C. FLAG-tagged proteins were immunoprecipitated from cell lysates (100 μg). Western blots were conducted to determine isolation of FLAG-tagged protein and its conjugation to GSH. Arrows point to protein-glutathione conjugates. (F) Purified Ahp1 variants (10 μM) were incubated with varying concentrations of GSSG for 20 min, prior to alkylation of free cysteines with N-ethylmaleimide and resolution by non-reducing SDS-PAGE. S-glutathionylation was monitored by Western blot. Ahp1 levels and oxidation were visualized with Coomassie blue. Results are representative of two-three independent experiments.
We further investigated whether DVSF-mediated GSH cross-linking to Ahp1 was dependent on the presence of either of its catalytic cysteines. We found that GSH conjugation to Ahp1 in DVSF-treated cells did not occur on a variant lacking the resolving cysteine (Cys31, Fig. 6E). In contrast, strong cross-linking to GSH was observed in the wild-type protein and in Ahp1 variants bearing cysteine substitutions at other sites. Moreover, purified Ahp1 bearing an alanine substitution at Cys31 did not undergo direct S-glutathionylation and/or further rearrangement to form the disulfide-linked dimer when incubated with oxidized GSH (GSSG, Fig. 6F), unlike the other forms of Ahp1 tested. Collectively, our results indicate that DVSF may be used as a proxy for detecting and predicting proteins subject to regulation by S-glutathionylation.
Discussion
In the present study, we used the thiol-reactive, bifunctional electrophile DVSF to determine whether a panel of previously described oxidant targets are subject to DVSF-mediated cross-linking. Several of the proteins screened underwent molecular weight shifts consistent with the formation of intermolecular cross-links (Tsa1, Tsa2, Ahp1, Mxr1, Mxr2, Ykg9, Srx1, Gpx1, Rnr1, and Leu1) and/or intramolecular cross-links (Mxr1, Ykg9, Gpx2, and Gpx3) upon treatment of cells with DVSF. Further study of a subset of these targets revealed that cross-linking depended upon the presence of their redox-active cysteines, many of which are functional cysteines used during catalysis. In investigating the potential cross-linked partners of candidate proteins, we found that DVSF cross-linked the cytosolic thioredoxin Trx2 to several of its known substrates (e.g., Tsa1, Tsa2, Ykg9, and Srx1). In other instances, DVSF covalently linked redox-active centers within individual proteins (e.g., Ahp1) or linked some S-glutathionylation-prone proteins (e.g., Tdh3) to GSH. Collectively, our results indicate that DVSF has utility as a chemical tool for trapping and mapping interactions between oxidant-sensitive network partners via irreversible modification of their oxidation-prone cysteines.
Overlapping Groups of Oxidant- and Electrophile-Sensitive Proteins
Although it has been reported that there are few commonalities between proteins undergoing diverse types of thiol post-translational modifications (e.g., sulfenylation, glutathionylation, nitrosylation, and acylation) [22], our results support a model wherein specific classes of oxidant- and electrophile-sensitive target proteins overlap [21, 31, 39]. In particular, the results of our screen indicate a prominent susceptibility of the cytosolic oxidant defense machinery to electrophilic modification by DVSF. Each cytosolic, thiol-dependent oxidoreductase involved in oxidant defense that we studied underwent a molecular weight shift in response to DVSF treatment, forming cross-links internally and/or with redox partners that function in the same network. Thus, our data support other findings wherein oxidant-defense enzymes that employ thiol-based mechanisms comprise a group of proteins targeted by alkylating agents, most likely due to the heightened reactivity of their active site thiols [31]. Such susceptibility of oxidant defense proteins to electrophilic modification commonly leads to irreversible inactivation of their function [4]. Indeed, many of these proteins are transcriptionally induced after exposure to electrophiles and oxidants, suggesting a common need for de novo versions of these proteins following thiol-targeted challenges [29, 58]. Moreover, the inhibition of multiple oxidant defense network proteins by electrophiles may account for the secondary oxidative stress component associated with electrophile-mediated stress [59-61]. Although many thiol-utilizing enzymes involved in oxidant defense are modified by both oxidants and electrophiles, further comparative work is needed to define more clearly the extent of overlap between electrophile- and oxidant-sensitive proteins throughout the entire proteome.
An Irreversible Trap for Redox-Active Cysteines That Form Disulfides
A number of global studies have yielded critical insights into the redox-regulated cysteine proteome, pinpointing which proteins and which cysteines within them are most susceptible to oxidation [7]. In addition, there have been several investigations aimed at identifying the thioredoxin substrate pool within the cell. The latter approaches frequently rely on trapping intermolecular disulfides between mutated thioredoxins (i.e., enzymes that lack a resolving cysteine) and their interaction partners [23, 25-28]. Despite major advances in the field from this earlier work, several technical challenges associated with understanding cysteine-based redox networks in cells remain. The reversibility and rearrangement of disulfides is advantageous from a standpoint of redox signaling and maintaining cellular homeostasis [62-64]. In support of the latter point, thioredoxin-deficient yeast are slow-growing normally and exhibit limited survival when exposed to low levels of oxidants (Supp. Fig. 8) [65]. However, the dynamic nature of disulfides can complicate proteomic analysis of redox partnerships within individual proteins and between two proteins in biological samples. Indeed, many proteomic approaches aimed at studying disulfide pairs in proteins require considerable sample work up ex vivo, often employing a step to block free thiols and, therefore, prevent undesirable disulfide rearrangements [7]. In addition, the limited bioavailability, bulkiness, and slow reactivity of many biorthogonal oxidation traps that react with cysteine sulfenic acids pose challenges in identifying oxidation-prone proteins in cells [30, 66, 67].
As a complement to existing approaches, we propose that DVSF may be a suitable reagent for studying interactions between specific cysteine residues that normally partner upon oxidation. DVSF is cell-permeable, has a small structure that could approximate the spatial constraints of a disulfide bond upon addition to thiols (unlike bulkier bifunctional agents), and yields cross-links within proteins over short treatment times (i.e., within one hour), suggesting relatively fast reactivity. Moreover, proteins cross-linked by DVSF are stable in the presence of standard thiol reductants, implying that they are incapable of rearrangement. While DVSF does not cross-link all redox partners to one another (e.g., Ahp1 to Trx2), the cross-linked complexes that form upon DVSF treatment may reflect a natural prioritization of partnerships in each of its target proteins. Thus, DVSF is of potential use in uncovering some, but not all, protein-based thiol partnerships in cells.
Competing Side Reactions in DVSF-Mediated Cross-Linking
One complication of our approach is that low molecular weight thiols (e.g., GSH via the action of GSH transferases) may react with one electrophilic center in DVSF and minimize intermolecular cross-linking between proteins in certain instances [68]. Indeed, many protein-GSH conjugates form upon DVSF treatment. GSH attachment, in one respect, negates the possibility of adding two different protein-derived thiols at the electrophilic centers in DVSF. However, such a conjugation to GSH may also be revealing about the ability of proteins with hyper-reactive cysteines to accommodate a GSH moiety in the site where DVSF is added, indicating that these proteins have the potential to be S-glutathionylated during oxidative stress. In support of this notion, several proteins that undergo S-glutathionylation in yeast (e.g., Tdh3) or that have orthologs susceptible to S-glutathionylation in other species (e.g., Ahp1, Tsa1, Ssa1) are among the proteins in which we observe GSH conjugation via DVSF [15, 16, 55]. Therefore, we propose that a DVSF-mediated cross-linking approach may be useful in predicting proteins that are prone to S-glutathionylation during oxidative stress in cells in some instances.
Applications for Using Bifunctional Electrophiles to Define Thiol Redox Networks
DVSF and other small bifunctional electrophiles cross-link proteins in a well-studied peroxide detoxification pathway at known sites of redox chemistry, supporting their use as tools in redox network biology [39-42]. The results presented herein expand these earlier findings and suggest a broader applicability of these reagents in studying additional redox partnerships, particularly in examining proteins involved in other aspects of oxidant defense. While our present work and earlier studies with cysteine cross-linkers have largely explored cross-linking reactions in established eukaryotic model systems and known redox networks, there are diverse, thiol-based redox homeostasis mechanisms in less-studied organisms wherein analytical approaches like the one we describe with DVSF may prove useful [6, 69, 70]. Moreover, we have mainly established this approach by studying redox partnerships localized within the cytosol; however, related disulfide relay mechanisms exist in other cellular compartments in both prokaryotes (e.g., periplasmic space) and eukaryotes (e.g., endoplasmic reticulum, chloroplasts, and mitochondria) that have not been interrogated fully [71-73]. In cases where redox interactions are poorly defined, cross-linking interacting proteins with DVSF or similar molecules may complement current methods for establishing key partnerships in thiol-based redox networks, thereby increasing our understanding of biological responses to oxidants during homeostasis and cellular stress.
Supplementary Material
Highlights.
A thiol-reactive cross-linker (DVSF) modifies many oxidation-prone proteins in cells.
DVSF traps interactions between thioredoxin and several known redox partners.
DVSF can be used to probe internal cysteine redox pairs within proteins.
DVSF may be used to predict proteins that undergo S-glutathionylation.
Acknowledgments
We thank Todd Lowther (Wake Forest School of Medicine) and Ah-lim Tsai (McGovern Medical School) for helpful suggestions and experimental assistance. Support for this work was provided by a Cottrell College Science Award from Research Corporation for Science Advancement (to J.D.W.), grant MF15-UMR06 from the Mindlin Foundation (to M.A.L. and J.D.W.), grant MF16-US03 from the Mindlin Foundation (to J.D.W.), funds from The College of Wooster (startup funds, Wilson Research Funds, the Copeland Fund for Independent Study, and Luce Funds for Distinguished Scholarship), and NIH Grant R01 GM074696 (to K.A.M.). K.M.A., M.G.K., M.A.L., A.H.W., J.M.P., and H.J.S. were supported by The College’s Sophomore Research Program, J.C. was supported by The College’s HHMI Science Education Program grant, and J.E.H. was supported by the Summer Undergraduate Research Program at the UT-Houston McGovern Medical School.
Abbreviations
- DVSF
divinyl sulfone
- ROS
reactive oxygen species
- GSH
glutathione
- TAP
tandom affinity purification
- GFP
green fluorescent protein
- TBHP
tert-butyl hydroperoxide
- WB
Western blot
- GSSG
oxidized glutathione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- [2].Dickinson BC, Chang CJ. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nathan C, Cunningham-Bussel A. Beyond oxidative stress: an immunologist's guide to reactive oxygen species. Nat. Rev. Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marnett LJ, Riggins JN, West JD. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J. Clin. Invest. 2003;111:583–593. doi: 10.1172/JCI18022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- [6].Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015;80:148–157. doi: 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem. Rev. 2013;113:4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pace NJ, Weerapana E. Diverse functional roles of reactive cysteines. ACS Chem. Biol. 2013;8:283–296. doi: 10.1021/cb3005269. [DOI] [PubMed] [Google Scholar]
- [9].Marino SM, Gladyshev VN. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012;287:4419–4425. doi: 10.1074/jbc.R111.275578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014;66:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- [12].Jonsson TJ, Lowther WT. The peroxiredoxin repair proteins. Subcell. Biochem. 2007;44:115–141. doi: 10.1007/978-1-4020-6051-9_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic. Biol. Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yang J, Carroll KS, Liebler DC. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics. 2016;15:1–11. doi: 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fratelli M, Demol H, Puype M, Casagrande S, Eberini I, Salmona M, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A, Vandekerckhove J, Gianazza E, Ghezzi P. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA. 2002;99:3505–3510. doi: 10.1073/pnas.052592699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shenton D, Grant CM. Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem. J. 2003;374:513–519. doi: 10.1042/BJ20030414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Le Moan N, Clement G, Le Maout S, Tacnet F, Toledano MB. The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J. Biol. Chem. 2006;281:10420–10430. doi: 10.1074/jbc.M513346200. [DOI] [PubMed] [Google Scholar]
- [18].Leichert LI, Gehrke F, Gudiseva HV, Blackwell T, Ilbert M, Walker AK, Strahler JR, Andrews PC, Jakob U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Brandes N, Reichmann D, Tienson H, Leichert LI, Jakob U. Using quantitative redox proteomics to dissect the yeast redoxome. J. Biol. Chem. 2011;286:41893–41903. doi: 10.1074/jbc.M111.296236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- [21].Marino SM, Li Y, Fomenko DE, Agisheva N, Cerny RL, Gladyshev VN. Characterization of surface-exposed reactive cysteine residues in Saccharomyces cerevisiae. Biochemistry. 2010;49:7709–7721. doi: 10.1021/bi100677a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gould NS, Evans P, Martinez-Acedo P, Marino SM, Gladyshev VN, Carroll KS, Ischiropoulos H. Site-specific proteomic mapping identifies selectively modified regulatory cysteine residues in functionally distinct protein networks. Chem. Biol. 2015;22:965–975. doi: 10.1016/j.chembiol.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Motohashi K, Kondoh A, Stumpp MT, Hisabori T. Comprehensive survey of proteins targeted by chloroplast thioredoxin. Proc. Natl. Acad. Sci. USA. 2001;98:11224–11229. doi: 10.1073/pnas.191282098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vignols F, Brehelin C, Surdin-Kerjan Y, Thomas D, Meyer Y. A yeast two-hybrid knockout strain to explore thioredoxin-interacting proteins in vivo. Proc. Natl. Acad. Sci. USA. 2005;102:16729–16734. doi: 10.1073/pnas.0506880102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lemaire SD, Guillon B, Le Marechal P, Keryer E, Miginiac-Maslow M, Decottignies P. New thioredoxin targets in the unicellular photosynthetic eukaryote Chlamydomonas reinhardtii. Proc Natl Acad Sci U S A. 2004;101:7475–7480. doi: 10.1073/pnas.0402221101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Go YM, Roede JR, Walker DI, Duong DM, Seyfried NT, Orr M, Liang Y, Pennell KD, Jones DP. Selective targeting of the cysteine proteome by thioredoxin and glutathione redox systems. Mol. Cell. Proteomics. 2013;12:3285–3296. doi: 10.1074/mcp.M113.030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ben-Lulu S, Ziv T, Admon A, Weisman-Shomer P, Benhar M. A substrate trapping approach identifies proteins regulated by reversible S-nitrosylation. Mol. Cell. Proteomics. 2014;13:2573–2583. doi: 10.1074/mcp.M114.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nakao LS, Everley RA, Marino SM, Lo SM, de Souza LE, Gygi SP, Gladyshev VN. Mechanism-based proteomic screening identifies targets of thioredoxin-like proteins. J. Biol. Chem. 2015;290:5685–5695. doi: 10.1074/jbc.M114.597245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rudolph TK, Freeman BA. Transduction of redox signaling by electrophile-protein reactions. Sci. Signal. 2009;2:re7. doi: 10.1126/scisignal.290re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Couvertier SM, Zhou Y, Weerapana E. Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta. 2014;1844:2315–2330. doi: 10.1016/j.bbapap.2014.09.024. [DOI] [PubMed] [Google Scholar]
- [31].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J. Biol. Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- [33].Fang J, Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J Am Chem Soc. 2006;128:1879–1885. doi: 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- [34].Moos PJ, Edes K, Cassidy P, Massuda E, Fitzpatrick FA. Electrophilic prostaglandins and lipid aldehydes repress redox-sensitive transcription factors p53 and hypoxia-inducible factor by impairing the selenoprotein thioredoxin reductase. J. Biol. Chem. 2003;278:745–750. doi: 10.1074/jbc.M211134200. [DOI] [PubMed] [Google Scholar]
- [35].Shibata T, Yamada T, Ishii T, Kumazawa S, Nakamura H, Masutani H, Yodoi J, Uchida K. Thioredoxin as a molecular target of cyclopentenone prostaglandins. J. Biol. Chem. 2003;278:26046–26054. doi: 10.1074/jbc.M303690200. [DOI] [PubMed] [Google Scholar]
- [36].Liu CX, Yin QQ, Zhou HC, Wu YL, Pu JX, Xia L, Liu W, Huang X, Jiang T, Wu MX, He LC, Zhao YX, Wang XL, Xiao WL, Chen HZ, Zhao Q, Zhou AW, Wang LS, Sun HD, Chen GQ. Adenanthin targets peroxiredoxin I and II to induce differentiation of leukemic cells. Nat. Chem. Biol. 2012;8:486–493. doi: 10.1038/nchembio.935. [DOI] [PubMed] [Google Scholar]
- [37].Muchowicz A, Firczuk M, Chlebowska J, Nowis D, Stachura J, Barankiewicz J, Trzeciecka A, Klossowski S, Ostaszewski R, Zagozdzon R, Pu JX, Sun HD, Golab J. Adenanthin targets proteins involved in the regulation of disulphide bonds. Biochem. Pharmacol. 2014;89:210–216. doi: 10.1016/j.bcp.2014.02.022. [DOI] [PubMed] [Google Scholar]
- [38].Soethoudt M, Peskin AV, Dickerhof N, Paton LN, Pace PE, Winterbourn CC. Interaction of adenanthin with glutathione and thiol enzymes: selectivity for thioredoxin reductase and inhibition of peroxiredoxin recycling. Free Radic. Biol. Med. 2014;77:331–339. doi: 10.1016/j.freeradbiomed.2014.09.025. [DOI] [PubMed] [Google Scholar]
- [39].Naticchia MR, Brown HA, Garcia FJ, Lamade AM, Justice SL, Herrin RP, Morano KA, West JD. Bifunctional electrophiles cross-link thioredoxins with redox relay partners in cells. Chem. Res. Toxicol. 2013;26:490–497. doi: 10.1021/tx4000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nguyen JB, Pool CD, Wong CY, Treger RS, Williams DL, Cappello M, Lea WA, Simeonov A, Vermeire JJ, Modis Y. Peroxiredoxin-1 from the human hookworm Ancylostoma ceylanicum forms a stable oxidized decamer and is covalently inhibited by conoidin A. Chem. Biol. 2013;20:991–1001. doi: 10.1016/j.chembiol.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jan YH, Heck DE, Malaviya R, Casillas RP, Laskin DL, Laskin JD. Cross-linking of thioredoxin reductase by the sulfur mustard analogue mechlorethamine (methylbis(2-chloroethyl)amine) in human lung epithelial cells and rat lung: selective inhibition of disulfide reduction but not redox cycling. Chem. Res. Toxicol. 2014;27:61–75. doi: 10.1021/tx400329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jan YH, Heck DE, Casillas RP, Laskin DL, Laskin JD. Thioredoxin cross-linking by nitrogen mustard in lung epithelial cells: formation of multimeric thioredoxin/thioredoxin reductase complexes and inhibition of disulfide reduction. Chem. Res. Toxicol. 2015;28:2091–2103. doi: 10.1021/acs.chemrestox.5b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Grant CM, MacIver FH, Dawes IW. Glutathione is an essential metabolite required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. Curr. Genet. 1996;29:511–515. doi: 10.1007/BF02426954. [DOI] [PubMed] [Google Scholar]
- [44].West JD, Stamm CE, Brown HA, Justice SL, Morano KA. Enhanced toxicity of the protein cross-linkers divinyl sulfone and diethyl acetylenedicarboxylate in comparison to related monofunctional electrophiles. Chem. Res. Toxicol. 2011;24:1457–1459. doi: 10.1021/tx200302w. [DOI] [PubMed] [Google Scholar]
- [45].Spencer MK, Radzinski NP, Tripathi S, Chowdhury S, Herrin RP, Chandran NN, Daniel AK, West JD. Pronounced toxicity differences between homobifunctional protein cross-linkers and analogous monofunctional electrophiles. Chem. Res. Toxicol. 2013;26:1720–1729. doi: 10.1021/tx400290j. [DOI] [PubMed] [Google Scholar]
- [46].Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB. A thiol peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell. 2002;111:471–481. doi: 10.1016/s0092-8674(02)01048-6. [DOI] [PubMed] [Google Scholar]
- [47].Mason JT, Kim SK, Knaff DB, Wood MJ. Thermodynamic basis for redox regulation of the Yap1 signal transduction pathway. Biochemistry. 2006;45:13409–13417. doi: 10.1021/bi061136y. [DOI] [PubMed] [Google Scholar]
- [48].Lian FM, Yu J, Ma XX, Yu XJ, Chen Y, Zhou CZ. Structural snapshots of yeast alkyl hydroperoxide reductase Ahp1 peroxiredoxin reveal a novel two-cysteine mechanism of electron transfer to eliminate reactive oxygen species. J. Biol. Chem. 2012;287:17077–17087. doi: 10.1074/jbc.M112.357368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- [50].Roussel X, Kriznik A, Richard C, Rahuel-Clermont S, Branlant G. Catalytic mechanism of Sulfiredoxin from Saccharomyces cerevisiae passes through an oxidized disulfide sulfiredoxin intermediate that is reduced by thioredoxin. J. Biol. Chem. 2009;284:33048–33055. doi: 10.1074/jbc.M109.035352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Le DT, Lee BC, Marino SM, Zhang Y, Fomenko DE, Kaya A, Hacioglu E, Kwak GH, Koc A, Kim HY, Gladyshev VN. Functional analysis of free methionine-R-sulfoxide reductase from Saccharomyces cerevisiae. J. Biol. Chem. 2009;284:4354–4364. doi: 10.1074/jbc.M805891200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kwak GH, Kim MJ, Kim HY. Cysteine-125 is the catalytic residue of Saccharomyces cerevisiae free methionine-R-sulfoxide reductase. Biochem. Biophys. Res. Commun. 2010;395:412–415. doi: 10.1016/j.bbrc.2010.04.036. [DOI] [PubMed] [Google Scholar]
- [53].Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- [54].Janssen-Heininger YM, Nolin JD, Hoffman SM, van der Velden JL, Tully JE, Lahue KG, Abdalla ST, Chapman DG, Reynaert NL, van der Vliet A, Anathy V. Emerging mechanisms of glutathione-dependent chemistry in biology and disease. J. Cell. Biochem. 2013;114:1962–1968. doi: 10.1002/jcb.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Fratelli M, Gianazza E, Ghezzi P. Redox proteomics: identification and functional role of glutathionylated proteins. Expert Rev. Proteomics. 2004;1:365–376. doi: 10.1586/14789450.1.3.365. [DOI] [PubMed] [Google Scholar]
- [56].Park JW, Mieyal JJ, Rhee SG, Chock PB. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 2009;284:23364–23374. doi: 10.1074/jbc.M109.021394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Peskin AV, Pace PE, Behring JB, Paton LN, Soethoudt M, Bachschmid MM, Winterbourn CC. Glutathionylation of the active site cysteines of peroxiredoxin 2 and recycling by glutaredoxin. J. Biol. Chem. 2016;291:3053–3062. doi: 10.1074/jbc.M115.692798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].West JD, Marnett LJ. Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem. Res. Toxicol. 2006;19:173–194. doi: 10.1021/tx050321u. [DOI] [PubMed] [Google Scholar]
- [59].Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Activation of stress signaling pathways by the end product of lipid peroxidation: 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J. Biol. Chem. 1999;274:2234–2242. doi: 10.1074/jbc.274.4.2234. [DOI] [PubMed] [Google Scholar]
- [60].Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J. Biol. Chem. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- [61].Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, Stern AM, Mandinova A, Schreiber SL, Lee SW. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [62].Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 2011;14:1065–1077. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Groitl B, Jakob U. Thiol-based redox switches. Biochim. Biophys. Acta. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Leichert LI, Dick TP. Incidence and physiological relevance of protein thiol switches. Biol. Chem. 2015;396:389–399. doi: 10.1515/hsz-2014-0314. [DOI] [PubMed] [Google Scholar]
- [65].Garrido EO, Grant CM. Role of thioredoxins in the response of Saccharomyces cerevisiae to oxidative stress induced by hydroperoxides. Mol. Microbiol. 2002;43:993–1003. doi: 10.1046/j.1365-2958.2002.02795.x. [DOI] [PubMed] [Google Scholar]
- [66].Gupta V, Carroll KS. Profiling the reactivity of cyclic C-nucleophiles towards electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 2016;7:400–415. doi: 10.1039/c5sc02569a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Baez NO, Reisz JA, Furdui CM. Mass spectrometry in studies of protein thiol chemistry and signaling: opportunities and caveats. Free Radic. Biol. Med. 2015;80:191–211. doi: 10.1016/j.freeradbiomed.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- [69].Van Laer K, Hamilton CJ, Messens J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013;18:1642–1653. doi: 10.1089/ars.2012.4964. [DOI] [PubMed] [Google Scholar]
- [70].Hemmis CW, Schildbach JF. Thioredoxin-like proteins in F and other plasmid systems. Plasmid. 2013;70:168–189. doi: 10.1016/j.plasmid.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mamathambika BS, Bardwell JC. Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 2008;24:211–235. doi: 10.1146/annurev.cellbio.24.110707.175333. [DOI] [PubMed] [Google Scholar]
- [72].Cho SH, Collet JF. Many roles of the bacterial envelope reducing pathways. Antioxid. Redox Signal. 2013;18:1690–1698. doi: 10.1089/ars.2012.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lu J, Holmgren A. The thioredoxin superfamily in oxidative protein folding. Antioxid. Redox Signal. 2014;21:457–470. doi: 10.1089/ars.2014.5849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.