

Abstract
Inflammatory bowel disease (IBD), including Crohn's disease (CD) and ulcerative colitis (UC), which is thought to result from immune-mediated inflammatory disorders, leads to high morbidity and health care cost. Fatty acid amide hydrolase (FAAH) is an enzyme crucially involved in the modulation of intestinal physiology through anandamide (AEA) and other endocannabinoids. Here we examined the effects of an FAAH inhibitor (FAAH-II), on dextran sodium sulphate (DSS)-induced experimental colitis in mice. Treatments with FAAH-II improved overall clinical scores by reversing weight loss and colitis-associated pathogenesis. The frequencies of activated CD4+ T cells in spleens, mesenteric lymph nodes (MLNs), Peyer's patches (PPs), and colon lamina propiria (LP) were reduced by FAAH inhibition. Similarly, the frequencies of macrophages, neutrophils, natural killer (NK), and NKT cells in the PPs and LP of mice with colitis declined after FAAH blockade, as did concentrations of systemic and colon inflammatory cytokines. Microarray analysis showed that 26 miRNAs from MLNs and 217 from PPs had a 1.5-fold greater difference in expression after FAAH inhibition. Among them, 8 miRNAs were determined by reverse-transcription polymerase chain reaction (RT-PCR) analysis to have anti-inflammatory properties. Pathway analysis demonstrated that differentially regulated miRNAs target mRNA associated with inflammation. Thus, FAAH-II ameliorates experimental colitis by reducing not only the number of activated T cells but also the frequency of macrophages, neutrophils, and NK/NKT cell, as well as inflammatory miRNAs and cytokine at effector sites in the colon. These studies demonstrate for the first time that FAAH-II inhibitor may suppress colitis through regulation of pro-inflammatory miRNAs expression.
Keywords: FAAH, microRNAs, inflammation, colitis, inflammatory bowel disease
1. Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn's disease (CD), are associated with aberrant regulation of the mucosal immune system, resulting in recruitment of inflammatory cells to the intestinal tract. IBD affects over a million people in the United States alone; its symptoms include abdominal pain, diarrhea, rectal bleeding, weight loss, and poor ability to digest food. The precise cause of IBD is unknown, but common agreement among scientists has converged on environmental and genetic factors, as well as immune dysregulation and, particularly the role of commensal microbiota in the intestine (Braegger and MacDonald, 1994; Dohi et al., 2000; Podolsky, 2002; Sartor, 2006; 2008). Abnormalities in the gut immune microenvironment, enteric persistent infection, trauma, and inflammation may initiate and cause the progression of IBD (Mayer and Collins, 2002).
Intestinal inflammation is normally associated with infiltration of immune cells, including macrophages, neutrophils, and type 1 T helper (Th1) cells into the colon, which secrete the proinflammatory cytokines TNF-α and IFN-γ (Strober et al., 2007; Strober et al., 2002). The available conventional treatments of IBD are useful and advance our understanding of the underlying pathologies, but patients are often resistant to these treatments and experience many side effects, justifying continued search for new, safe, and effective therapeutic approaches.
In recent years, there has been increasing recognition of the role of endogenous cannabinoid receptors 1-2 (CB1 and CB2) in the regulation of inflammation-associated with colitis. Anandamide (AEA) is the endogenous signaling lipid that binds and activates CB1 and CB2, receptors and AEA levels are tightly regulated by the catabolic enzyme FAAH. Further, FAAH is an integral membrane hydrolase with a single n-terminus transmembrane domain. Many distinct classes of FAAH inhibitors have been reported (Salaga et al., 2014). A study focusing on FAAH and intestinal inflammation demonstrated that CB1 receptor-deficient mice are more sensitive to experimental colitis than are control mice and that a CB1 antagonist increased the severity of experimental colitis (Massa et al., 2004). Other studies (Cluny et al.; Engel et al.; Kimball et al., 2006; Massa et al., 2004; Storr et al., 2008; Storr et al., 2009) have suggested that endogenous CB receptor activation may ameliorate colitis. Storr et al. (2008) showed that FAAH mRNA increased 3 days after trinitrobenzene sulfonic acid (TNBS) induction. Further, elevated levels of endogenous AEA resulting from deficiency or inhibition of FAAH are significantly more resistant to TNBS-induced colitis than are controls (Izzo and Sharkey, 2010). AEA can activate both CB1 and CB2 receptors and decrease the level of proinflammatory cytokines (Izzo and Camilleri, 2009).
MicroRNAs (miRNAs) are endogenous small RNA molecules 20-25 nucleotides in length; they regulate multiple genes by binding to target mRNAs, thereby controlling the stability and translation of protein coding mRNAs (Esteller, 2011; Guo et al., 2010). It has been established that cells of the innate and adaptive immune system express more than 100 miRNAs that are involved in mediating many functions, including cell proliferation and apoptosis. In addition, miRNAs are expressed in many developing tissues and are active in regulating inflammation (Ambros, 2004). Recent evidence suggests that miRNAs are differentially expressed in UC and have a key role as negative regulators of inflammation and innate immunity (Wu et al., 2008). miRNAs mir-21 and mir-216 are differentially expressed in active versus inactive UC. Further, it has been shown that induced miR-101b and miR-455 miRNAs mediates anti-inflammatory properties in colitis and associated colon cancer (Altamemi et al., 2014; Feng et al., 2012). However, the differential expression of miRNAs and their role in experimental colitis have not been investigated.
IBD is a major burden to both patients and society and there is an always need for safe and effective therapeutic options. The main hypothesis of this study is to determine the cellular and epigenetic mechanism of FAAH inhibitors mediated abrogation of experimental colitis. In this study, we examined changes in the severity of inflammation, cytokine levels, changes in the expression of miRNAs and immune function after FAAH inhibition using a dextran sodium sulphate (DSS) induced model of experimental colitis. Our studies demonstrated that FAAH-II inhibitor ameliorates colitis by suppressing inflammation through intervention of pro and or anti-inflammatory miRNAs at mucosal sites.
2. Materials and Methods
2.1 Animals
Female wild-type mice on C57BL/6 background aged 8~10 weeks were purchased from Jackson Laboratories (Bar Harbor, ME). The animals were housed and maintained in microisolator cages under conventional housing conditions at the University of South Carolina School of Medicine animal facility. Care and use of animals was overseen and approved by the Animal Resource Facility (ARF). The ARF is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care-International (AAALAC). Experimental groups consisted of six mice each; all studies were repeated three times.
2.2 Colitis induction by DSS and FAAH treatment
The FAAH-II inhibitor was purchased from EMD Millipore (Billercia, MA). Experimental colitis was induced using DSS as described previously (Singh et al., 2014; Singh et al., 2010). In brief, eight-week-old C57BL/6 naive group of mice received water alone, while mice in the colitic group received drinking water containing 3% DSS (MP Biomedical, LLC, Ohio) ad libitum for seven days. In the following seven days, mice in the colitic group received water without DSS. Initially, we did a dose-response experiment using 5, 10, 20, or 40 mg/kg body weight doses of FAAH-II, determining that 10 mg/kg, as compared to higher or lower doses, effectively suppresses colitis symptoms. Therefore, 10 mg/kg doses of FAAH-II were used throughout this study. We not only used the same lot of FAAH-II in all experiments, but we used same lot and dose of DSS as described earlier in our experimental model of colitis (Singh et al., 2014; Singh et al., 2010).
Mice were given either 100 μl of FAAH-II (10 mg/kg body weight; DSS+FAAH) or phosphate buffered saline (PBS; DSS+vehicle) beginning on the day after DSS induction and continuing every day until Day 14, the experimental end-point. The body weight of mice was monitored every day after DSS induction. During the 14 days of the experiment, we also monitored mice for clinical symptoms of colitis (diarrhea, stool consistency, and blood in fecal matter). At the end of the experiment, animals were sacrificed by ether vapor and their blood collected for measurement of cytokines and chemokines. Colon samples were isolated, washed with PBS, cut longitudinally, one part immediately kept in −80°C to make tissue homogenate for cytokine measurement and other part fixed in 10% formalin, then embedded in paraffin for histological analysis.
2.3 Cell isolation
At the end of the experiment, single-cell suspensions were prepared from the spleens, MLNs, PPs and colon lamina propiria (LP) from each group of mice. Cells were dissociated and RBCs lysed with lysis buffer (Sigma St. Louis, MO). After centrifugation, single-cell suspensions from spleens, MLNs, and PPs were passed through a sterile filter 70 μm (Sigma St. Louis, MO) to remove any cell debris. Cells were washed twice in RPMI 1640 (Sigma St. Louis, MO) and stored in media containing 5% fetal bovine serum (FBS) on ice until their use, which occurred within two hours for flow cytometry. The cells from colon LP were isolated as described previously (Singh et al., 2003). In brief, the colon was cut into 1-cm stripes and stirred in PBS containing 1mM EDTA at 37 °C for 30 min. Next, LP was isolated by digesting intestinal tissue with collagenase type IV (Sigma St. Louis, MO) in RPMI 1640 (collagenase solution) for 45 min at 37 °C with moderate stirring. After each 45 min interval, the released cells were centrifuged, stored in complete media and mucosal pieces were replaced with fresh collagenase solution for at least three times. LP cells were further purified using a discontinuous Percoll (Pharmacia, Uppsala, Sweden) gradient collecting at the 40–75% interface. The cells were maintained in complete medium consisting of RPMI 1640 supplemented with 10 ml/L of nonessential amino acids (Mediatech, Washington, DC), 1 mM sodium pyruvate (Sigma), 10 mM HEPES (Mediatech), 100 U/ml penicillin, 100 μg/ml streptomycin, 40 μg/ml gentamycin (Elkins-Sinn, Inc., Cherry Hill, NJ), 50 μM mercaptoethanol (Sigma) and 10 % FCS (Atlanta Biologicals).
2.4 Flow cytometry analysis
Cells were pre-blocked for Fc receptors (Becton Dickinson San Diego, CA) for 15 min at 4° C before cell-surface antigen staining. The cells were then washed with FACS staining buffer consisting of PBS with 1% fetal bovine serum (FBS). The cells were then stained with the appropriate antibodies (Abs). Cells were stained for 45 min with occasional shaking at 4° C with fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (H129.19), FITC-conjugated CD11b (M1/70), PE-conjugated anti-CD69 (H1.2F3), FITC-conjugated Ly6G (neutrophils), PE-conjugated anti-mouse CD11c (HL3), NK1.1, PE-conjugated anti-CD3 (145-2C11) (BD-PharMingen, San Diego, CA), and FITC-conjugated F4/80 (BM8), all of which were obtained from Biolegend (San Diego, CA). The cells were washed two times with FACS staining buffer resuspended in 500 μl of FACS buffer, then analyzed by flow cytometry (FC 500, Beckman Coulter Fort Collins, CO).
2.5 Colon anatomy and histology
Colon length was used to determine the protective effects of FAAH-II on morphology of the colon. Colons were isolated and the total length measured from naïve mice, vehicle-treated DSS colitis mice, and DSS colitis mice treated with FAAH-II. Tissue samples from colons were fixed in 10% formalin, dehydrated through ethanol, and embedded in paraffin. Fixed tissues were sectioned at 6 μm and stained with hematoxylin and eosin. Two independent trained pathologists, blind to the study, determined the histological changes and mucosal inflammation by assigning an inflammation score to each group of mice. Each colon segment, ascending, transverse, and descending, was scored from 0 to 4, following well-established criteria (Singh et al., 2003). In brief, grade 0 indicated no change from normal tissue; grade 1, one or a few multifocal mononuclear cell infiltrates in the lamina propria (LP); grade 2, intestinal lesion involving several multifocal cellular infiltrates in the LP; grade 3, lesions involving moderate inflammation and epithelial hyperplasia; grade 4, inflammation involving most of the segment. The summation of scores in the three regions of the colon per mouse provided a total colonic disease score.
2.6 Cytokine measurement by Luminex™ analysis
Levels of T helper-cell-derived cytokines IL-2, IL-3, and IL-10 (anti-inflammatory), as well as IL-6, IL-1β, MCP-1, RANTES, KC, and G-CSF (pro-inflammatory) in serum and colon tissue homogenate were determined by a luminex bioplex ELISA assay kit (Bio Rad, Hercules). The detailed method has been previously described (Singh et al., 2014). In brief, filter bottom ELISA plates (Bio Rad Hercules, CA) were rinsed with 100 μl of bioplex assay buffer and removed using a Millipore™ Multiscreen Separation Vacuum Manifold System (Bedford, MA), set at 5 mm Hg. All cytokine beads in the assay buffer were added to pre-wet vacuum wells plate followed by 50 μl of assay beads. The assay buffer was then removed and the plate washed with wash buffer. Following the wash, 50 μl of standard serum and blank were added to each well and incubated for 1 h with continuous shaking at setting #3, using a Lab-Line™ Instrument Titer Plate Shaker (Melrose, IL). The filter bottom plates were washed and vortexed at 300 × g for 30 sec, after which 25 μl of anti-mouse detection Abs was added to each well and incubated for 30 min at room temperature. Next, 50 μl of streptavidin-phycoerythrin solution was added to each well and incubated with continuous shaking for 10 min at room temperature, after which 125μl of assay buffer was added to each well. Readings were measured using a Luminex™ System (Austin, TX) and calculated using BioRad software (Bio-Plex manager 6.1). The Ab BioRad assays are capable of detecting >15 pg/ml for each analytes: IL-2, IL-3, IL-6, IL-10, IL-1β, MCP-1, RANTES, KC, and G-CSF.
2.7 miRNAs isolation and microarray analysis
Cells from MLNs and PPs were immediately placed in TRIzol reagent from a miRNeasy mini-kit (QIAGEN, Valencia, CA). miRNAs were isolated as described by the manufacturer's instructions. Samples were stored at −80° C until they were sent to the Johns Hopkins University sequencing core facility for miRNAs microarray analysis. Total RNA, including miRNAs from MLNs and PPs, was isolated and hybridized on an Affymetrix GeneChip high-throughput miR array containing 609 murine probes (Affymetrix, Santa Clara, CA). The data generated from the array were analyzed using hierarchical clustering. By use of ingenuity pathway analysis (IPA) software (Qiagen; www.ingenuity.com), results from the miRNA microarray were analyzed to identify molecular pathways potentially altered by single or multiple miRNAs target genes. In brief, this analysis compares each set of miRNAs to all available pathways in the database and assigns priority scores based on the predicted strength of miRNAs interaction with components of the target pathway.
2.8 Validation of miRNAs data by RT-PCR analysis
To validate key results from the microarray analysis, three representative miRNAs that were significantly increased in PPs (miRNA-145-5p, miRNA-182-5p, and miRNA-200b-5p), and miR-217, which changed in both MLNs and PPs, were subjected to further RT-PCR analysis (CFX connect Bio-Rad, Hercules). From PPs, we also validated the downstream targets of forkhead-box transcription factor (FOXO1) and SMAD-2, which are relevant to inflammation and direct targets of these miRNAs. miRNAs were reverse-transcribed (miScript II; Qiagen) to make cDNA, and RT-PCR was carried out with primers (5′GCAGUCCACGGGCAUAUACAC-3′ and 5′-UACAGUACUGUGAUAGCUGAA-3′ for miRNAs 455 and 101b, respectively). Reactions were done with SYBR Green Master Mix (Qiagen). Initiation was done for 15 min at 95°C. The samples were then run for 40 cycles each at 94° C for 5 sec, 55° C for 30 sec, and 70° C for 30 sec. All samples were run in triplicate.
2.9 Statistics
Several response variables were measured for each of the 18 experimental units within each of the three treatment groups (Naive, DSS + Vehicle, and DSS + FAAH-II). The change in body weight over time measured longitudinally is demonstrated in Figure 1 for the three groups, which depicts the mean ± the standard error of the sample mean (SEM). There was no significant change for the naive group, whereas the comparisons of statistically significant changes in two latter groups over time were performed using a t-test at each observation time. The colon length summaries were provided in Figure 1. One-way analysis of variance (ANOVA) was used to compare the three treatment groups. The inflammation score and comparisons among the three groups were performed using ANOVA. These statistical analyses were implemented using StatView II (Abacus Concepts, Berkeley, CA) and/or Microsoft Excel (Microsoft, Seattle, WA). Comparisons using ANOVA were also performed with respect to Cell Counts, which are pictorially summarized in Table 1, and graphical depictions are presented in Figure 2, 3. Statistical significance is assessed at the 5% level of significance, so significance is declared when the p-value was < 0.05 and indicated by the symbol ✠. The main comparisons were between the colitis group (DSS+Vehicle) and the DSS+FAAH-II group.
Figure 1.
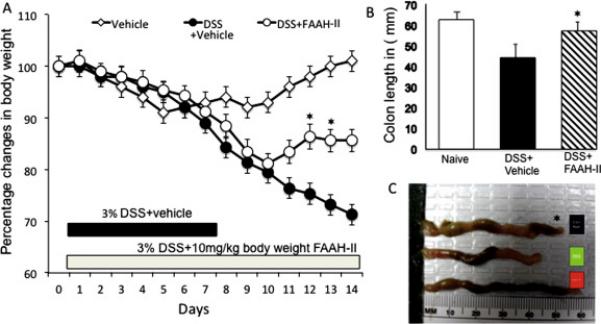
Change in body weight and colon length after FAAH-II treatment in DSS induced colitis. C57BL/6 mice (△) naive received normal water, (●) 3% DSS+vehicle, (○) 3% DSS+ 10mg/kg body weight FAAH-II every day beginning at same time on day 1 when mice received DSS+vehicle and continued till day 14. Body weight of the mice was recorded every day, and the change from initial body weight was expressed as a percentage change in body weight (Fig. 1A). Mice were sacrificed after two weeks and the length of colons from the three groups of mice at day 14 are presented (as described in Fig 1b and C). Asterisks (*) indicate statistically significant differences (p < 0.01) between DSS+vehicle and DSS+ FAAH-II treated groups.
Table 1.
Changes in the frequency of macrophages, as well as dendritic and NK1.1cells and neutrophils in the mice after DSS induction and FAAH-II treatmenta
| Cell types | Group | Spleen (%) Changes | MLNs (%) Changes | PPs (%) Changes | Colon LP (%) Changes |
|---|---|---|---|---|---|
| Macrophage (CD11b) | Naïve mice | 2.8 ± 0.3 | 4.6 ± 0.9 | 4.7 ± 1.1 | 1.9 ± 0.2 |
| DSS+Vehicle | 8.9 ± 1.1 ✠ | 7.1 ± 1.1 ✠ | 7.8 ± 2.1✠ | 4.6 ± 0.8 ✠ | |
| DSS+FAAH-II | 3.5 ± 0.4 | 4.7 ± 0.4 | 3.9 ± 1.6 | 2.4 ± 0.3 | |
| NK1.1 | Naïve mice | 5.4 ± 1.4 | 3.7 ± 2.4 | 3.1 ± 0.4 | 1.8 ± 0.2 |
| DSS+ Vehicle | 9.8 ± 0.9 ✠ | 4.8 ± 1.4 | 14.9 ± 2.3 ✠ | 2.9 ± 0.8 | |
| DSS+FAAH-II | 8.5 ±1.1 | 3.9 ± 1.2 | 4.0 ± 0.6 | 2.1 ± 0.4 | |
| Neutrophils (LY6G) | Naïve mice | 1.4 ± 0.4 | 4.4 ± 0.2 | 3.2 ± 0.4 | 2.4 ± 0.4 |
| DSS+Vehicle | 6.9 ± 0.7 ✠ | 5.1 ± 0.3 | 8.1 ± 1.1 ✠ | 9.2 ± 1.2 ✠ | |
| DSS+FAAH-II | 4.6 ± 0.3 | 3.7 ± 0.1 | 3.1 ± 0.2 | 3.7 ± 0.3 | |
| Dendritic cells (CD11c) | Naïve mice | 4.5 ± 0.2 | 7.7 ± 0.5 | 5.1± 0.3 | 3.8 ± 0.2 |
| DSS+Vehicle | 7.5 ± 0.8 | 11.3 ± 0.7 ✠ | 7.2± 0.7 ✠ | 4.4 ± 0.8 | |
| DSS+FAAH-II | 7.6 ± 0.3 | 6.3 ± 1.2 | 4.2± 0.6 | 3.5 ± 0.4 | |
| NKT cells | Naïve mice | 2.1 ± 0.4 | 2.1 ± 0.3 | 2.7 ± 0.1 | 3.1 ± 0.2 |
| DSS+Vehicle | 3.2 ± 0.1 | 1.7 ± 0.2 | 7.6 ± 0.2 ✠ | 8.1 ± 0.6 ✠ | |
| DSS+FAAH-II | 2.7 ± 0.2 | 2.1 ± 0.4 | 3.6 ± 0.1 | 3.7± 0.5 | |
Change in the frequency of macrophages, neutrophils, NK, NKT and dendritic cells after FAAH-II inhibition during DSS-induced colitis. Vehicle or FAAH-II inhibitor was administered every day after DSS induction. Fourteen days after FAAH-II treatment, cells from spleens, MLNs, PPs and LP were isolated, stained for various expressions markers, and analyzed by flow cytometry. Studies were repeated 3 times. The data presented are the mean percentage of changes ± SEM of these experiments. Differences between experimental groups were considered significant when p < 0.01 (✠).
Figure 2.
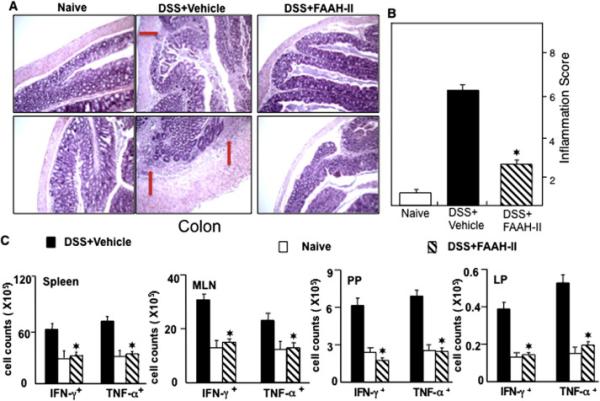
Changes in colitis scores, cytokines level and disease severity of DSS-induced colitis after FAAH-II treatment. Histological sections of colons from the three groups of mice are shown (Panel A). Mice in the untreated group with DSS+ vehicle showed significant lymphocyte infiltration and distortion of glands (arrow) and mice with DSS-induced colitis that were treated with FAAH-II showed colon lumina having markedly decreased lymphocyte infiltrations. Panel B shows the persistence or improvement of colitis inflammation scores after FAAH-II treatment. Panel C shows changes in frequency of CD4+ T cells expressing TNF-α and IFN-γ after FAAH-II treatment. The statistical significance between values of each group was assessed by Student's t test. Data represent the mean of three experiments involving 6 mice per group.
Figure 3.
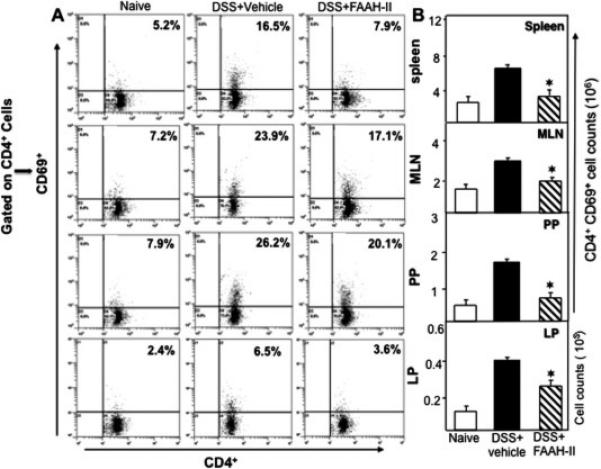
FAAH-II reduces the induction of activated T cells during experimental colitis. Spleens, MLNs, PPs and LP lymphocytes were isolated from the three groups of mice on day 14 and stained for CD4+ and CD69+ T cells. Panel A shows a representative experiment indicating the percentages of activated T cells. Panel B shows the absolute numbers of these cells in various lymphoid organs. Panel B data represent the total number of cells ± SEM from three independent experiments. Asterisks (*) indicate statistically significant differences (p < 0.01) between DSS+ vehicle and DSS+FAAH- II treated groups.
3. Results
3.1 FAAH-II reverses body weight loss associated with colitis
We examined the effect of FAAH-II inhibitor on body weight after DSS-induced colitis. We found no significant change in naïve mouse body weight during the entire study. Therefore, we compared the difference in body weight between mice given DSS+vehicle (colitic) versus mice given DSS+ FAAH-II. As shown in Figure 1A, the DSS+vehicle mice developed acute colitis, as shown by significant (P< 0.01) weight loss and diarrhea. Their body weight in this group progressively declined until the end of the experiment. Treatment of DSS colitic mice with FAAH-II resulted in significant (P<0.05) improvement in body weight as compared to the vehicle treated mice (Fig. 1A). These results indicate that in mice 10 mg/kg doses of FAAH-II protect against colitis-induced weight loss.
3.2 FAAH-II treatment reduces inflammatory cytokines and the severity of colitis
The effect of FAAH-II on DSS-induced colitis was assessed by measuring changes in the gross morphology of the colon (length measurements) and, at the end of experiments, by tissue pathology. The mean colon length of mice after DSS-induced colitis was significantly (P<0.05) shorter than the colons of colitic mice treated with FAAH-II (Fig. 1B, C). This suggested that administration of FAAH-II has a protective effect in maintaining normal colon length and morphology. Indeed, the colons of DSS colitic mice showed hypertrophied epithelial layers at multiple sites, as well as increases in cellular infiltrates, predominantly macrophages, neutrophils, and CD4+T cells. In contrast, as measured by positive changes in the mean histological scores of these mice compared to those with severe colitis receiving DSS+vehicle, the administration of FAAH-II resulted in improved colon histology (Fig. 2 A, B). Moreover, the tissue pathology associated with colitis, which includes massive cellular infiltrates in the colon, was reduced following FAAH-II treatment (Fig. 2A).
Next, we enumerated the number of IFN-γ− and TNF-α-expressing CD4+ T cells from the spleens, MLNs, PPs as well as LP lymphocytes, in colitic mice versus those given FAAH-II treatment. We found a decline in the number of IFN-γ− or TNF-α-expressing CD4+ T cells in the spleens, MLNs, PPs and LP (P<0.05) of mice treated with FAAH-II as compared with the numbers of such cells in vehicle-treated colitic mice (Fig. 2C, lower panel). Taken together, these findings suggested that FAAH-II protects mice against DSS-induced colitis by suppressing cytokine-expressing inflammatory cell infiltration from systemic and mucosal effector organs.
3.3 FAAH-II reduces systemic and mucosal-activated CD4+ T helper lymphocytes
Previous studies support the notion that CD4+ Th1 cells mediate inflammation and therefore, we examined the effect of FAAH-II on populations of activated and infiltrating CD4+ T cells in the spleens, MLNs, PPs, and LP using flow cytometry analysis (Fig. 3). The frequency of activated T cells (CD4+CD69+) in the spleens of colitic mice was significantly (P<0.05) reduced by FAAH-II treatment as compared to the occurrence of these cells in spleens of mice in the vehicle-treated group. Similarly, in MLNs, PPs, and LP the percentage and number of activated CD4+T cells decreased (P<0.05) in FAAH-II treated groups as compared to the vehicle-treated group (Fig. 3). Together, these findings indicated that FAAH-II treatment significantly reduces the frequency of activated Th1 lymphocytes in the spleen, MLNs, PPs, and LP which are systemic, inductive, and effector sites, respectively, thus protecting mice against DSS-induced colitis.
3.4 FAAH-II inhibits macrophage, neutrophil, NK1.1, NKT, and dendritic cell infiltration
Macrophage, neutrophil, natural killer and dendritic cells play a role in colitis progression. Therefore, we determined changes in the frequency of mucosal and systemic macrophages, NK1.1, NKT, neutrophils and dendritic cells after FAAH-II treatment in DSS induced colitis. As shown in Table 1, macrophage frequency was significantly increased in the spleens, MLNs, PPs and LP of mice with DSS-induced colitis as compared to their frequency in naïve mice. However, FAAH-II treatment decreased the frequency of macrophages in spleens, MLNs, PPs and LP as compared to the numbers of macrophages in DSS colitic mice (P<0.05; Table 1). These results indicated that FAAH-II treatment reduces the elevated numbers of macrophages in the spleen, MLNs, PPs and LP that are characteristic of colitis.
There also were significant (P<0.05) increases in the numbers of neutrophils in the spleens, PPs, and LP of vehicle-treated DSS-induced mice as compared to naïve mice (Table 1). Again, treatment with FAAH-II significantly reduced the numbers of neutrophils in the spleen, PPs and LP as compared with those in the DSS-treated group. Thus, the increased numbers of neutrophils that occur during colitis at both systemic and mucosal sites were significantly reduced by FAAH-II treatment.
We also examined the effect of FAAH-II on NK 1.1, dendritic, and NKT cells. After DSS induction of colitis, the percentage of splenic and LP NK 1.1 cells increased. Treatment of these colitic mice with FAAH-II reduced the percentages of NK1.1 cells in spleens and LP (Table 1). Further, this treatment significantly (P<0.05) decreased NKT cells in PPs, as well as dendritic cells localized in the MLNs and LP of colitic mice (Table 1). Taken together, the results indicated that FAAH-II treatment reduces macrophages, neutrophils, NKT, dendritic and NK1.1 cells from systemic and effector sites, resulting in suppression of colitis.
3.5 FAAH-II regulates systemic and colon cytokine levels in DSS-induced colitis
We next, examined whether FAAH-II treatment leads to changes in effector (colon site) and systemic pro- and anti-inflammatory cytokine levels in DSS-induced colitis. In DSS colitic mice, colon tissue and systemic IL-6, MCP-1, IL-1β, RANTES, G-CSF, and KC were increased as compared with levels in naive mice (Fig. 4 and Supplemental Fig. 1). Administration of FAAH-II to DSS colitic mice decreased (P<0.05) the serum levels of all these cytokines and increased IL-2, IL-3 and IL-10 compared to their levels in DSS-induced colitis (Supplemental Fig. 1). Thus, FAAH-II administration can suppress the levels of systematic and local inflammatory cytokines and concurrently increase levels of systemic anti-inflammatory cytokines in mice with DSS colitis. This suggests a possible pathway for manipulating cytokine levels, using FAAH-II inhibitor to provide protection from experimental colitis.
Figure 4.
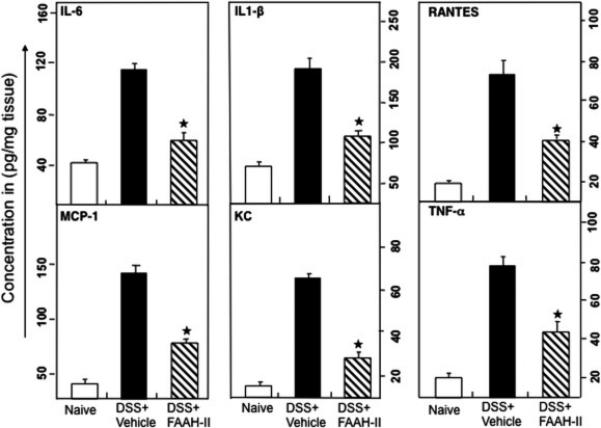
FAAH-II treatment reduces systemic pro-inflammatory cytokines and increases anti-inflammatory cytokines. After mice were sacrificed, colon tissue homogenate levels of IL-6, IL-1β, MCP-1, RANTES, KC, G-CSF, IL-2, IL-3 and IL-10 were determined by a Bio-Rad ELISA multiplex kit capable of detecting >10 pg/ml of these analytes. The data presented are the mean concentrations of these cytokines ± in three separate experiments. Asterisks (*) indicate statistically significant differences (p< 0.01) between DSS+ vehicle and FAAH-II-treated groups.
3.6 Effect of FAAH inhibition on miRNA expression patterns during colitis
Differential expression and changes in miRNAs was found in immune cells of naïve mice, colitic mice (DSS+ vehicle), and colitic mice treated with FAAH-II inhibitor. Since, we did not get enough cells to isolate miRNAs from LP, we analyzed miRNAs from MLNs and PPs immune cells from the three groups. The heat map of miRNAs from mice with DSS-induced colitis and the colitic group treated with FAAH-II showed different expression of a total 26 miRNAs from MLNs and 217 miRNAs from PPs, with >1.5-fold changes between the two groups (Fig. 5). The miRNAs that showed an increase or decrease more than 1.5-folds in MLNs and PPs of colitis+FAAH-II versus colitis+vehicle were further plotted (Supplemental Fig. 2). We identified 8 miRNAs that were upregulated or downregulated in MLNs and PPs after FAAH-II treatment (Supplemental Fig. 2) based on their ability to target inflammation and regulation of immune cells.
Figure 5.
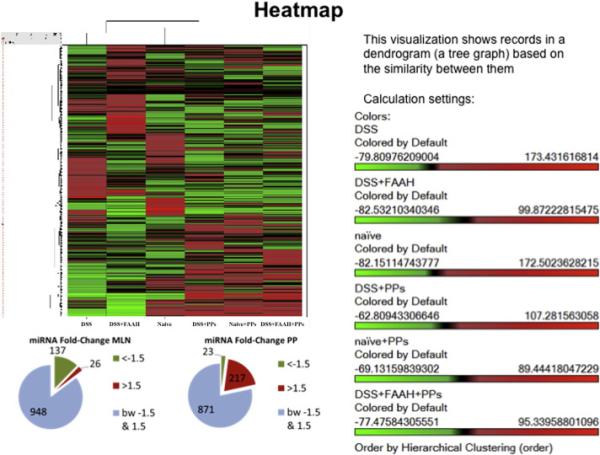
The changes in miRNAs expression profiles after DSS induction in DSS+vehicle or DSS+FAAH-II treated group. (A) Heat map of the miRNA array done on immune cells from MLNs and PPs from the naïve, DSS-induced (DSS +vehicle or DSS +FAAH-II) groups. A cluster of miRNAs is differentially expressed in mice treated with FAAH-II. Red color indicates a positive change, green a negative change, and black no change. (B) Pie charts (B-1) showing proportion of genes with <1.5-fold change (light green) and > 1.5 fold up- or downregulation (red) for MLNSs and PPs.
Of these, 3 miRNAs that were upregulated including, miR-145-5p, miR-200b-5p, and miR-182-5p from PPs and their downstream target (FOXO1, SMAD2) were validated using quantitative RT-PCR analysis (Fig. 6). The results were highly consistent with the microarray data in that miR-145-5p, miR-182-5P, and miR-200b-5p were significantly (P <0.02) upregulated by FAAH-II treatment in colitic mice as compared with those in DSS +vehicle mice. The other miRNA-217 levels increased in MLNs and decreased in PPs (Fig. 7) thereby suggesting that there were differences at inductive and effector sites after FAAH-II treatment. miRNA-141-3p decreased at both sites after FAAH-II treatment (Fig. 7). Taken together, the microarray data correlated with the RT-PCR analysis results.
Figure 6.
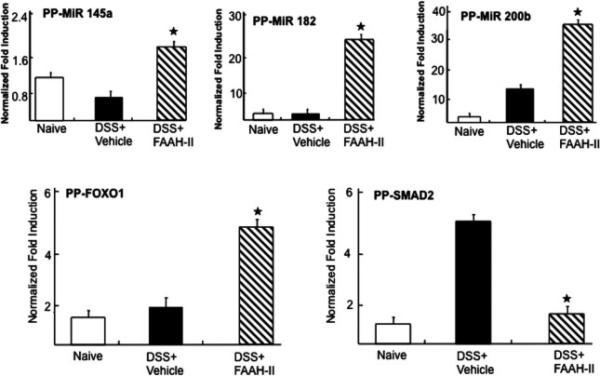
Validation of selected miRNAs involved in inflammatory pathways by RT-PCR analysis after FAAH-II treatment. Figure shows quantitative RT-PCR verifying upregulation of miRNAs-145-5p, −182-5p, and −200b-5p, as well as target FOXO1 and SMAD2 in PPs in DSS-induced mice treated with FAAH-II. Data are expressed as mean ± S.E.M. Statistical significance was calculated using the Student's t test. *P< 0.05 DSS+vehicle versus DSS+ FAAH-II treated group.
Figure 7.
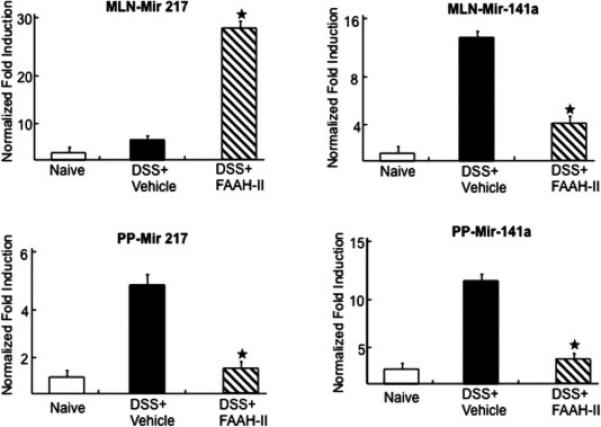
miRNAs (miR-141-3p) and (miR-217) validation by RT-PCR analysis after FAAH-II treatment in PPs and MNLs. Quantitative RT-PCR verifying upregulation of miRNA-217 in MLNs and downregulation of mir-217 in PPs, and upregulation of miRNA-141-3p expression in both MLNs and PPs in DSS-induced mice treated with FAAH-II. Data are expressed as mean ± S.E.M. Statistical significance was calculated using the Student's t test. *P< 0.05 in DSS+Vehicle vs. DSS+FAAH-II treated group.
Deep analysis using IPA software was used to delineate a possible miRNA-mediated pathway to suppress DSS-induced inflammation (Supplemental Fig. 3). Of the miRNAs that were upregulated or downregulated by > 1.5-fold in response to FAAH-II treatment, 8 had direct association with an inflammatory pathway and immune modulation target. The direct association of the upregulated or downregulated miRNAs was mapped with targets of interest by red and green color and indirect pathways by pink (Supplemental Fig. 3). The upregulation of miRNAs from PPs, such as miR-182-5p (FOXO1), miR-145-5p (FOXO1, SMAD2), and miR-200b-5p (SMAD2) suggested decreases in FOXO1 and SMAD2 that are involved in the induction of IL-6, thereby correlating with decreased IL-6 levels following FAAH-II treatment (Supplemental Fig. 1). The changes in miRNAs also indicated how they might regulate other inflammatory markers such as IL-1, IL-10, CCL2, CCL3, CCL5, and FOXP3 (Supplemental Fig. 3). Interestingly, in the MLN, majority of the miRNAs were down regulated when compared to PPs (Supplemental Fig. 4). Moreover, as shown in Fig 10, we also were able to identify some upregulated miRNAs in MLNs such as miR-217-5p that may target inflammatory pathways such as (IL-6 and CXCL2), miR-3084-3p and miR-762 (IL-1 or IL-17). Also, some of the down-regulated miRNAs such as miR-146a-5p may induce more IL-10 or miR-141-3p may induce STAT4, which regulates IL-10 (Supplemental Fig. 4). Taken together, the pathway analysis showed that FAAH-II treatment alters the expression of many miRNAs that together may downregulate inflammatory markers to ameliorate colitis.
4. Discussion
We have identified the effectiveness of FAAH-II inhibitor in ameliorating inflammation in a well-established DSS-induced model of colitis. Considerable immunological evidence and similarities in the pathology of experimental mouse models that mimic with IBD suggest that this model can be used to explore novel therapeutics. Here, we demonstrated that administration of select FAAH-II inhibitor reversed the severity of DSS-induced colitis. Treatment of colitic mice with FAAH-II reversed weight loss, improved inflammation scores, and decreased disease severity. The increased frequency of mucosal and systemic activated T cells, macrophages, neutrophils, and NKT cells during experimental colitis were also significantly reduced by FAAH-II treatment. Taken together, these results indicate that FAAH-II abrogates DSS-induced colitis by suppressing the activation of CD4+ T cells and reducing the frequency of macrophages and NKT cells, thereby reducing other inflammatory cells at sites of inflammation.
Recent studies suggest that both exogenous and endogenous cannabinoids inhibit the inflammatory response and suppress severity of colitis (Stella et al., 1997; Storr et al., 2008). Storr et al.'s study suggests that inhibition of FAAH at a later stage may alleviate disease symptoms by activating endogenous cannabinoids. To this end, mice lacking CB1-, CB2-, or both receptors, showed augmented inflammation in a TNBS-induced model of colitis (Engel et al., 2010). Further, blocking the degradation of endocannabinoids or cellular reuptake inhibited inflammation and maintained normal colon length (Storr et al., 2008). Furthermore, CB-receptor-knockout mice in which levels of anandamide were elevated by deficiency in or inhibition of FAAH showed significant resistance to TNBS-induced colitis (Izzo and Sharkey, 2010). The results of these studies have been supported by clinical reports of increased levels of an endocannabinoid, AEA, in biopsies of the submucosal layer, but not the mucosal layer of the inflamed colon of human UC patients (D'Argenio et al., 2006). Another study indicated that anandamide modulates the CB receptor to mediate a decrease in contractility and secretion using CB1 and decreasing proinflammatory cytokines by CB2 receptors (Izzo and Camilleri, 2009). Further, FAAH inhibitors have already been tested in preclinical and clinical trials and may be an important potential target for the treatment of IBD (Kimball et al., 2006; Massa et al., 2004; Singh et al., 2012; Storr et al., 2008; Storr et al., 2009). These findings, as well as the results of this study, clearly demonstrate that FAAH inhibition in DSS-induced colitis can improve disease severity, recover body weight, prevent disease progression and ameliorate colitis.
Several IBD models of experimental colitis indicate not only that CD4+ T cells are important in the induction of IBD, that much of the intestinal damage caused by this disease is a result of T-cell-mediated injury (Elson et al., 1996; Singh et al., 2012; Singh et al., 2008a). These findings are consistent with those of the current study, in which we found that the percentage of activated CD4+ T cells in spleens, MLNs, PPs and LP increased significantly during experimental colitis and decreased after FAAH-II treatment. Earlier, we showed in spontaneous model that during severe colitis the numbers of IFN-γ+ expressing CD4+ T cells are elevated in the LP (Singh et al., 2008b). Based on these findings, we suggest that both systemic and mucosal site decreases in activated T cells might decrease the inflammatory cytokine environment to amend the colitis symptoms. These observations suggest that inhibition of FAAH suppresses colitis by downregulating T-cell activation responses.
Macrophage populations are prominent in active IBD (Strober and James, 1986) as compared to healthy donors. Further, percentages of macrophages are increased in DSS-induced colitis (Grose et al., 2001; Ogawa et al., 2004). IBD is also linked to an influx of neutrophils and depletion of neutrophils by monoclonal antibodies has been shown to decrease several parameters of DSS-induced colitis (Natsui et al., 1997). In the present study, we observed increases in macrophages, neutrophils, NK1.1, and NKT cells in the spleens, MLNs, PPs and LP of DSS-induced colitic mice. FAAH-II treatment of colitic mice reversed the increase in number of inflammatory cells to normal levels in spleens, MLNs, PPs and LP.
There is increased expression and level of IL-6 and IL-1β in IBD patient systemic and mucosal biopsies (Casini-Raggi et al., 1995; Reimund et al., 1996). Also, patients’ IL-6 serum levels correlate with disease activity (Holtkamp et al., 1995). RANTES has been shown to be elevated in several inflammatory conditions, including lupus, rheumatoid arthritis, and multiple sclerosis (Boiardi et al., 1999; Vila et al., 2007). MCP-1 levels are elevated in experimental models of colitis and in the mucosal tissues of IBD patients (Keates et al., 1997; McCormack et al., 2001; Scheerens et al., 2001). In the present study, we observed significant decreases in both systemic and local colon tissue MCP-1, RANTES, IL-6, and IL-1β in colitic mice treated with FAAH-II as compared to colitic mice given DSS alone. Similarly, we observed a significant increase in systemic anti-inflammatory cytokine IL-10 in the FAAH-II treated group as compared to mice given DSS alone. These results show that reduction of these cytokines and chemokines, from colon site along with increases in systemic anti-inflammatory cytokines, which correlate with the severity of colon inflammation after FAAH-II treatment, can lead to the amelioration of colitis.
In the present study, we have demonstrated that FAAH-II inhibitor suppresses several proinflammatory cytokines, which is linked to alterations in the expression of several miRNAs. After FAAH-II treatment, we found changes in expression of 26 miRNAs from MLNs and 217 miRNAs from PPs. Several putative targets of these miRNAs are related to the inflammatory process. After FAAH-II treatment, miR-145-5p, miR-182-p, and miR-200b-5p were increased in the PPs. Based on pathway analysis, they targeted FOXO1 and SMAD2, which are involved in IL-6 induction thereby suggesting how these miRs may down, regulate IL-6. miR-217 has also been shown to downregulate Sirt-1 expression in ethanol-induced fat accumulation in hepatocytes (Yin et al., 2012). Treatment with FAAH-II was followed by decreased miR-217 expression in PPs. Thus, in our study, miR-217 may induce Sirt-1 and provide protection against intestinal inflammation in as much as, we have shown previously that Sirt-1 induction by resveratrol protects mice from gut inflammation (Singh et al., 2010). Also, some of the down regulated miRNAs in MLNs such as miR-146a-5p seen following treatment with FAAH-II may induce more IL-10 or miR-141-3p may induce STAT4 which regulates IL-10, thereby promoting anti-inflammatory cytokines.
Significant evidence indicates that microRNAs play a critical role in the pathogenesis and progression of IBD. However, it is not known how alteration in miRNAs expression specifically contributes to IBD pathobiology. Several studies have identified miRNAs that are associated with IBD in intestinal tissues and peripheral blood specimens (Coskun et al., 2012; Wu et al., 2011; Wu et al., 2008). To this end, studies using interleukin-10 knockout (IL-10−/−) mice, a model of chronic intestinal inflammation, have shown selective dysregulation of miRNAs in colon tissue and peripheral blood leukocytes (Schaefer et al., 2011). In the present study, we found an increase in miR-200b-3p after FAAH treatment in PP. This was consistent with the earlier finding of (Zidar et al., 2016) that downregulation of members of the miR-200 family supports the possibility that miR-200 has a role in pathogenesis of fibrosis in IBD. Further, it has also been shown that has-miR-141-3p is upregulated in the rectosigmoid area, as compared to the ascending colon area, of UC patients (Ranjha et al., 2015). This supports our present finding of an increase in miR-141 expression in the MLN after FAAH treatment. Similarly, it has been shown that changes in miR-141 expression in TNBS induced or IL-10−/− colitis models regulate leukocyte infiltration and mediate colitis (Huang et al., 2014). Taken together, our results and those of others clearly support the role of miRNAs in models of acute and experimental colitis.
It has been shown that mice lacking SMAD2 in T cells do not develop spontaneous lymphoproliferative autoimmunity (Malhotra et al., 2010). Further, FOXO1 is known to activate proinflammatory MCP-1 and IL-6 (Ito et al., 2009). To this end, we also validated the downstream target FOXO1 and SMAD2 after FAAH inhibition. Interestingly, FAAH inhibition increased FOXO1 and decreased SMAD2, suggesting a mechanism that suppresses the inflammatory response in acute colitis. Taken together, changes in the expression of these miRNAs in response to FAAH-II treatment are considered to be a potential molecular mechanism, which can lead to the decreased cytokine and SMAD2 expression and increased FOXO1 expression observed in this study.
In summary, the current study convincingly demonstrated that FAAH inhibitors act as anti-inflammatory agents by targeting multiple cellular pathways during colitis. We have showed that inhibition of FAAH induces an anti-inflammatory state by inhibiting activated T cells, reducing inflammatory immune cells, systemic and effector site cytokines, and changing the expression of miRNAs in experimental model of colitis. However, further studies are necessary by using mimics for downregulated miRNAs or antagomirs for upregulated miRNAs prior to drawing any firm conclusions on FAAH treatment induced miRNAs mediated suppression of experimental colitis.
Supplementary Material
❖ Fatty acid amide hydrolase (FAAH) is an enzyme crucially involved in the modulation of intestinal physiology
❖ FAAH-II improved overall clinical scores by reversing weight loss and colitis-associated pathogenesis.
❖ FAAH-II ameliorates experimental colitis by reducing the frequency of activated T cells, macrophages, neutrophils, and NK/NKT cells in the colon.
❖ FAAH-II mediates inflammatory cytokine and miRNAs at mucosal sites.
Acknowledgments
This study was supported in part by grants from R56DK087836 (UPS) and P01AT003961 (MN and PN) and the Intramural Research Program, NIA, NIH. We thank Prof. Edsel Pena, director of the Biometry Core of the Center for Colon Cancer Research (CCCR) for his advice on statistical analysis.
Abbreviations
- FAAH
Fatty acid amide hydrolase
- DSS
Dextran sodium sulphate
- MLNSS
Mesenteric lymph nodes
- PPs
Peyer's patches
- CB1-2
Cannabinoid receptors 1-2
- AEA
Anandamide
- TNF-α
Tumor Necrosis Factor-Alpha
- IBD
Inflammatory Bowel Disease
- UC
Ulcerative Colitis
- CD
Crohn's Disease
- TNBS
Trinitrobenzene sulfonic acid
- miRNAs
MicroRNAs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altamemi I, Murphy EA, Catroppo JF, Zumbrun EE, Zhang J, McClellan JL, Singh UP, Nagarkatti PS, Nagarkatti M. Role of microRNAs in resveratrol-mediated mitigation of colitis-associated tumorigenesis in Apc(Min/+) mice. The Journal of Pharmacology and Experimental Therapeutics. 2014;350:99–109. doi: 10.1124/jpet.114.213306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- Boiardi L, Macchioni P, Meliconi R, Pulsatelli L, Facchini A, Salvarani C. Relationship between serum RANTES levels and radiological progression in rheumatoid arthritis patients treated with methotrexate. Clinical and Experimental Rheumatology. 1999;17:419–425. [PubMed] [Google Scholar]
- Braegger CP, MacDonald TT. Immune mechanisms in chronic inflammatory bowel disease. Annals of Allergy. 1994;72:135–141. [PubMed] [Google Scholar]
- Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal imbalance of IL-1 and IL-1 receptor antagonist in inflammatory bowel disease. A novel mechanism of chronic intestinal inflammation. Journal of Immunology. 1995;154:2434–2440. [PubMed] [Google Scholar]
- Cluny NL, Keenan CM, Duncan M, Fox A, Lutz B, Sharkey KA. Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone (SAB378), a peripherally restricted cannabinoid CB1/CB2 receptor agonist, inhibits gastrointestinal motility but has no effect on experimental colitis in mice. The Journal of Pharmacology and Experimental Therapeutics. 2010;334:973–980. doi: 10.1124/jpet.110.169946. [DOI] [PubMed] [Google Scholar]
- Coskun M, Bjerrum JT, Seidelin JB, Nielsen OH. MicroRNAs in inflammatory bowel disease--pathogenesis, diagnostics and therapeutics. World Journal of Gastroenterology. 2012;18:4629–4634. doi: 10.3748/wjg.v18.i34.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Argenio G, Valenti M, Scaglione G, Cosenza V, Sorrentini I, Di Marzo V. Up-regulation of anandamide levels as an endogenous mechanism and a pharmacological strategy to limit colon inflammation. FASEB Journal. 2006;20:568–570. doi: 10.1096/fj.05-4943fje. [DOI] [PubMed] [Google Scholar]
- Dohi T, Fujihashi K, Kiyono H, Elson CO, McGhee JR. Mice deficient in Th1- and Th2-type cytokines develop distinct forms of hapten-induced colitis. Gastroenterology. 2000;119:724–733. doi: 10.1053/gast.2000.16500. [DOI] [PubMed] [Google Scholar]
- Elson CO, Beagley KW, Sharmanov AT, Fujihashi K, Kiyono H, Tennyson GS, Cong Y, Black CA, Ridwan BW, McGhee JR. Hapten-induced model of murine inflammatory bowel disease: mucosa immune responses and protection by tolerance. Journal of Immunology. 1996;157:2174–2185. [PubMed] [Google Scholar]
- Engel MA, Kellermann CA, Burnat G, Hahn EG, Rau T, Konturek PC. Mice lacking cannabinoid CB1-, CB2-receptors or both receptors show increased susceptibility to trinitrobenzene sulfonic acid (TNBS)-induced colitis. J Physiol Pharmacol. 2010;61:89–97. [PubMed] [Google Scholar]
- Esteller M. Non-coding RNAs in human disease. Nature Reviews Genetics. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Feng X, Wang H, Ye S, Guan J, Tan W, Cheng S, Wei G, Wu W, Wu F, Zhou Y. Up-regulation of microRNA-126 may contribute to pathogenesis of ulcerative colitis via regulating NF-kappaB inhibitor IkappaBalpha. PLoS One. 2012;7:e52782. doi: 10.1371/journal.pone.0052782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grose RH, Howarth GS, Xian CJ, Hohmann AW. Expression of B7 costimulatory molecules by cells infiltrating the colon in experimental colitis induced by oral dextran sulfate sodium in the mouse. J Gastroenterol Hepatol. 2001;16:1228–1234. doi: 10.1046/j.1440-1746.2001.02558.x. [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466:835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp W, Stollberg T, Reis HE. Serum interleukin-6 is related to disease activity but not disease specificity in inflammatory bowel disease. Journal of Clinical Gastroenterology. 1995;20:123–126. doi: 10.1097/00004836-199503000-00010. [DOI] [PubMed] [Google Scholar]
- Huang Z, Shi T, Zhou Q, Shi S, Zhao R, Shi H, Dong L, Zhang C, Zeng K, Chen J, Zhang J. miR-141 Regulates colonic leukocytic trafficking by targeting CXCL12beta during murine colitis and human Crohn's disease. Gut. 2014;63:1247–1257. doi: 10.1136/gutjnl-2012-304213. [DOI] [PubMed] [Google Scholar]
- Ito Y, Daitoku H, Fukamizu A. Foxo1 increases pro-inflammatory gene expression by inducing C/EBPbeta in TNF-alpha-treated adipocytes. Biochemical and Biophysical Research Communications. 2009;378:290–295. doi: 10.1016/j.bbrc.2008.11.043. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Camilleri M. Cannabinoids in intestinal inflammation and cancer. Pharmacological Research : The Official Journal of the Italian Pharmacological Society. 2009;60:117–125. doi: 10.1016/j.phrs.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacology & Therapeutics. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Keates S, Keates AC, Mizoguchi E, Bhan A, Kelly CP. Enterocytes are the primary source of the chemokine ENA-78 in normal colon and ulcerative colitis. Am J Physiol. 1997;273:G75–82. doi: 10.1152/ajpgi.1997.273.1.G75. [DOI] [PubMed] [Google Scholar]
- Kimball ES, Schneider CR, Wallace NH, Hornby PJ. Agonists of cannabinoid receptor 1 and 2 inhibit experimental colitis induced by oil of mustard and by dextran sulfate sodium. American Journal of Physiology - Gastrointestinal & Liver Physiology. 2006;291:G364–371. doi: 10.1152/ajpgi.00407.2005. [DOI] [PubMed] [Google Scholar]
- Malhotra N, Robertson E, Kang J. SMAD2 is essential for TGF beta-mediated Th17 cell generation. J Biol Chem. 2010;285:29044–29048. doi: 10.1074/jbc.C110.156745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B. The endogenous cannabinoid system protects against colonic inflammation. Journal of Clinical Investigation. 2004;113:1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer EA, Collins SM. Evolving pathophysiologic models of functional gastrointestinal disorders. Gastroenterology. 2002;122:2032–2048. doi: 10.1053/gast.2002.33584. [DOI] [PubMed] [Google Scholar]
- McCormack G, Moriarty D, O'Donoghue DP, McCormick PA, Sheahan K, Baird AW. Tissue cytokine and chemokine expression in inflammatory bowel disease. Inflammation Research : Official Journal of the European Histamine Research Society [et al] 2001;50:491–495. doi: 10.1007/PL00000223. [DOI] [PubMed] [Google Scholar]
- Natsui M, Kawasaki K, Takizawa H, Hayashi SI, Matsuda Y, Sugimura K, Seki K, Narisawa R, Sendo F, Asakura H. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. Journal of Gastroenterology & Hepatology. 1997;12:801–808. doi: 10.1111/j.1440-1746.1997.tb00375.x. [DOI] [PubMed] [Google Scholar]
- Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol. 2004;110:55–62. doi: 10.1016/j.clim.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Podolsky DK. Inflammatory bowel disease. New England Journal of Medicine. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- Ranjha R, Aggarwal S, Bopanna S, Ahuja V, Paul J. Site-Specific MicroRNA Expression May Lead to Different Subtypes in Ulcerative Colitis. PLoS One. 2015;10:e0142869. doi: 10.1371/journal.pone.0142869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimund JM, Wittersheim C, Dumont S, Muller CD, Kenney JS, Baumann R, Poindron P, Duclos B. Increased production of tumour necrosis factor-alpha interleukin-1 beta, and interleukin-6 by morphologically normal intestinal biopsies from patients with Crohn's disease. Gut. 1996;39:684–689. doi: 10.1136/gut.39.5.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salaga M, Sobczak M, Fichna J. Inhibition of fatty acid amide hydrolase (FAAH) as a novel therapeutic strategy in the treatment of pain and inflammatory diseases in the gastrointestinal tract. Eur J Pharm Sci. 2014;52:173–179. doi: 10.1016/j.ejps.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407. doi: 10.1038/ncpgasthep0528. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- Schaefer JS, Montufar-Solis D, Vigneswaran N, Klein JR. Selective upregulation of microRNA expression in peripheral blood leukocytes in IL-10−/−mice precedes expression in the colon. Journal of Immunology. 2011;18:5834–5841. doi: 10.4049/jimmunol.1100922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheerens H, Hessel E, de Waal-Malefyt R, Leach MW, Rennick D. Characterization of chemokines and chemokine receptors in two murine models of inflammatory bowel disease: IL- 10[r]−/−[/r] mice and Rag-2[r]−/−[/r] mice reconstituted with CD4+CD45RBhigh T cells. European Journal of Immunology. 2001;31:1465–1474. doi: 10.1002/1521-4141(200105)31:5<1465::AID-IMMU1465>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Singh UP, Murphy AE, Enos RT, Shamran HA, Singh NP, Guan H, Hegde VL, Fan D, Price RL, Taub DD, Mishra MK, Nagarkatti M, Nagarkatti PS. miR-155 deficiency protects mice from experimental colitis by reducing Th1/Th17 responses. Immunology. 2014;143:479–489. doi: 10.1111/imm.12328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh NP, Singh B, Hofseth LJ, Price RL, Nagarkatti M, Nagarkatti PS. Resveratrol (trans-3,5,4′-trihydroxystilbene) induces silent mating type information regulation-1 and down-regulates nuclear transcription factor-kappaB activation to abrogate dextran sulfate sodium-induced colitis. Journal of Pharmacology & Experimental Therapeutics. 2010;332:829–839. doi: 10.1124/jpet.109.160838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh NP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Cannabinoid receptor- 2 (CB2) agonist ameliorates colitis in IL-10([r]−/−[/r]) mice by attenuating the activation of T cells and promoting their apoptosis. Toxicology & Applied Pharmacology. 2012;258:256–267. doi: 10.1016/j.taap.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh R, Singh S, Karls RK, Quinn FD, Taub DD, Lillard JW., Jr. CXCL10[r]+[/r] T cells and NK cells assist in the recruitment and activation of CXCR3+ and CXCL11[r]+[/r] leukocytes during Mycobacteria-enhanced colitis. BMC immunology. 2008a;9:25. doi: 10.1186/1471-2172-9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh S, Singh R, Cong Y, Taub DD, Lillard JW., Jr. CXCL10-producing mucosal CD4+ T cells, NK cells, and NKT cells are associated with chronic colitis in IL-10([r]−/−[/r]) mice, which can be abrogated by anti-CXCL10 antibody inhibition. Journal of Interferon & Cytokine Research. 2008b;28:31–43. doi: 10.1089/jir.2007.0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh UP, Singh S, Taub DD, Lillard JW., Jr. Inhibition of IFN-gamma-inducible protein-10 abrogates colitis in IL-10([r]−/−[/r]) mice. Journal of Immunology. 2003;171:1401–1406. doi: 10.4049/jimmunol.171.3.1401. [DOI] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long- term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Emmerdinger D, Zhang H, Yuce B, Sibaev A, Massa F, Buckley NE, Lutz B, Goke B, Brand S, Patel KD, Sharkey KA. Targeting endocannabinoid degradation protects against experimental colitis in mice: involvement of CB1 and CB2 receptors. Journal of Molecular Medicine. 2008;86:925–936. doi: 10.1007/s00109-008-0359-6. [DOI] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA. Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflammatory Bowel Diseases. 2009;15:1678–1685. doi: 10.1002/ibd.20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. Journal of Clinical Investigation. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annual review of Immunology. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- Strober W, James SP. The immunologic basis of inflammatory bowel disease. J Clin Immunol. 1986;6:415–432. doi: 10.1007/BF00915248. [DOI] [PubMed] [Google Scholar]
- Vila LM, Molina MJ, Mayor AM, Cruz JJ, Rios-Olivares E, Rios Z. Association of serum MIP-1alpha, MIP-1beta, and RANTES with clinical manifestations, disease activity, and damage accrual in systemic lupus erythematosus. Clinical Rheumatology. 2007;26:718–722. doi: 10.1007/s10067-006-0387-y. [DOI] [PubMed] [Google Scholar]
- Wu F, Guo NJ, Tian H, Marohn M, Gearhart S, Bayless TM, Brant SR, Kwon JH. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn's disease. Inflammatory Bowel Diseases. 2011;17:241–250. doi: 10.1002/ibd.21450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F, Zikusoka M, Trindade A, Dassopoulos T, Harris ML, Bayless TM, Brant SR, Chakravarti S, Kwon JH. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology. 2008;135:1624–1635. e1624. doi: 10.1053/j.gastro.2008.07.068. [DOI] [PubMed] [Google Scholar]
- Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. 2012;287:9817–9826. doi: 10.1074/jbc.M111.333534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidar N, Bostjancic E, Jerala M, Kojc N, Drobne D, Stabuc B, Glavac D. Down-regulation of microRNAs of the miR-200 family and up-regulation of Snail and Slug in inflammatory bowel diseases - hallmark of epithelial-mesenchymal transition. J Cell Mol Med. 2016 doi: 10.1111/jcmm.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.