

Abstract
Significant therapeutic progress has been made in treating prostate cancer in recent years. Drugs such as enzalutamide, abiraterone, and cabazitaxel have expanded the treatment armamentarium, although it is not completely clear which of these drugs are the most-effective option for individual patients. Moreover, such advances have been tempered by the development of therapeutic resistance. The purpose of this review is to summarize the current literature pertaining to the biochemical effects of AR variants and their consequences on prostate cancer therapies at both the molecular level and in clinical treatment. We address how these AR splice variants and mutations affect tumor progression and therapeutic resistance and discuss potential novel therapeutic strategies under development. It is hoped that these therapies can be administered with increasing precision as tumor genotyping methods become more sophisticated, thereby lending clinicians a better understanding of the underlying biology of prostate tumors in individual patients.
Keywords: Androgen receptor, prostate cancer, AR-V7, pharmacology, mutation
Graphical Abstract
INTRODUCTION
Prostate cancer is the second leading cause of death from cancer and the most prevalent cancer in men; currently, 14% of the men in America diagnosed with prostate cancer over their lifetime (1). The androgen receptor (AR) is quintessential to prostate carcinogenesis, progression, and treatment. Metastatic prostate cancer is therefore treated with androgen deprivation therapy (ADT), which includes surgical or chemical castration that deprives tumor cells of testicular androgens thereby slowing growth. Typically, ADT initially proves to be effective, but in most cases the patient progresses to castration resistant prostate cancer (CRPC). Treatment for CRPC may include continuing ADT concurrent with immunotherapy, radiotherapy, cytotoxic chemotherapy, and/or other hormone manipulations.
Therapeutic development has recently improved upon two classes of anti-androgens. AR ligands (e.g., bicalutamide) inhibit AR (AR) signaling by binding to the AR itself and preventing the transcription of AR effectors. More recently, more potent inhibitors have been developed that simultaneously inhibit both AR ligand binding and the DNA-binding capacity of the AR (e.g., enzalutamide). Androgen synthesis inhibitors (e.g., ketoconazole) block the synthesis of androgens from their many precursors. Newer androgen synthesis inhibitors more-specifically inhibit these enzymes at lower concentrations (e.g., abiraterone). While such innovations have greatly improved prostate cancer treatment, patients inevitably acquire resistance toward newer therapies as well. The purpose of this review is to summarize current knowledge about how AR splice variants and mutations affect tumor progression and therapeutic resistance, address precision treatment options for patients harboring such AR variants, and discuss emerging therapies to target these variants.
ANDROGEN RECEPTOR SIGNALING
AR Signaling in the Normal Prostate
The AR is encoded by the AR gene (located at chromosome Xq12), and the full-length transcription product has a molecular weight of 100 kDa. In normal cells, the AR consists of four domains: a transactivation domain (encoded by exon 1), a DNA-binding domain (exons 2–3), a hinge region (encoded by the 5′ portion of exon 4), and a ligand-binding domain (exons 4–8), as shown in Figure 1 (2).
Figure 1.
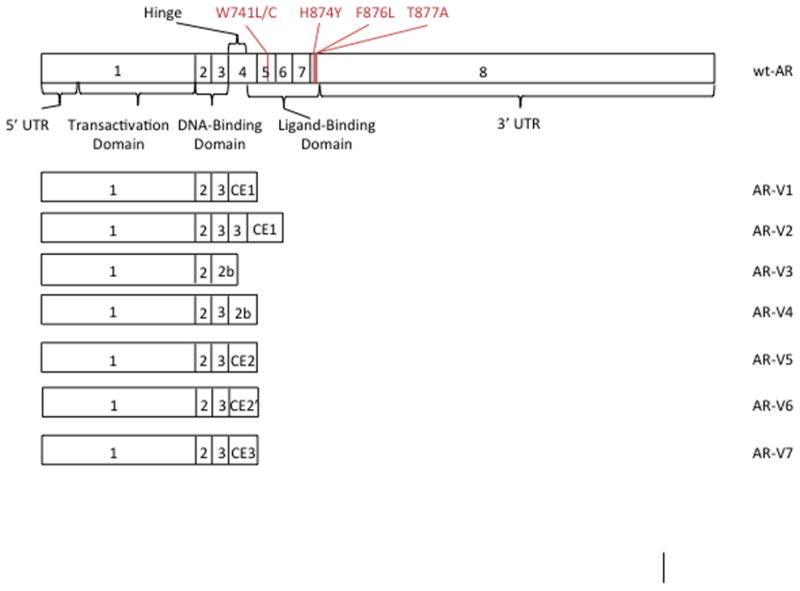
Wt-AR and selected splice variant transcripts. The wt-AR includes important AR functional domains and locations of clinically relevant missense mutations. CE1: cryptic exon 1. 2b: 11 C-terminal amino acids spliced downstream of either exon 2 or exon 3. CE2: cryptic exon 2. CE2′: cryptic exon 2 utilizing a different splice site downstream of exon 3. CE3: cryptic exon 3. (11, 25, 93).
Cytosolic AR is sequestered by heat shock proteins (HSPs) until it binds to androgens (3, 4). Ligand binding is a function of the conformation of the ligand-binding pocket, which prefers dihydrotestosterone (DHT) and testosterone (to a lesser extent) while excluding weaker androgens and non-androgens (3, 4). Following ligand binding, the AR undergoes a conformational change in which helix 12 covers the hormone-binding pocket, causing the AR to adopt the active conformation (5). The AR then forms a homodimer that is transported to the nucleus where it binds to DNA and activates gene transcription (Figure 2) (6).
Figure 2.
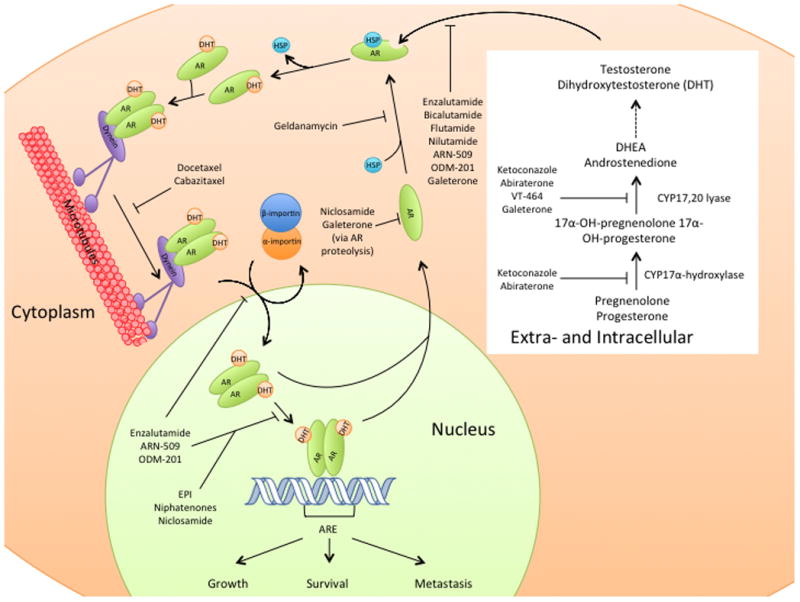
AR ligand binding, nuclear localization, and signaling in the normal prostate. Relevant drugs are shown inhibiting their respective target pathways. ARE: androgen response element. HSP: heat shock proteins.
The full-length AR contains a bipartite nuclear localization sequence that runs from the C-terminal end of the DNA-binding domain to the N-terminal end of the hinge region (Figure 1), which is necessary for regulation of nuclear transport by alpha and beta importin (7). In addition to the regulation by alpha and beta importins, the full-length AR also relies upon cytoskeletal nuclear transport to translocate to the nucleus (8, 9). In this modified version of nuclear transport, a portion the nuclear localization signal binds to dynein, which moves along microtubules toward the nucleus and enhances nuclear transport by alpha and beta importin (9). DNA binding results in the subsequent transactivation of various genes that contain AR elements in their promoter regions (10). Such genes are responsible for a range of functions, including cell growth and proliferation (Figure 2) (11).
AR signaling in metastatic prostate cancer progression and CRPC
Therapeutic resistance typically develops through several mechanisms that confer a selective advantage to tumor cells in a low-androgen environment: reliance on non-AR signaling pathways, intratumoral androgen biosynthesis, androgen scavenging, AR overexpression, AR splicing variation, and/or AR mutation (12). Whereas ADT resistance was thought to be a function of increased AR copy number in most cases, it has recently been proven that clinically relevant AR splice variants also contribute to progression on ADT (13, 14). Mutations that uncouple AR signaling from ligand binding are very-often involved in resistance to other classes of antiandrogens (13, 14). Taxanes also affect the AR pathway, which may be responsible for certain taxane-resistant tumors (13, 14). As these therapy-resistant cells grow, they become the dominant cell population that eventually progress in spite of treatment (13, 14). There are several biochemical mechanisms by which CRPC and/or therapeutic resistance arise; those that are caused by genomic alterations to the AR are summarized below.
BIOCHEMICAL EFFECTS OF ANDROGEN RECEPTOR VARIANTS
AR mutations
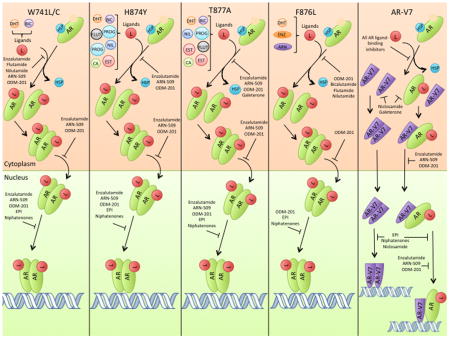
AR mutations, particularly those affecting the ligand-binding domain, contribute to prostate cancer progression and resistance to anti-androgens. Marcelli et al. indicated that while none of the study patients with early stage prostate cancer had mutations in their AR coding sequence, 21% of patients with advanced disease did (13). In general, somatic mutations that substitute an amino acid with a large size difference from the amino acid encoded by the germline in the ligand-binding pocket allow the AR to be more easily activated by alternative ligands (15). The AR is normally activated when DHT binds the ligand-binding pocket and helix 12 then moves into the active position (5). In the mutated state, amino acids that have a stronger affinity for helix 12 than the original amino acid can pull helix 12 closer to the active position and make the AR less reliant on ligand binding for activation (15). Alternatively, smaller amino acid substitutions in the ligand-binding domain result in a larger ligand-binding pocket that can accommodate more ligands. Although over 70 different AR missense mutations have been identified, H874Y, F876L, T877A, and W741L/C (Figure 3) are known to cause drug resistance and disease progression (16, 17).
Figure 3.

AR ligand binding, nuclear localization, and signaling in missense-mutated ARs and AR-V7. Treatments are included only if they remain effective for the respective variant shown. The potential ligands for each AR variant are shown and abbreviated as follows, DHT: dihydroxytestosterone. BIC: bicalutamide. FLUT: flutamide. NIL: nilutamide. PROG: progestins. EST: estradiol. CA: cyproterone acetate. HSP: heat shock protein. ENZ: enzalutamide. ARN: apalutamide/ARN-509.
The H874Y mutation is common in clinical prostate cancer and was first observed in the 22Rv1 cell line (18). This mutation is found in the C-terminus of helix 10/11 and is part of a sequence of 8 highly conserved amino acids in the AR (19). Because the histidine between helix 11 and 12 is switched for a larger, more hydrophobic residue (tyrosine), helix 12 is pushed closer to the ligand-binding pocket and the interaction between these two domains is increased. By strengthening this interaction, the H874Y mutation confers promiscuous binding to other ligands, such as estradiol and progesterone, which can now induce helix 12 to shift into the active position (Figure 3) (16, 19, 20). H874Y also allows the AR to better associate with p160 co-activators, further increasing AR activity (19, 20).
F876L is another missense mutation found in the ligand-binding domain of the AR that substitutes phenylalanine for leucine, a much smaller amino acid. Consequently, helix 12 can move into the active position in the presence of antiandrogens, such as enzalutamide (Figure 3) (21). This mutation is most relevant in the context of enzalutamide therapy and resistance, which will be discussed later.
The T877A mutation is frequently detected in CRPC and is the primary AR mutation in the LNCaP cell line (18, 22, 23). Located in helix 11 of the AR ligand-binding domain, T877 directly interacts with the AR ligand. Due to the smaller size of alanine, T877A results in a larger amount of space in the ligand-binding pocket thereby accommodating more ligands with different shapes (e.g., estradiol, progestins, cyproterone acetate) (Figure 3) (24). Like H874Y, T877A similarly increases AR promiscuity, but T877A specifically increases the preference for estradiol binding over that of progestins (19, 24).
The W741L/C mutation alters the tertiary structure of helix 12 such that it is nearer to the ligand-binding domain and closer to the active position (15). While this mutation lowers the affinity for the ligand-binding domain to interact with androgens, it also causes bicalutamide to act as an agonist of AR, rather than an antagonist (Figure 3) (15).
AR Splice Variants
While many different AR splice variants exist, AR-V1 through AR-V7 each have alternative splicing of exons that result in prematurely truncated ligand domains with the potential to confer resistance to ADT (Figure 1) (25). Of these, AR-V2 and AR-V4 are only observed in 22Rv1 cells, AR-V3 contains a stop codon within cryptic exon 4 that would exclude the second zinc finger of the AR DNA binding domain, and AR-V5 and AR-V6 are generally in low abundance (25). AR-V1 and AR-V7 are more frequently expressed in clinical CRPC (25). Increased expression of AR-V1 is not associated with treatment outcome however, since the AR-V1 splice variant is missing basic amino acids in the bipartite nuclear localization sequence of the protein. The lack of these critical amino acids results in shorter nuclear-retention of AR-V1. Since AR-V7 has a longer nuclear residence time, it appears to have the greatest impact on clinical prostate cancer (25).
Clinical data have demonstrated that AR-V7 and other AR splice variants contribute to prostate cancer disease progression. In a study by Hornberg et al., AR-V7 RNA transcripts were detected in 77% of primary prostate tumors, 80% of hormone-naïve bone metastases, and 100% of CRPC bone metastases (26). Patients expressing high levels of AR-V7 transcripts also had significantly shorter survival than those with low expression (26).
AR-V7 results from the contiguous splicing of AR exons 1/2/3/CE3, the latter of which is 16 amino acids from exon 3b (see Figure 1) (2, 11, 25). Because this alternative splicing results in the exclusion of the ligand-binding domain, AR-V7 can remain constitutively active in the absence of androgens (25, 27). This alternative splicing pattern may be caused by specific heterogenous nuclear ribonucleoproteins that favor AR-V7 over the normal AR (28). Heterogenous nuclear ribonucleoproteins (hnRNP) bind to mRNA to repress splicing events, and thereby regulate which splice variants are formed (29). hnRNPA1 and hnRNPA2 are the best characterized of the hnRNPs; both are up-regulated in some tumor tissues and are used as biomarkers for certain cancers (30). Down-regulation of hnRNPA1 and hnRNPA2 also decreases expression of AR variant mRNA in both 22Rv1 and VCaP cells, and down-regulation of hnRNPA1 specifically reduces transcript levels of AR-V7 (28). Overexpression of hnRNPA1 resulted in significant up-regulation of AR-V7 protein and other AR variants (28).
A study performed by Watson et al. discovered that AR-V7 relies on full-length AR signaling to enhance its own transcription. Ligand-dependent AR signaling reduces transcription of wild-type AR (wt-AR), thereby decreasing AR-V7 transcription as well (31). Therefore, ADT provides a selective advantage for cells expressing AR-V7 and is unintended consequence of therapy.
AR-V7 also maintains the ability to dimerize, which is necessary for transactivation of its effectors. AR-V7 has recently been shown to dimerize with itself, full length AR, and other AR splice variants, which increases the chances it will be transported into the nucleus to promote tumor growth (Figure 2) (32). Homodimerization and heterodimerization are a function of the interaction between the DNA binding domains of AR-V7 and its counterpart (32). Consequently, the AR-V7 splice variant’s ability to dimerize with multiple AR variants allows it to contribute significantly to tumor growth and the transition to CRPC.
While the transcriptional profile of the wild-type AR and AR-V7 ostensibly overlap, there are differences in key pathways that promote tumor growth. For example, wt-AR and AR-V7 both increase expression of genes involved in glycolysis, but AR-V7 differentially affects genes that control TCA cycle intermediates. Not only does AR-V7 increase citrate production by increasing expression of MDH1 (forming citrate from malate) and reducing OGDH overexpression (shunting α-ketoglutarate to citrate instead of succinate), but it also promotes citrate formation through GLUD1 overexpression by reductive carboxylation (forming α-ketoglutarate from glutamate). Increased citrate formation results in an increased formation of amino acids and steroids in AR-V7-expressing cells, ultimately causing lower intracellular citrate concentrations than would otherwise be present in cells expressing wt-AR (2). These findings, in part, explain why patients with AR-V7-positive tumors have reduced intratumoral citrate concentrations when compared to those tumors expressing wt-AR (2). Differential regulation of enzymes also results in an accumulation of oxaloacetate, which the cell can use to make additional TCA metabolites or amino acids (2). Furthermore, increased levels of GLUD1 allow AR-V7-positive cells to rely more heavily on reductive carboxylation to form α-ketoglutarate and drive the TCA cycle forward in spite of a low-oxygen environment (2).
AR MUTATIONS IN ANTI-ANDROGEN AND CHEMOTHERAPY RESISTANCE
Resistance to Surgical and Chemical Castration
While ADT initially slows prostate tumor growth, resistance occurs as tumors develop the capability to thrive in a low-androgen environment, and patients must then be treated with other therapies (33, 34). Cellular models and tumor histology suggest that resistance often develops due to AR overexpression, including wt-AR and AR-V7 (34). Furthermore, Guo et al. found that AR-V7 was the most frequently and abundantly expressed splice variant in CRPC patient samples, suggesting that AR-V7 may act as a biomarker for the transition to CRPC (35). These results support the theory that increased AR copy number and constitutively activated AR-V7 both play substantial roles in CRPC development before antiandrogen treatment is employed. In a clinical study, only one out of 17 patients treated with androgen deprivation had a somatic point mutation in their AR coding sequence (36). Conversely, 5 of the 16 patients treated with the androgen antagonist, flutamide, had the same T877A missense mutation present in their AR (36). Another clinical study found that while AR copy number increased in only 6% of pre-castration patients, 35% and 57% of plasma and tumor samples from patients with CRPC, respectively, had increased AR expression (37). Although AR mutations have been heavily implicated in developing resistance to anti-hormonal therapies such as abiraterone and enzalutamide, their role in resistance to primary castration treatment appears minor in comparison with increased copy number of AR and AR-V7 (38).
Resistance to Bicalutamide, Flutamide, and Nilutamide
Bicalutamide, flutamide, and nilutamide are competitive first-generation AR inhibitors, which prevent DHT binding and subsequent activation of the AR. However, various AR mutations arise during treatment that may render these drugs ineffective (15).
The missense mutation, W741L/C, may arise during bicalutamide therapy and contribute to drug resistance (15). While this mutant has less binding affinity for DHT than wild type AR, W741L/C causes greater AR-bicalutamide binding affinity and conversion of bicalutamide from an AR antagonist to an agonist (15, 39). Cells harboring this mutation are still sensitive to enzalutamide and nilutamide (40, 41).
Clinical data for the presence of mutations in mCRPC are difficult to obtain because removal of biopsies from common metastatic bone lesions or deep abdominal lymph nodes can be extremely invasive. However, a limited number of studies have performed deep sequencing of circulating cell-free DNA in an attempt to genotype metastatic tumors. In a study by Carreira et al., plasma samples from 16 patients were sequenced and tumor biopsies were collected to study the relationship between drug resistance and common genomic aberrations (37). In one patient, samples of circulating tumor DNA indicated an increase in AR copy number and identified the W741C mutation, which was confirmed by testing a liver biopsy from the patient. This mutation was observed in a liver metastasis that had developed 3 months after the patient had first started treatment with bicalutamide. When the liver tumor regressed, the W741C mutation was no longer detected. This is the only known clinical data supporting that W741L/C plays a role in bicalutamide resistance (37).
Flutamide resistance is conferred via the T877A AR missense mutation, which commonly occurs in prostate tumors (21). The promiscuous bindng properties of the T877A-mutated AR result in the ability of other ligands to bind to the AR and evade flutamide-mediated AR repression. In a clinical study, 5 of the 16 patients treated with flutamide had AR mutations, and all of them tested positive for T877A (36). Flutamide can also activate the H874Y-mutated AR, but it remains effective against the W741L/C mutant (41, 42). Although it may play a role in resistance, there is not yet any data relating AR-V7 expression levels with resistance to flutamide.
Nilutamide resistance can occur through the H874Y and T877A somatic mutations (42). Both of these mutations can switch nilutamide from an antagonist to an agonist of AR signaling (42, 43). In in vitro studies, nilutamide was still effective at suppressing AR signaling in cells containing W741C-mutated ARs (41). No clinical data providing deep-sequencing information to link nilutamide treatment efficacy with the expression AR mutants or splice variants has been gathered.
Resistance to Enzalutamide
Enzalutamide is a dual function AR antagonist and signaling inhibitor that both inhibits androgen binding to the AR, and decreases the affinity of the AR for DNA (44, 45). Treatment resistance to enzalutamide emerges after approximately 3.4 months (44, 46, 47). Some degree of enzalutamide resistance may be caused by AR-V7, which lacks the ligand-binding domain (48). Unlike flutamide, bicalutamide, and nilutamide, the DNA binding domain of AR-V7 is still inhibited by enzalutamide (45). In a study by Li et al., AR-V7-expressing cells were able to maintain transcription of AR-induced genes in the absence of androgens, and during treatment with enzalutamide (49). Cell lines that were resistant to enzalutamide and bicalutamide were only able overcome this resistance when AR-V7 levels were decreased (49).
Clinical studies support the theory that AR-V7 plays a central role in developing resistance to enzalutamide. Of 31 enzalutamide-treated patients, those with detectable expression of AR-V7 had lower PSA response rates at 0% compared to 53% without AR-V7 expression. These AR-V7-positive patients had a median overall survival of 5.5 months, whereas 50% overall survival was not reached in AR-V7-negative patients (50).
More studies have also indicated that the AR splice variants have their own specific transcriptional profile when exposed to enzalutamide. In particular, AR-V7 up-regulates genes, such as UBE2C, that regulate the cell cycle and promote growth after treatment with enzalutamide or other suppressors of AR signaling (51). Based on these data, Hu et al. concluded that differential AR signaling by AR-V7 might contribute to the development of enzalutamide resistance (51).
Enzalutamide resistance also arises due to the AR mutation, F876L. Korpal et al. found this mutation in all enzalutamide resistant cell lines, but not in weakly resistant or control lines (21). The F876L mutation allows enzalutamide to extend into the space left by the substitution of leucine for phenylalanine rather than blocking helix 12, thereby promoting AR activation (21). When AR activity was assessed using the synthetic androgen R1881, F876L-mutated AR was the only form that exhibited significant activity when combined with enzalutamide treatment, suggesting that the F876L mutation switches enzalutamide from an AR antagonist to an agonist (21).
In a clinical trial where patients were treated with ARN-509 (a second-generation anti-androgen that is highly similar to enzalutamide), deep sequencing revealed that rising levels of F876L-mutated AR were correlated with increased PSA and chronic exposure to ARN-509 (52). Based on preclinical data, the F876L mutation is thought to have the same effect on enzalutamide treatment, although no deep sequencing analysis has yet shown this (53). These data support the hypothesis that the F876L mutation plays a role in resistance to second-generation anti-androgens.
Resistance to Abiraterone
Abiraterone inhibits the hydroxylase and lyase functions of CYP17A1 (54). CYP17-hydroxylase converts pregnenolone and progesterone to 17a-hydroxypregnenolone and 17a-hydroxyprogesterone, whereas CYP17A1-lyase converts these metabolites into dehydroepiandrosterone (DHEA) and androstenedione, respectively. DHEA is the major circulating androgen that is primarily synthesized by the adrenals, whereas androsteinedione is more similar in structure to testosterone and is synthesized by the adrenals, gonads, and certain tumors that perform de novo testosterone biosynthesis. By inhibiting the formation of these two testosterone precursors, abiraterone significantly reduces the amount of adrenal and intratumoral testosterone that can promote tumor growth.
While abiraterone treatment is initially successful (44, 46), almost all patients eventually develop resistance after approximately 4.8 months (47). Although the AR is downstream of CYP17, AR-V7 is often indirectly involved in abiraterone resistance since its constitutive activity evades abiraterone-mediated restriction of androgen synthesis (48). In a clinical trial of abiraterone, AR-V7-expressing patients had a PSA response rate of 0% while AR-V7-negative patients had a PSA response rate of 68% (50). The progression free survival (PFS) of AR-V7 expressers was only 1.3 months, while 50% of patients not expressing AR-V7 had not yet progressed on abiraterone when the study was published (50). Similarly, overall survival was only 10.6 months in AR-V7-positive patients, while 50% overall survival was not reached AR-V7-negative patients (50).
Resistance to abiraterone can also occur through the H874Y and T877A missense mutations, which allow the AR to promiscuously bind and be activated by non-androgens such as progesterone and estradiol (19). Indeed, a clinical study detected both H874Y and T877A mutations in circulating cell-free DNA of abiraterone-resistant patients (18).
VT-464 is a CYP17 inhibitor highly similar to abiraterone, although it selectively inhibits the 17,20-lyase activity of CYP17 and does not affect the hydroxylase activity (55). While in vitro data suggest VT-464 could be a promising replacement for abiraterone, Phase II clinical trials are ongoing (55). No research has yet been done to assess how VT-464 treatment interacts with AR mutants or splice variants; however, because its activity is so similar to abiraterone, it is highly likely that VT-464 is susceptible to the same resistance mechanisms.
Taxane Resistance
Docetaxel and cabazitaxel are members of the taxane family of drugs, which bind beta-tubulin to stabilize microtubule networks (56). Such stabilization interrupts microtubule-dependent transport to the nucleus and causes mitotic arrest resulting in cell death (57). Taxanes also slow tumor growth by phosphorylating and inactivating Bcl-2, a protein that prevents apoptotic cell death (58).
While the full length AR has been shown to rely on microtubule association to be transported into the nucleus, AR-V7 does not display the same dependence on microtubules and can still be translocated to the nucleus after treatment with docetaxel by an unknown mechanism (8). In a study of 37 patients with mCRPC that had been treated with either docetaxel or cabazitaxel (n=30 and n=7, respectively), PSA response rate was not different between AR-V7-positive and AR-V7-negative patients (59, 60). Still, AR-V7-negative patients did have a slightly higher PFS (6.2 months vs 4.5 months, p=0.06) than AR-V7-positive patients (59, 60). Patients who were AR-V7-positive responded better to taxane treatment than to treatment with abiraterone and enzalutamide, suggesting AR-V7 is a valuable diagnostic tool when deciding which treatment to utilize (59, 60). Individuals who received docetaxel after treatment with abiraterone had lower docetaxel activity than anticipated, while abiraterone-resistant patients were completely unresponsive to docetaxel (61). Taken together, these data suggest that abiraterone and docetaxel resistance may occur by overlapping mechanisms.
FUTURE DIRECTIONS
Targeting Interacting Pathways
In prostate cancer cells, AR signaling regulates the insulin-like growth factor-1 (IGF-1) receptor and increases IGF-1 binding capacity (62). Through pathway crosstalk, IGF-1 also activates the AR in the absence of androgens (63). This positive feedback loop significantly contributes to both increased tumor growth and uncoupling of androgen signaling from androgens themselves (63). IGF-1 silencing with siRNA reduces PSA response in-vitro by 69% (64). Inhibition of IGF-1R also produces a significant PSA response in AR-V7-transfected PC-3 cells (64). Therefore, IGF-1 signaling up-regulates both AR and AR-V7, marking it as a drug target. Recently, metformin has been used to inhibit this pathway in prostate cancer, with limited success. Although in vitro studies have shown the ability of metformin to inhibit the growth and proliferation of PC-3 cells, clinical trials have indicated no significant difference in prostate cancer risk between metformin-treated and untreated patients (65, 66).
As previously mentioned, the AR is sequestered in the cytosol by HSPs (Figure 2) before ligand binding and subsequent nuclear translocation and held in a ligand-receptive conformation (3, 4). Without HSPs, AR folding is disrupted and the AR is targeted for degradation (67). Several drugs derived from geldanamycin are currently under investigation as HSP inhibitors, which act by binding the ATP-binding pocket of HSPs and preventing them from associating with AR (67, 68). While these drugs have enjoyed some limited success, cells harboring AR-V7 are resistant to HSP inhibitors since AR-V7 lacks the ligand-binding domain (67, 69).
HIF-1α signaling also appears to participate in a positive feedback loop with the AR (70). When HIF-1α was inhibited, either through siRNA silencing or treatment with the HIF-1α inhibitor, chetomin, the inhibitory effect of enzalutamide on cell growth was significantly increased (70). Furthermore, HIF-1α inhibition was able to restore enzalutamide sensitivity to the enzalutamide-resistant 22Rv1 prostate cancer cell line, suggesting a possible effect on AR-V7/AR signaling (70). However, no studies have yet been done to directly assess the effect of HIF-1α signaling on AR-V7 levels.
Numerous other pathways (including PI3/AKT, mTOR, TMPRSS2, MMP2 and FGF) interact with the AR signaling pathway and are therefore under investigation as drug targets to inhibit or down-regulate AR transcription and activity. Currently, there is limited information with respect to how AR somatic mutations or splice variants will affect these pathways and treatment options.
Targeting AR-V7 & Its Splicing Regulation
In a study by Qu et al., increased expression of AR-V7 in primary tumors predicted progression to CRPC and corresponded with poor prognosis in patients with CRPC (71). Because AR-V7 has been so heavily implicated in the transition to ADT-resistant prostate cancer, targeting splicing factors that selectively favor AR-V7 transcription over full length AR may help prevent prostate cancer progression and improve the prognosis of patients with CRPC. When AR pre-mRNA in enzalutamide-resistant 22Rv1 cells was analyzed, there was increased recruitment of hnRNPA1 to AR-V7 splice sites, suggesting hnRNPA1 plays an important role in the transition to mCRPC (28). When matched tumor and benign tissues from prostate cancer patients were analyzed, hnRNPA1 and hnRNPA2 levels were elevated in 44% of tumor tissues. The elevated levels of hnRNPA1 and 2 were also positively correlated with AR-V7 protein expression (28). When 22Rv1 cells resistant to enzalutamide were treated with hnRNPA1 siRNA silencing, they were re-sensitized to enzalutamide (28). The regulatory pathway involving NF-kB2/p52, c-Myc, and hnRNPA1 plays a central role in the generation of AR splice variants and enzalutamide sensitivity, uncovering a signal axis with potential drug targets (28).
Niclosamide
Niclosamide is a potential AR-V7 inhibitor that is relatively potent (GI50=0.5umol/L), has no effect on normal prostate epithelial cells, and is already FDA-approved for treatment of tapeworm infections (72). Unlike enzalutamide, niclosamide appears to act on the AR by binding both wt-AR and AR-V7, reducing their ability to bind DNA, and targeting them for degradation via the proteasome (72). Enzalutamide-resistant cells remain sensitive to niclosamide treatment and the combination of niclosamide and enzalutamide results in significant additive effects (72). Niclosamide also reduces tumor cell migration and invasion (73), perhaps by inhibiting phosphorylation of Stat3 by IL6, which mitigates activation of c-Myc and other downstream target genes that promote tumor cell metastasis (72, 74–77). While preliminary data on niclosamide seems promising, the bioavailability of this compound is quite low (78). Overcoming this will prove a formidable challenge for effectively implementing niclosamide in a clinical setting.
Galeterone
Galeterone simultaneously inhibits CYP17 and the AR, and it is currently in Phase III clinical trials (79). Compared with abiraterone, galeterone is three-times more potent in CYP17 enzyme activity assays (80). In in vitro competitive binding studies, galeterone successfully inhibited steroid binding to both the wild-type AR and the T877A AR mutant (79, 80). While galeterone showed slightly lower (but comparable) PSA reduction than enzalutamide in LNCaP cells, it showed greater PSA reduction in VCaP cells (81). Similar to enzalutamide, galeterone was also able to prevent AR chromatin binding (81).
Yu et al. also found that galeterone treatment increases degradation of T877A-mutated AR in LNCaP cells (81). Unfortunately, this effect was not replicated with the wt-AR, which demonstrated no difference in protein degradation after galeterone treatment (81). These data conflict with a more recent study by Kwegyir-Afful et al. that suggested galeterone treatment enhanced wt-AR degradation (82). Protein degradation of the T877A mutant is thought to occur through galeterone binding to the larger ligand-binding pocket provided by the T877A amino acid substitution. Unlike other compounds that can fit into this enhanced ligand-binding pocket, galeterone contains a bulky benzimidazole group that is hypothesized to distort the pocket and target the protein for degradation by E3 ubiquitin ligases that normally mediate the activated AR (81).
In addition to degrading AR mutants, galeterone also degrades the AR-V7 splice variant in vitro. When both LNCaP and 22Rv1 cells were concurrently treated with a proteasome inhibitor and galeterone, protein levels of AR-V7 increased when compared to treatment of these cells with galeterone alone. The level of AR-V7 ubiquitination was also increased with galeterone treatment, further suggesting that galeterone enhances degradation of these proteins by the 26S proteasome pathway (82). In in vivo studies, treatment with galeterone was shown to inhibit the growth of LNCaP cells in a dose-dependent manner (80). Furthermore, treatment with galeterone twice-daily decreased tumor size by 85% compared to controls in LAPC4 xenografts grown in SCID mice (80).
In a Phase II clinical trial of galeterone, 51 patients were dosed with varying amounts of galeterone for 12 weeks. Across all dosing levels, 82% of patients experienced a 30% decrease in PSA and 75% had at least a 50% decrease in PSA (83). Previous clinical trials have shown that galeterone has limited and minor side affects, making this an attractive treatment option (83). While most of the recent data indicates that galeterone is promising, clinical trials to determine whether galeterone is more effective than enzalutamide are currently ongoing. There are currently no clinical studies that ascertain the effects of AR splice variants or mutations on galeterone.
Apalutamide (ARN-509)
Apalutamide (ARN-509) is an AR antagonist that is structurally similar to enzalutamide and also prevents AR ligand binding, AR nuclear translocation, and AR-DNA binding (84). When compared to enzalutamide, apalutamide was shown to be a more potent AR inhibitor in vivo (30mg/Kg/day vs 100mg/Kg/day) and has 4-fold less brain penetration (which causes convulsions in some patients by binding the GABA-gated chloride ion channel) than enzalutamide (84–86). The toxicity profile and potency of apalutamide may be more desirable, as indicated by early results in a Phase I clinical trial (86).
In an in vivo study involving mice injected with LNCaP tumors expressing F876L-mutated AR, neither apalutamide nor enzalutamide were able to decrease tumor growth and actually showed agonist activity for this AR mutant (52). Therefore, the F876L AR missense mutation is associated with apalutamide resistance, which was expected based on the structural similarity between apalutamide and enzalutamide. Apalutamide also binds to the AR ligand-binding domain, although no studies have examined if apalutamide is effective for the treatment of AR-V7-positive tumors.
ODM-201
ODM-201 is an AR antagonist similar to enzalutamide that is currently in Phase III clinical trials for prostate cancer treatment (87). When tested against enzalutamide and apalutamide, ODM-201 demonstrated more potent AR inhibition (87). In earlier Phase I and II clinical trials, ODM-201 showed minimal side effects (88). And, unlike enzalutamide and apalutamide, even the highest doses of ODM-201 did not produce seizures in any patients studied (88). It has been suggested by results from in vivo rat studies that, possibly due to its unique structure, ODM-201 is not able to bypass the blood-brain barrier to a significant extent (87, 88). Phase III clinical trials are currently in progress to determine if ODM-201 provides more effective treatment for prostate cancer than currently FDA-approved drugs. While bicalutamide is converted to an agonist when bound to W741L/C-mutated AR and enzalutamide/apalutamide activate the F876L-mutated AR, ODM-201 inhibited both of these AR mutants (as well as T877A) without any reported agonist activity (87).
Targeting the AR’s N-terminus
While most therapies targeting the AR have been focused on the C-terminal ligand-binding domain, the fact that this is altered or excluded in many drug-resistant AR phenotypes has motivated the development of drugs targeting the N-terminal region of the AR (89). Recently, EPI has been shown to bind successfully to the N-terminal region of the AR and prevent it from activating genes (89). EPI was shown to have the same effect on a constitutively active AR-V7, making it an attractive therapy option even in the presence of AR ligand-binding domain mutants or splice variants (89). Furthermore, when combined with docetaxel treatment, EPI significantly inhibited prostate cancer cell growth both in vitro and in vivo (90).
Niphatenones, small molecules derived from a sea sponge, have also been shown to inhibit the AR by binding the N-terminal domain (91). When niphatenones bind the N-terminal domain of the AR, they prevent the AR from binding with proteins needed for activation and subsequent transcriptional regulation, successfully inhibiting its activity (92). These compounds have also been shown to inhibit the proliferation of LNCaP cells (92). However, the AR-inhibiting action of these compounds is mediated by observed off-target effects, such as the formation of glutathione adducts (92). These off-target effects were not observed in EPI and as a result niphatenones have less potential as a treatment option for prostate cancer (92).
CONCLUSION
Overall, mutability of the AR has impeded therapeutic development in prostate cancer. While second-generation anti-androgens and taxanes provide patients with some extended survival benefit, future treatments directly targeting AR variants are underway to overcome the acquired resistance seen with these therapies. As clinical tumor-genotyping methods improve, future clinical trials should identify AR mutations and take them into account when developing novel agents to increase precision in drug development and clinical treatment of prostate cancer.
Acknowledgments
This study was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Bethesda, MD, USA.
Footnotes
Disclaimer
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: a cancer journal for clinicians. 2016;66(1):7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, Coarfa C, Frigo DE, Putluri N, Sreekumar A, Weigel NL. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6(31):31997–32012. doi: 10.18632/oncotarget.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azad AA, Zoubeidi A, Gleave ME, Chi KN. Targeting heat shock proteins in metastatic castration-resistant prostate cancer. Nature reviews Urology. 2015;12(1):26–36. doi: 10.1038/nrurol.2014.320. [DOI] [PubMed] [Google Scholar]
- 4.Srinivas-Shankar U, Wu FC. Drug insight: testosterone preparations. Nature clinical practice Urology. 2006;3(12):653–665. doi: 10.1038/ncpuro0650. [DOI] [PubMed] [Google Scholar]
- 5.Zhou J, Geng G, Shi Q, Sauriol F, Wu JH. Design and synthesis of androgen receptor antagonists with bulky side chains for overcoming antiandrogen resistance. Journal of medicinal chemistry. 2009;52(17):5546–5550. doi: 10.1021/jm801218k. [DOI] [PubMed] [Google Scholar]
- 6.Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. Journal of carcinogenesis. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clinckemalie L, Vanderschueren D, Boonen S, Claessens F. The hinge region in androgen receptor control. Molecular and cellular endocrinology. 2012;358(1):1–8. doi: 10.1016/j.mce.2012.02.019. [DOI] [PubMed] [Google Scholar]
- 8.Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR, Giannakakou P. Androgen receptor splice variants determine taxane sensitivity in prostate cancer. Cancer research. 2014;74(8):2270–2282. doi: 10.1158/0008-5472.CAN-13-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagstaff KM, Jans DA. Importins and beyond: non-conventional nuclear transport mechanisms. Traffic. 2009;10(9):1188–1198. doi: 10.1111/j.1600-0854.2009.00937.x. [DOI] [PubMed] [Google Scholar]
- 10.Gordon V, Bhadel S, Wunderlich W, Zhang J, Ficarro SB, Mollah SA, Shabanowitz J, Hunt DF, Xenarios I, Hahn WC, Conaway M, Carey MF, Gioeli D. CDK9 regulates AR promoter selectivity and cell growth through serine 81 phosphorylation. Molecular endocrinology. 2010;24(12):2267–2280. doi: 10.1210/me.2010-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocrine-related cancer. 2011;18(5):R183–196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sissung TM, Reece KM, Spencer S, Figg WD. Contribution of the OATP1B subfamily to cancer biology and treatment. Clinical pharmacology and therapeutics. 2012;92(5):658–660. doi: 10.1038/clpt.2012.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcelli M, Ittmann M, Mariani S, Sutherland R, Nigam R, Murthy L, Zhao Y, DiConcini D, Puxeddu E, Esen A, Eastham J, Weigel NL, Lamb DJ. Androgen receptor mutations in prostate cancer. Cancer research. 2000;60(4):944–949. [PubMed] [Google Scholar]
- 14.Taplin ME, Bubley GJ, Shuster TD, Frantz ME, Spooner AE, Ogata GK, Keer HN, Balk SP. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. The New England journal of medicine. 1995;332(21):1393–1398. doi: 10.1056/NEJM199505253322101. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, An X, Li S, Wang Y, Li J, Liu H. Interaction mechanism exploration of R-bicalutamide/S-1 with WT/W741L AR using molecular dynamics simulations. Molecular bioSystems. 2015;11(12):3347–3354. doi: 10.1039/c5mb00499c. [DOI] [PubMed] [Google Scholar]
- 16.Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Current genomics. 2009;10(1):18–25. doi: 10.2174/138920209787581307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB): 2004 update. Human mutation. 2004;23(6):527–533. doi: 10.1002/humu.20044. [DOI] [PubMed] [Google Scholar]
- 18.Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, Anderson SA, McConeghy B, Shukin R, Bazov J, Youngren J, Paris P, Thomas G, Small EJ, Wang Y, Gleave ME, Collins CC, Chi KN. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21(10):2315–2324. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 19.Duff J, McEwan IJ. Mutation of histidine 874 in the androgen receptor ligand-binding domain leads to promiscuous ligand activation and altered p160 coactivator interactions. Molecular endocrinology. 2005;19(12):2943–2954. doi: 10.1210/me.2005-0231. [DOI] [PubMed] [Google Scholar]
- 20.Brooke GN, Parker MG, Bevan CL. Mechanisms of androgen receptor activation in advanced prostate cancer: differential co-activator recruitment and gene expression. Oncogene. 2008;27(21):2941–2950. doi: 10.1038/sj.onc.1210955. [DOI] [PubMed] [Google Scholar]
- 21.Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, Monahan JE, Stegmeier F, Roberts TM, Sellers WR, Zhou W, Zhu P. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer discovery. 2013;3(9):1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 22.Mahmoud AM, Zhu T, Parray A, Siddique HR, Yang W, Saleem M, Bosland MC. Differential effects of genistein on prostate cancer cells depend on mutational status of the androgen receptor. PloS one. 2013;8(10):e78479. doi: 10.1371/journal.pone.0078479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. The Journal of steroid biochemistry and molecular biology. 1992;41(3–8):665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 24.Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. International journal of cancer Journal international du cancer. 2002;100(3):309–317. doi: 10.1002/ijc.10495. [DOI] [PubMed] [Google Scholar]
- 25.Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, Bova GS, Luo J. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer research. 2009;69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, Bergh A, Wikstrom P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS one. 2011;6(4):e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer research. 2008;68(13):5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nadiminty N, Tummala R, Liu C, Lou W, Evans CP, Gao AC. NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Molecular cancer therapeutics. 2015;14(8):1884–1895. doi: 10.1158/1535-7163.MCT-14-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchette M, Green RE, MacArthur S, Brooks AN, Brenner SE, Eisen MB, Rio DC. Genome-wide analysis of alternative pre-mRNA splicing and RNA-binding specificities of the Drosophila hnRNP A/B family members. Molecular cell. 2009;33(4):438–449. doi: 10.1016/j.molcel.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, Karni R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer research. 2011;71(13):4464–4472. doi: 10.1158/0008-5472.CAN-10-4410. [DOI] [PubMed] [Google Scholar]
- 31.Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(39):16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, Gambhir SS, Lee P, Sartor O, Flemington EK, Zhang H, Hu CD, Dong Y. Androgen Receptor Splice Variants Dimerize to Transactivate Target Genes. Cancer research. 2015;75(17):3663–3671. doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamaoka M, Hara T, Kusaka M. Overcoming persistent dependency on androgen signaling after progression to castration-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(17):4319–4324. doi: 10.1158/1078-0432.CCR-10-0255. [DOI] [PubMed] [Google Scholar]
- 34.Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, Balk SP. Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(6):1590–1600. doi: 10.1158/1078-0432.CCR-13-1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, Kung HJ, Brodie AM, Edwards J, Qiu Y. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research. 2009;69(6):2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taplin ME, Bubley GJ, Ko YJ, Small EJ, Upton M, Rajeshkumar B, Balk SP. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer research. 1999;59(11):2511–2515. [PubMed] [Google Scholar]
- 37.Carreira S, Romanel A, Goodall J, Grist E, Ferraldeschi R, Miranda S, Prandi D, Lorente D, Frenel JS, Pezaro C, Omlin A, Rodrigues DN, Flohr P, Tunariu N, SdBJ, Demichelis F, Attard G. Tumor clone dynamics in lethal prostate cancer. Science translational medicine. 2014;6(254):254ra125. doi: 10.1126/scitranslmed.3009448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, Jarosz M, Lipson D, Tagawa ST, Nanus DM, Stephens PJ, Mosquera JM, Cronin MT, Rubin MA. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. European urology. 2013;63(5):920–926. doi: 10.1016/j.eururo.2012.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(17):6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Neill D, Jones D, Wade M, Grey J, Nakjang S, Guo W, Cork D, Davies BR, Wedge SR, Robson CN, Gaughan L. Development and exploitation of a novel mutant androgen receptor modelling strategy to identify new targets for advanced prostate cancer therapy. Oncotarget. 2015;6(28):26029–26040. doi: 10.18632/oncotarget.4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Urushibara M, Ishioka J, Hyochi N, Kihara K, Hara S, Singh P, Isaacs JT, Kageyama Y. Effects of steroidal and non-steroidal antiandrogens on wild-type and mutant androgen receptors. The Prostate. 2007;67(8):799–807. doi: 10.1002/pros.20542. [DOI] [PubMed] [Google Scholar]
- 42.Fenton MA, Shuster TD, Fertig AM, Taplin ME, Kolvenbag G, Bubley GJ, Balk SP. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 1997;3(8):1383–1388. [PubMed] [Google Scholar]
- 43.Marhefka CA, Moore BM, 2nd, Bishop TC, Kirkovsky L, Mukherjee A, Dalton JT, Miller DD. Homology modeling using multiple molecular dynamics simulations and docking studies of the human androgen receptor ligand binding domain bound to testosterone and nonsteroidal ligands. Journal of medicinal chemistry. 2001;44(11):1729–1740. doi: 10.1021/jm0005353. [DOI] [PubMed] [Google Scholar]
- 44.Scher HI, Beer TM, Higano CS, Anand A, Taplin ME, Efstathiou E, Rathkopf D, Shelkey J, Yu EY, Alumkal J, Hung D, Hirmand M, Seely L, Morris MJ, Danila DC, Humm J, Larson S, Fleisher M, Sawyers CL Prostate Cancer Foundation/Department of Defense Prostate Cancer Clinical Trials C. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1–2 study. Lancet. 2010;375(9724):1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324(5928):787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, Messiou C, Molife LR, Maier G, Thompson E, Olmos D, Sinha R, Lee G, Dowsett M, Kaye SB, Dearnaley D, Kheoh T, Molina A, de Bono JS. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27(23):3742–3748. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nadal R, Tsai HL, Sinibaldi VJ, Paller CJ, Antonarakis ES, Denmeade SR, Carducci MA, Eisenberger MA. Prognostic factors for clinical outcomes in patients with metastatic castration resistant prostate cancer treated with sequential novel androgen receptor-directed therapies. The Prostate. 2015 doi: 10.1002/pros.23141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crona DJ, Milowsky MI, Whang YE. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clinical pharmacology and therapeutics. 2015;98(6):582–589. doi: 10.1002/cpt.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer research. 2013;73(2):483–489. doi: 10.1158/0008-5472.CAN-12-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine. 2014;371(11):1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu R, Lu C, Mostaghel EA, Yegnasubramanian S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E, Plymate SR, Luo J. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer research. 2012;72(14):3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer discovery. 2013;3(9):1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 53.Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Donnell A, Judson I, Dowsett M, Raynaud F, Dearnaley D, Mason M, Harland S, Robbins A, Halbert G, Nutley B, Jarman M. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. British journal of cancer. 2004;90(12):2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Toren PJ, Kim S, Pham S, Mangalji A, Adomat H, Guns ES, Zoubeidi A, Moore W, Gleave ME. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Molecular cancer therapeutics. 2015;14(1):59–69. doi: 10.1158/1535-7163.MCT-14-0521. [DOI] [PubMed] [Google Scholar]
- 56.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa ST, Bander NH, Nanus DM, Giannakakou P. Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer research. 2011;71(18):6019–6029. doi: 10.1158/0008-5472.CAN-11-1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu ML, Horbinski CM, Garzotto M, Qian DZ, Beer TM, Kyprianou N. Tubulin-targeting chemotherapy impairs androgen receptor activity in prostate cancer. Cancer research. 2010;70(20):7992–8002. doi: 10.1158/0008-5472.CAN-10-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kraus LA, Samuel SK, Schmid SM, Dykes DJ, Waud WR, Bissery MC. The mechanism of action of docetaxel (Taxotere) in xenograft models is not limited to bcl-2 phosphorylation. Investigational new drugs. 2003;21(3):259–268. doi: 10.1023/a:1025436307913. [DOI] [PubMed] [Google Scholar]
- 59.Maughan BL, Antonarakis ES. Clinical Relevance of Androgen Receptor Splice Variants in Castration-Resistant Prostate Cancer. Current treatment options in oncology. 2015;16(12):57. doi: 10.1007/s11864-015-0375-z. [DOI] [PubMed] [Google Scholar]
- 60.Antonarakis ES, Lu C, Luber B, Wang H, Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA oncology. 2015;1(5):582–591. doi: 10.1001/jamaoncol.2015.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mezynski J, Pezaro C, Bianchini D, Zivi A, Sandhu S, Thompson E, Hunt J, Sheridan E, Baikady B, Sarvadikar A, Maier G, Reid AH, Mulick Cassidy A, Olmos D, Attard G, de Bono J. Antitumour activity of docetaxel following treatment with the CYP17A1 inhibitor abiraterone: clinical evidence for cross-resistance? Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2012;23(11):2943–2947. doi: 10.1093/annonc/mds119. [DOI] [PubMed] [Google Scholar]
- 62.Pandini G, Mineo R, Frasca F, Roberts CT, Jr, Marcelli M, Vigneri R, Belfiore A. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer research. 2005;65(5):1849–1857. doi: 10.1158/0008-5472.CAN-04-1837. [DOI] [PubMed] [Google Scholar]
- 63.Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer research. 1994;54(20):5474–5478. [PubMed] [Google Scholar]
- 64.Zengerling F, Azoitei A, Herweg A, Jentzmik F, Cronauer MV. Inhibition of IGF-1R diminishes transcriptional activity of the androgen receptor and its constitutively active, C-terminally truncated counterparts Q640X and AR-V7. World journal of urology. 2015 doi: 10.1007/s00345-015-1674-5. [DOI] [PubMed] [Google Scholar]
- 65.Kato H, Sekine Y, Furuya Y, Miyazawa Y, Koike H, Suzuki K. Metformin inhibits the proliferation of human prostate cancer PC-3 cells via the downregulation of insulin-like growth factor 1 receptor. Biochemical and biophysical research communications. 2015;461(1):115–121. doi: 10.1016/j.bbrc.2015.03.178. [DOI] [PubMed] [Google Scholar]
- 66.Franciosi M, Lucisano G, Lapice E, Strippoli GF, Pellegrini F, Nicolucci A. Metformin therapy and risk of cancer in patients with type 2 diabetes: systematic review. PloS one. 2013;8(8):e71583. doi: 10.1371/journal.pone.0071583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen Y, Sawyers CL, Scher HI. Targeting the androgen receptor pathway in prostate cancer. Current opinion in pharmacology. 2008;8(4):440–448. doi: 10.1016/j.coph.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shafi AA, Yen AE, Weigel NL. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacology & therapeutics. 2013;140(3):223–238. doi: 10.1016/j.pharmthera.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 69.He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, Proia DA. Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. International journal of oncology. 2013;42(1):35–43. doi: 10.3892/ijo.2012.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fernandez EV, Reece KM, Ley AM, Troutman SM, Sissung TM, Price DK, Chau CH, Figg WD. Dual targeting of the androgen receptor and hypoxia-inducible factor 1alpha pathways synergistically inhibits castration-resistant prostate cancer cells. Molecular pharmacology. 2015;87(6):1006–1012. doi: 10.1124/mol.114.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qu Y, Dai B, Ye D, Kong Y, Chang K, Jia Z, Yang X, Zhang H, Zhu Y, Shi G. Constitutively active AR-V7 plays an essential role in the development and progression of castration-resistant prostate cancer. Scientific reports. 2015;5:7654. doi: 10.1038/srep07654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(12):3198–3210. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu C, Lou W, Armstrong C, Zhu Y, Evans CP, Gao AC. Niclosamide suppresses cell migration and invasion in enzalutamide resistant prostate cancer cells via Stat3-AR axis inhibition. The Prostate. 2015;75(13):1341–1353. doi: 10.1002/pros.23015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin TH, Izumi K, Lee SO, Lin WJ, Yeh S, Chang C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell death & disease. 2013;4:e764. doi: 10.1038/cddis.2013.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chu XY, Chen LB, Wang JH, Su QS, Yang JR, Lin Y, Xue LJ, Liu XB, Mo XB. Overexpression of survivin is correlated with increased invasion and metastasis of colorectal cancer. Journal of surgical oncology. 2012;105(6):520–528. doi: 10.1002/jso.22134. [DOI] [PubMed] [Google Scholar]
- 76.Tai CJ, Chin-Sheng H, Kuo LJ, Wei PL, Lu HH, Chen HA, Liu TZ, Liu JJ, Liu DZ, Ho YS, Wu CH, Chang YJ. Survivin-mediated cancer cell migration through GRP78 and epithelial-mesenchymal transition (EMT) marker expression in Mahlavu cells. Annals of surgical oncology. 2012;19(1):336–343. doi: 10.1245/s10434-011-1692-5. [DOI] [PubMed] [Google Scholar]
- 77.Knight BB, Oprea-Ilies GM, Nagalingam A, Yang L, Cohen C, Saxena NK, Sharma D. Survivin upregulation, dependent on leptin-EGFR-Notch1 axis, is essential for leptin-induced migration of breast carcinoma cells. Endocrine-related cancer. 2011;18(4):413–428. doi: 10.1530/ERC-11-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews P, Thyssen J, Lorke D. The biology and toxicology of molluscicides, Bayluscide. Pharmacology & therapeutics. 1982;19(2):245–295. doi: 10.1016/0163-7258(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 79.Bruno RD, Vasaitis TS, Gediya LK, Purushottamachar P, Godbole AM, Ates-Alagoz Z, Brodie AM, Njar VC. Synthesis and biological evaluations of putative metabolically stable analogs of VN/124-1 (TOK-001): head to head anti-tumor efficacy evaluation of VN/124-1 (TOK-001) and abiraterone in LAPC-4 human prostate cancer xenograft model. Steroids. 2011;76(12):1268–1279. doi: 10.1016/j.steroids.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Handratta VD, Vasaitis TS, Njar VC, Gediya LK, Kataria R, Chopra P, Newman D, Jr, Farquhar R, Guo Z, Qiu Y, Brodie AM. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. Journal of medicinal chemistry. 2005;48(8):2972–2984. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- 81.Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20(15):4075–4085. doi: 10.1158/1078-0432.CCR-14-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kwegyir-Afful AK, Ramalingam S, Purushottamachar P, Ramamurthy VP, Njar VC. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget. 2015;6(29):27440–27460. doi: 10.18632/oncotarget.4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. Journal of medicinal chemistry. 2015;58(5):2077–2087. doi: 10.1021/jm501239f. [DOI] [PubMed] [Google Scholar]
- 84.Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y, Grillot K, Bischoff ED, Cai L, Aparicio A, Dorow S, Arora V, Shao G, Qian J, Zhao H, Yang G, Cao C, Sensintaffar J, Wasielewska T, Herbert MR, Bonnefous C, Darimont B, Scher HI, Smith-Jones P, Klang M, Smith ND, De Stanchina E, Wu N, Ouerfelli O, Rix PJ, Heyman RA, Jung ME, Sawyers CL, Hager JH. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer research. 2012;72(6):1494–1503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bambury RM, Rathkopf DE. Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urologic oncology. 2015 doi: 10.1016/j.urolonc.2015.05.025. [DOI] [PubMed] [Google Scholar]
- 86.Rathkopf D, Liu G, Carducci MA, Eisenberger MA, Anand A, Morris MJ, Slovin SF, Sasaki Y, Takahashi S, Ozono S, Fung NK, Cheng S, Gan J, Gottardis M, Obermeier MT, Reddy J, Zhang S, Vakkalagadda BJ, Alland L, Wilding G, Scher HI Prostate Cancer Clinical Trials C. Phase I dose-escalation study of the novel antiandrogen BMS-641988 in patients with castration-resistant prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(4):880–887. doi: 10.1158/1078-0432.CCR-10-2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykanen PS, Tormakangas OP, Palvimo JJ, Kallio PJ. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Scientific reports. 2015;5:12007. doi: 10.1038/srep12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fizazi K, Massard C, Bono P, Jones R, Kataja V, James N, Garcia JA, Protheroe A, Tammela TL, Elliott T, Mattila L, Aspegren J, Vuorela A, Langmuir P, Mustonen M group As. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. The Lancet Oncology. 2014;15(9):975–985. doi: 10.1016/S1470-2045(14)70240-2. [DOI] [PubMed] [Google Scholar]
- 89.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer cell. 2010;17(6):535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 90.Martin SK, Banuelos CA, Sadar MD, Kyprianou N. N-terminal targeting of androgen receptor variant enhances response of castration resistant prostate cancer to taxane chemotherapy. Molecular oncology. 2014 doi: 10.1016/j.molonc.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Meimetis LG, Williams DE, Mawji NR, Banuelos CA, Lal AA, Park JJ, Tien AH, Fernandez JG, de Voogd NJ, Sadar MD, Andersen RJ. Niphatenones, glycerol ethers from the sponge Niphates digitalis block androgen receptor transcriptional activity in prostate cancer cells: structure elucidation, synthesis, and biological activity. Journal of medicinal chemistry. 2012;55(1):503–514. doi: 10.1021/jm2014056. [DOI] [PubMed] [Google Scholar]
- 92.Banuelos CA, Lal A, Tien AH, Shah N, Yang YC, Mawji NR, Meimetis LG, Park J, Kunzhong J, Andersen RJ, Sadar MD. Characterization of niphatenones that inhibit androgen receptor N-terminal domain. PloS one. 2014;9(9):e107991. doi: 10.1371/journal.pone.0107991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu R, Isaacs WB, Luo J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. The Prostate. 2011;71(15):1656–1667. doi: 10.1002/pros.21382. [DOI] [PMC free article] [PubMed] [Google Scholar]