

Abstract
Gigantism and acromegaly are rare disorders that are caused by excessive GH secretion and/or high levels of its mediator, IGF-1. Gigantism occurs when excess GH or IGF-1 lead to increased linear growth, before the end of puberty and epiphyseal closure. The majority of cases arise from a benign GH-secreting pituitary adenoma, with an incidence of pituitary gigantism and acromegaly of approximately 8 and 11 per million person-years, respectively. Over the past two decades, our increasing understanding of the molecular and genetic etiologies of pituitary gigantism and acromegaly yielded several genetic causes, including multiple endocrine neoplasia type 1 and 4, McCune-Albright syndrome, Carney complex, familial isolated pituitary adenoma, pituitary adenoma association due to defects in familial succinate dehydrogenase genes, and the recently identified X-linked acrogigantism. The early diagnosis of these conditions helps guide early intervention, screening, and genetic counseling of patients and their family members. In this review, we provide a concise and up-to-date discussion on the genetics of gigantism and acromegaly.
Keywords: Acromegaly, Gigantism, IGF-1, GH, Genetics, X-LAG, Pituitary adenoma
Introduction
Gigantism and acromegaly are distinguished by the status of the epiphyseal growth plates with respect to GH and/or IGF-1 excess; otherwise, the two disorders represent a continuum of clinical manifestations: it is not uncommon for acromegalics to be tall, and most giants have acromegalic features. Nevertheless, it is important to remember that gigantism occurs only when the epiphyseal growth plates are not fused, whereas acromegaly affects individuals with normal height. In both conditions, most of the time, the cause is a benign GH-secreting pituitary tumor (co-secretion of prolactin is called mammosomatotropic), and less likely from pituitary hyperplasia, ectopic GH or growth hormone releasing hormone (GHRH) secretion. The incidence of pituitary gigantism and acromegaly is likely under reported; approximately 8 and 11 cases per million person-years, respectively.1-3 The term acromegaly was coined in 1886 using the Greek words “akron” and “mega” to describe the typical large features of this condition.1,2 The childhood onset of gigantism and the fact that both giants and acromegalics were frequently known to have inherited a condition that run in their families, suggested early that at least pituitary-dependent GH excess could be a genetic disease. Indeed, over the past three decades, genetic investigations have revealed a number of genetic defects predisposing to gigantism or acromegaly (Tables 1 and 2, Fig. 1).4 These syndromes have been widely studied and include multiple endocrine neoplasia (MEN) syndromes types 1 and 4 (MEN1 and MEN4), McCune-Albright syndrome (MAS), Carney complex (CNC), the 3Ps (paraganglioma, pheochromocytoma and pituitary adenoma) association (3PA) due to defects in genes coding for succinate dehydrogenase (SDHx), and familial isolated pituitary adenoma (FIPA).5,6 Recently, we described a form of familial and sporadic acromegaly and gigantism that is caused by microduplications on chromosome Xq26.3, a disorder that we named X-LAG for X-linked acrogigantism.7 X-LAG may be responsible for as many as 80% of the cases of pre-pubertal gigantism. Together, FIPA and X-LAG may explain a substantial number of patients with gigantism and acromegaly as shown by a recent study of 208 patients (78.4% males), mostly with macroadenomas (84%) causing GH excess: 29% of the patients had AIP mutations, whereas Xq26.3 microduplications were seen in two families previously diagnosed with FIPA and in 10 sporadic patients (10%).8 In this report, we review the recent data on gigantism and acromegaly and provide recommendations for genetic screening of patients with GH excess.
Table 1.
Nonstandard abbreviations, acronyms and definitions
| 3PA: pituitary adenoma association |
| AC: adenyl cyclase |
| (c) AMP: cyclic adenosine monophosphate |
| CNC: Carney complex |
| FIPA: familial isolated pituitary adenoma |
| GPCRs: G protein-coupled receptors |
| MAS: McCune-Albright syndrome |
| MEN: Multiple endocrine neoplasia |
| NF1: neurofibromatosis 1 |
| PKA: protein kinase A |
| PPNAD: primary pigmented nodular adrenocortical disease |
| PRKAR1A: protein kinase A regulatory subunit type 1 |
| SOCS: suppressors of cytokine signaling |
| X-LAG: X-linked acrogigantism |
Table 2.
Genetic syndromes associated with gigantism and acromegaly.
| Syndrome | Gene | Chromosome | Function | Inheritance |
|---|---|---|---|---|
| Familial isolated pituitary adenoma (FIPA) |
AIP | 11q13.3 | Tumor suppressor | Autosomal dominant or sporadic |
| X-linked acrogigantism (X- LAG) |
GPR101 | Xq26.3 | Putative oncogene | X-linked dominant or sporadic |
| Carney complex (CNC) | PRKAR1A | 17q24.2 | Tumor suppressor | Autosomal dominant |
| Multiple endocrine neoplasia type 1 (MEN1) |
MEN1 | 11q13.1 | Tumor suppressor | Autosomal dominant or sporadic |
| Multiple endocrine neoplasia type 4 (MEN4) |
CDKN1B | 12p13.1 | Tumor suppressor | Autosomal dominant |
| McCune-Albright Syndrome (MAS) |
GNAS1 | 20q13.32 | Oncogene | Sporadic |
| Paraganglioma, pheochromocytoma, and pituitary adenoma association (3PA) |
SDHA SDHB SDHC SDHD* |
5p15.33 1p36.13 1q23.3 11q23.1 |
Tumor suppressor | Autosomal dominant or sporadic *SDHD with genomic imprinting |
Figure 1.
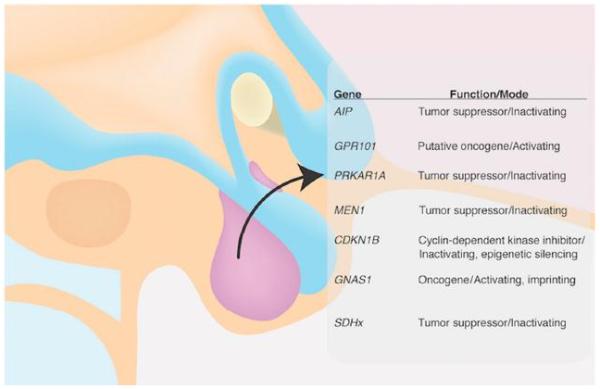
Genetic mutations associated with gigantism and acromegaly.
1. Familial isolated pituitary adenoma (FIPA)
FIPA (OMIM #605555) represents the largest group of familial gigantism and acromegaly. Mutations in AIP are identified in approximately 15-20% of familial FIPA and predominantly cause a truncated or missing protein. The gene AIP is mapped to chromosome 11q13.3 and functions as a tumor suppressor gene.9,10 Typically, mutations in AIP affect young patients, with or without a family history, with a low penetrance (approximately 15-30%). Gigantism is a relative manifestation of AIP mutations and occurs in about one-third of patients with a somatotropinoma.6,8
In a 2006 population-based series from Northern Finland, Vierimaa et al.10 identified the first AIP mutations implicated in the pathogenesis of acromegaly. A founder Gln14-to-Ter (p.Q14*) substitution in exon 1 accounted for 16% of acromegalics.10 In a subsequent study of 73 FIPA families (156 patients with pituitary adenomas, including somatotropinomas, GH and prolactin co-secreting tumors, prolactinomas, and non-secreting tumors), 11 families were reported to harbor 10 different germline AIP mutations.5 Tumors with mutation-positive AIP (FIPA representing 15%, familial somatotropinomas representing 50%) were significantly larger (P = 0.0005) and diagnosed at a younger age (P = 0.0006) than their mutation-negative counterparts.5 Other studies confirmed the causative role for AIP point mutations and large deletions in pituitary tumorigenesis in the setting of FIPA and, more rarely, sporadic acromegaly.9,11-14 The molecular pathway(s) affected by the defective AIP protein are being heavily investigated.15-19 Interestingly, other mechanisms leading to reduced AIP protein expression in sporadic acromegaly in the absence of AIP mutations have been reported,20,21 including the identification of potential modifier loci.22
Two recently published studies confirmed and extended the initial clinical and genetic spectrum for FIPA. Daly et al.23 compared 96 patients with germline AIP mutations and pituitary adenomas to 232 AIP mutation-negative acromegalics in order to identify statistically significant features associated with AIP defects. They showed that AIP mutation-positive acromegalics were predominantly young males with approximately 50% of affected cases presenting during childhood or adolescence. Moreover, when compared to controls, their tumors secreted higher levels of GH and prolactin, were more likely to undergo surgical intervention, and were less responsive to somatostatin analogues.
The study by Hernandez-Ramirez et al.6 also reported several important findings. The AIP variants were studied in 216 FIPA families and 404 sporadic acromegalics; pathogenic and likely pathogenic mutations were reported in 17.1% and 8.4% of patients, respectively. A positive genotype-phenotype correlation was found between truncating AIP mutations and younger age of onset. Screening of family members led to the diagnosis of a pituitary tumor in approximately 25% of the mutation carriers, highlighting the importance of prospective screening. The authors also examined the role of two putative disease modifying genes (GNAS1 and FGFR4) in mediating the final phenotype of AIP mutation-positive patients and showed that neither of these genes were involved in tumorigenesis. In contrast to the study by Daly et al.24, this large investigation showed no male predominance among AIP mutation-positive FIPA patients.
AIP defects that have been reported in association with GH excess include promoter, splice-site, and missense mutations; given the fact that AIP is highly polymorphic in the population it is essential that functional studies are performed with each newly identified sequence variant. Beyond protein truncation, missense and even silent sequence changes have been linked to abnormal splicing, reduced AIP expression, and loss of crucial protein interactions.9,11,17,25
The prevalence of AIP mutations in patients with sporadic pituitary adenomas is approximately 4%26, and there are no known somatic mutations of AIP reported to date.
2. X-linked acrogigantism (X-LAG)
X-LAG (OMIM #300942) is a rare cause of early childhood-onset gigantism resulting from GH oversecretion due to a pituitary adenoma or hyperplasia. The median age of onset is 12 months. Nearly all of the patients identified so far also had hyperprolactinemia at the time of presentation.7,27 Unlike most AIP mutations, germline microduplications on chromosome Xq26.3 causing X-LAG mainly arise de novo. Since most index cases are females with a de novo defect, it is likely that most affected male hemizygous embryos are not viable (although not all), as it is true in other X-linked dominant disorders. Only two instances of X-LAG inheritance have been reported so far, and in both kindreds the affected mother transmitted the microduplication to their affected sons. While females present with germline mutations, male patients harbor the mutation in a mosaic state, which could be missed from a peripheral leukocyte-, saliva-, and buccal cell-derived DNA for microduplication in Xq26.3 or GPR101.28, 29 In such circumstances, DNA isolated from the pituitary tissue and forearm skin proved essential to show a duplicated dosage of GPR10129.
The common duplicated genomic region shared by the first reported patients covered approximately 500 kb, including CD40LG, ARHGEF6, RBMX, and GPR101. To date, no pathogenic point mutations were identified to cause X-LAG in any of the duplicated genes. Among the coding sequences from this region, only GPR101, which codes for an orphan G-protein coupled receptor (GPCR), was highly over-expressed in the pituitary lesions of the affected patients. This finding, along with the fact that GPR101 strongly activates cyclic (c) AMP generation through GNAS1, a pathway known to be involved in GH-producing tumors, suggested that it might be the causative gene for X-LAG.7,27 A rare GPR101 missense variant (p.E308D) was identified in approximately 4% of patients with sporadic acromegaly; it was also seen at least once to occur de novo at the somatic level. In a GH-secreting cell line model, over-expression of this variant led to an increase in GH secretion.7 Screening for GPR101 point variants was recently carried out in two other cohorts of patients with pituitary adenomas, reporting conflicting results with regards to the p.E308D variant.30,31 In one study, patients with sporadic familial acromegaly did not show an increased prevalence of the c.924G > C (p.E308D) GPR101 variant when compared to public databases.28 Although the specific role of GPR101 in stimulating GH secretion remains elusive, recent evidence supports an effect on GHRH secretion.7,32 Whether this is the main mechanism leading to pituitary tumorigenesis in XLAG, or that there are additional roles of GPR101 in the pituitary somatomammotroph cells remains to be seen.
3. Carney complex (CNC)
CNC (OMIM #160980) is an autosomal dominant (AD) disorder that consists of skin pigmentation, cardiac myxomas, GH and prolactin-secreting pituitary tumors or hyperplasia, and Cushing syndrome from primary pigmented nodular adrenocortical disease (PPNAD).33,34 CNC is mainly caused by mutations of PRKAR1A, a tumor suppressor gene on chromosome 17q22-24 (the CNC1 locus); a second locus (CNC2) on chromosome 2p16 has not led to the identification of a specific genetic defect, whereas a single case of CNC has been described in association with PRKACB amplification.34-36 The most common PRKAR1A mutations causing CNC are protein-truncating; in approximately 32% of the cases, these defects are de novo.34,36 Acromegaly is seen in approximately 79% of patients with CNC and is usually due to somatotroph cell hyperplasia, and less likely from microadenomas, which can be multiple.34 Somatic alterations in PRKAR1A or PRKACB are infrequently found in sporadic GH-secreting pituitary adenomas.37
4. Multiple endocrine neoplasia 1 (MEN1)
MEN1 (OMIM #131100) describes the association of pituitary, parathyroid, and pancreatic neuroendocrine tumors caused by germline (and rarely somatic) mutations in MEN1.38 MEN1 is located on chromosome 11q13 and consists of 10 exons that encode for the tumor suppressor nuclear protein menin.39 Over 1300 germline mutations have been discovered to date, and are scattered throughout its entire 1,830 bp coding region; many are at splice sites of the gene.40 The majority of these inactivating mutations are frameshift deletions or insertions.39 The incidence of GH-secreting pituitary adenomas in MEN1 is approximately 10% by age 40.38
5. Multiple endocrine neoplasia 4 (MEN4)
MEN4 (MIM #610755) describes the association of pituitary and parathyroid neoplasms with pheochromocytomas, thyroid and other tumors. MEN4 is caused by heterozygous loss-of-function germline mutations in CDKN1B, which is located on chromosome 12p13.41,42,43 Mutations in the non-coding regions of CDKN1B have also been reported.44 In MEN1 mutation-negative cases, which are seen in less than 10% of suspected MEN-1,40 the possibility of MEN4 should be sought. Although gigantism or acromegaly is rare in MEN4, one study found a novel heterozygous mutation in the CDKN1B 5'-UTR region (c.-29_-26delAGAG) causing gigantism.45 This mutation was associated with a reduction in CDKN1B mRNA levels and reduced transcriptional activity in vitro.45 CDKN1B mutations in sporadic gigantism or acromegaly appear to be rare.46
6. McCune-Albright Syndrome (MAS)
MAS (OMIM #174800) is characterized by poly or monostotic fibrous dysplasia, caféau-lait spots, peripheral precocious puberty and hyperfunctioning endocrinopathies as a result of gain-of-function mutations in GNAS1 in the mosaic state.47 The mutation renders the gene functionally constitutive, which affects the stimulatory subunit (Gs alpha) of the heterotrimeric G protein complex responsible for intracellular signaling of GPCRs. Acromegaly is seen in 20–30% of MAS patients, with a mean age at diagnosis of 24.4 years (range 3-64 years), and accompanying hyperprolactinemia in the majority of cases.48 Somatotroph hyperplasia involves the entire pituitary gland, with or without development of somatotroph adenoma.49
7. Paraganglioma, pheochromocytoma, and pituitary adenoma association (3PA)
Germline and sporadic defects in SDHx have been implicated in 3PA, a recently identified form of familial paragangliomas and pheochromocytomas (PPGL) and other tumors.50 The first description of an acromegalic with paraganglioma due to a pathogenic mutation in SDHD was reported by Xekouki et al.51 Dénes et al.52 sequenced the known genes of PPGL and pituitary adenomas in 39 patients with sporadic or familial 3PA and found 11 germline mutations (5 SDHB, 1 SDHC, 1 SDHD, 2 VHL, and 2 MEN1) and 4 variants of unknown significance (2 SDHA, 1 SDHB, and 1 SDHAF2).52 Further genetic studies are required to better elucidate the molecular and genetic underpinning of this recently described syndrome which points to the involvement of the mitochondrial oxidation pathway in the pathogenesis of GH-producing tumors.
Other genetic conditions and molecular pathways associated with GH excess
Neurofibromatosis type-1 (NF1, OMIM #162200) is a multiple tumor syndrome characterized by café-au-lait spots, neurofibromas, freckling in the inguinal and axillary regions and ocular Lisch nodules with an extremely variable clinical presentation.53 NF1 commonly arises from point mutations in the neurofibromin (NF1) gene, including splicing, nonsense, frameshift, and missense variants, while whole-gene deletions are present in approximately 5% of patients.53,54 GH excess has been reported in children and adults with NF1. Rarely, patients with NF1 and GH excess may have a co-existing pituitary adenoma; in a patient we recently saw, one such tumor stained negative for GH, GHRH, and somatostatin expression suggesting an unlikely association (unpublished data). Loss of somatostatinergic inhibition from infiltrating optic tract gliomas (OTG) leading to a dysregulated GH secretion has been suggested as a possible mechanism of GH excess in patients with NF1.55,56 A recent study described a series of five children with OTG (3 due to NF1) and GH excess without a clear etiology for gigantism.57
A signaling pathway previously not suspected to be involved in gigantism or acromegaly is that of the suppressors of cytokine signaling (SOCS) family of proteins. The JAKSTAT pathway mediates the intracellular signaling of GH, and is regulated by SOCS, including SOCS2. Polymorphisms in SOCS2 have been implicated in GH sensitivity throughout the body.58 Although there appears to be a genetic association between SOCS2 and longitudinal height growth from birth to adulthood,59 further studies to help determine the association between SOCS2 and gigantism or acromegaly may be of interest.
Recently, a possibly pathogenic germline variant (p.N604T) in IGSF1, a plasma membrane protein of the immunoglobulin superfamily (OMIM #300137), was identified in patients with somatomammotroph lesions from the same family.60 This variant was associated with increased IGSF1 staining in pituitary tissue when compared with a GH-secreting adenoma from a patient that was negative for the IGSF1 variant, and with a normal control pituitary tissue. Since the affected family was later reported to be affected by the X-LAG syndrome,7 the findings may imply that IGSF1, while not playing a direct causative role in GH excess, could act synergistically in pituitary tumorigenesis.
Summary and genetic counseling
Over the last three decades, advances in genetics have led to the characterization of several molecular defects leading to gigantism or acromegaly. Although more than half of sporadic GH-producing tumors do not have an identifiable germline or somatic genetic defect, most cases of gigantism and acromegaly in young adults are in the context of the conditions described in this review (AIP mutations and/or FIPA, X-LAG, CNC, MAS, MEN1, MEN4, 3PA). Thus, dealing with a young patient with gigantism or acromegaly should always involve genetic counseling regardless of family history, as many of these conditions (FIPA, MEN1, and MEN4, in particular) have decreased penetrance and first-degree relatives that are carriers may not be affected. These patients should also be carefully screened for the defects described in the review. An older patient with sporadic acromegaly (over the age of 30 years) and no family history of any condition may not necessarily need to be screened for any of these genetic defects. However, the clinician should bear in mind that the occasional patient with sporadic acromegaly could also have a genetic mutation and have a low threshold for exploring genetic testing if the clinical phenotype warrants it.
Highlights.
Over the past two decades, our increasing understanding of the molecular and genetic etiologies of pituitary gigantism and acromegaly yielded several genetic causes, including multiple endocrine neoplasia type 1 and 4, McCune-Albright syndrome, Carney complex, familial isolated pituitary adenoma, pituitary adenoma association due to defects in familial succinate dehydrogenase genes, and X-linked acrogigantism.
Recently, we described a form of familial and sporadic acromegaly and gigantism that is caused by microduplications on chromosome Xq26.3, a disorder that we named X-LAG for X-linked acrogigantism. X-LAG may be responsible for as many as 80% of the cases of pre-pubertal gigantism.
Dealing with a young patient with gigantism or acromegaly should always involve genetic counseling regardless of family history, as many of these conditions (FIPA, MEN1, and MEN4, in particular) have decreased penetrance and first-degree relatives that are carriers may not be affected.
The early diagnosis of these conditions helps guide early intervention, screening, and genetic counseling of patients and their family members.
Acknowledgments
This work was supported by the Intramural Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement:
The authors declare that the research was conducted in absence of any potential conflict of interest.
Author and Contributors
All authors contributed equally to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work; AND drafting the work or revising it critically for important intellectual content; AND final approval of the version to be published; AND agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Other (non-NIH) Financial support and sponsorship
None.
References
- 1.Sotos JF, Argente J. Overgrowth disorders associated with tall stature. Advances in pediatrics. 2008;55:213–254. doi: 10.1016/j.yapd.2008.07.019. [DOI] [PubMed] [Google Scholar]
- 2.Sotos JF. Overgrowth. Hormonal Causes. Clinical pediatrics. 1996 Nov;35(11):579–590. [PubMed] [Google Scholar]
- 3.Burton T, Le Nestour E, Neary M, Ludlam WH. Incidence and prevalence of acromegaly in a large US health plan database. Pituitary. 2016 Jan 20; doi: 10.1007/s11102-015-0701-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xekouki P, Azevedo M, Stratakis CA. Anterior pituitary adenomas: inherited syndromes, novel genes and molecular pathways. Expert review of endocrinology & metabolism. 2010 Sep 1;5(5):697–709. doi: 10.1586/eem.10.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daly AF, Vanbellinghen JF, Khoo SK, et al. Aryl hydrocarbon receptor-interacting protein gene mutations in familial isolated pituitary adenomas: analysis in 73 families. J Clin Endocrinol Metab. 2007 May;92(5):1891–1896. doi: 10.1210/jc.2006-2513. [DOI] [PubMed] [Google Scholar]
- 6.Hernandez-Ramirez LC, Gabrovska P, Denes J, et al. Landscape of Familial Isolated and Young-Onset Pituitary Adenomas: Prospective Diagnosis in AIP Mutation Carriers. J Clin Endocrinol Metab. 2015 Sep;100(9):E1242–1254. doi: 10.1210/jc.2015-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trivellin G, Daly AF, Faucz FR, et al. Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. N Engl J Med. 2014 Dec 18;371(25):2363–2374. doi: 10.1056/NEJMoa1408028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rostomyan L, Daly AF, Petrossians P, et al. Clinical and genetic characterization of pituitary gigantism: an international collaborative study in 208 patients. Endocrine-related cancer. 2015 Oct;22(5):745–757. doi: 10.1530/ERC-15-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leontiou CA, Gueorguiev M, van der Spuy J, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab. 2008 Jun;93(6):2390–2401. doi: 10.1210/jc.2007-2611. [DOI] [PubMed] [Google Scholar]
- 10.Vierimaa O, Georgitsi M, Lehtonen R, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006 May 26;312(5777):1228–1230. doi: 10.1126/science.1126100. [DOI] [PubMed] [Google Scholar]
- 11.Igreja S, Chahal HS, King P, et al. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Human mutation. 2010 Aug;31(8):950–960. doi: 10.1002/humu.21292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Occhi G, Trivellin G, Ceccato F, et al. Prevalence of AIP mutations in a large series of sporadic Italian acromegalic patients and evaluation of CDKN1B status in acromegalic patients with multiple endocrine neoplasia. European journal of endocrinology / European Federation of Endocrine Societies. 2010 Sep;163(3):369–376. doi: 10.1530/EJE-10-0327. [DOI] [PubMed] [Google Scholar]
- 13.Stratakis CA, Tichomirowa MA, Boikos S, et al. The role of germline AIP, MEN1, PRKAR1A, CDKN1B and CDKN2C mutations in causing pituitary adenomas in a large cohort of children, adolescents, and patients with genetic syndromes. Clinical genetics. 2010 Nov;78(5):457–463. doi: 10.1111/j.1399-0004.2010.01406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Georgitsi M, Heliövaara E, Paschke R, et al. Large genomic deletions in AIP in pituitary adenoma predisposition. The Journal of clinical endocrinology and metabolism. 2008;93:4146–4151. doi: 10.1210/jc.2008-1003. [DOI] [PubMed] [Google Scholar]
- 15.Chahal HS, Trivellin G, Leontiou CA, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. The Journal of clinical endocrinology and metabolism. 2012 Aug;97(8):E1411–1420. doi: 10.1210/jc.2012-1111. [DOI] [PubMed] [Google Scholar]
- 16.Formosa R, Xuereb-Anastasi A, Vassallo J. Aip regulates cAMP signalling and GH secretion in GH3 cells. Endocrine-related cancer. 2013 Aug;20(4):495–505. doi: 10.1530/ERC-13-0043. [DOI] [PubMed] [Google Scholar]
- 17.Morgan RM, Hernandez-Ramirez LC, Trivellin G, et al. Structure of the TPR domain of AIP: lack of client protein interaction with the C-terminal alpha-7 helix of the TPR domain of AIP is sufficient for pituitary adenoma predisposition. PloS one. 2012;7(12):e53339. doi: 10.1371/journal.pone.0053339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raitila A, Lehtonen HJ, Arola J, et al. Mice with inactivation of aryl hydrocarbon receptor-interacting protein (Aip) display complete penetrance of pituitary adenomas with aberrant ARNT expression. The American journal of pathology. 2010 Oct;177(4):1969–1976. doi: 10.2353/ajpath.2010.100138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuominen I, Heliovaara E, Raitila A, et al. AIP inactivation leads to pituitary tumorigenesis through defective Galphai-cAMP signaling. Oncogene. 2015 Feb 26;34(9):1174–1184. doi: 10.1038/onc.2014.50. [DOI] [PubMed] [Google Scholar]
- 20.Denes J, Kasuki L, Trivellin G, et al. Regulation of aryl hydrocarbon receptor interacting protein (AIP) protein expression by MiR-34a in sporadic somatotropinomas. PloS one. 2015;10(2):e0117107. doi: 10.1371/journal.pone.0117107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trivellin G, Butz H, Delhove J, et al. miR-107 Is Overexpressed in Pituitary Adenomas and Inhibits the Expression of Aryl Hydrocarbon Receptor-Interacting Protein in vitro. Am. J. Physiol. Endocrinol. Metab. 2012 Sep 15;303(6):E708–E719. doi: 10.1152/ajpendo.00546.2011. [DOI] [PubMed] [Google Scholar]
- 22.Toledo RA, Lourenco DM, Jr., Toledo SP. Familial isolated pituitary adenoma: evidence for genetic heterogeneity. Front Horm Res. 2010;38:77–86. doi: 10.1159/000318497. [DOI] [PubMed] [Google Scholar]
- 23.Daly AF, Tichomirowa Ma, Petrossians P, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. The Journal of clinical endocrinology and metabolism. 2010;95:E373–383. doi: 10.1210/jc.2009-2556. [DOI] [PubMed] [Google Scholar]
- 24.Daly AF, Tichomirowa MA, Petrossians P, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. The Journal of clinical endocrinology and metabolism. 2010;95:E373–383. doi: 10.1210/jc.2009-2556. [DOI] [PubMed] [Google Scholar]
- 25.Martucci F, Trivellin G, Korbonits M. Familial isolated pituitary adenomas: an emerging clinical entity. Journal of endocrinological investigation. 2012;35:1003–1014. doi: 10.1007/BF03346742. [DOI] [PubMed] [Google Scholar]
- 26.Lecoq AL, Kamenicky P, Guiochon-Mantel A, Chanson P. Genetic mutations in sporadic pituitary adenomas--what to screen for? Nature reviews. Endocrinology. 2015 Jan;11(1):43–54. doi: 10.1038/nrendo.2014.181. [DOI] [PubMed] [Google Scholar]
- 27.Beckers A, Lodish MB, Trivellin G, et al. X-linked acrogigantism syndrome: clinical profile and therapeutic responses. Endocr Relat Cancer. 2015 Jun;22(3):353–367. doi: 10.1530/ERC-15-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iacovazzo D, Caswell R, Bunce B, et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta neuropathologica communications. 2016;4(1):56. doi: 10.1186/s40478-016-0328-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodd C, Millette M, Iacovazzo D, et al. Somatic GPR101 Duplication Causing X-Linked Acrogigantism (XLAG)-Diagnosis and Management. J Clin Endocrinol Metab. 2016 May;101(5):1927–1930. doi: 10.1210/jc.2015-4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrau F, Romeo PD, Puglisi S, et al. Analysis of GPR101 and AIP genes mutations in acromegaly: a multicentric study. Endocrine. 2016 Jan 27; doi: 10.1007/s12020-016-0862-4. [DOI] [PubMed] [Google Scholar]
- 31.Lecoq AL, Bouligand J, Hage M, et al. Very low frequency of germline GPR101 genetic variation and no biallelic defects with AIP in a large cohort of patients with sporadic pituitary adenomas. European journal of endocrinology / European Federation of Endocrine Societies. 2016 Jan 20; doi: 10.1530/EJE-15-1044. [DOI] [PubMed] [Google Scholar]
- 32.Daly AF, Lysy PA, Desfilles C, et al. GHRH excess and blockade in X-LAG syndrome. Endocr Relat Cancer. 2016 Mar;23(3):161–170. doi: 10.1530/ERC-15-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine. 1985 Jul;64(4):270–283. doi: 10.1097/00005792-198507000-00007. [DOI] [PubMed] [Google Scholar]
- 34.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001 Sep;86(9):4041–4046. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 35.Forlino A, Vetro A, Garavelli L, et al. PRKACB and Carney complex. N Engl J Med. 2014 Mar 13;370(11):1065–1067. doi: 10.1056/NEJMc1309730. [DOI] [PubMed] [Google Scholar]
- 36.Correa R, Salpea P, Stratakis CA. Carney complex: an update. Eur J Endocrinol. 2015 Oct;173(4):M85–97. doi: 10.1530/EJE-15-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandrini F, Kirschner LS, Bei T, et al. PRKAR1A, one of the Carney complex genes, and its locus (17q22-24) are rarely altered in pituitary tumours outside the Carney complex. Journal of medical genetics. 2002 Dec;39(12):e78. doi: 10.1136/jmg.39.12.e78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012 Sep;97(9):2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- 39.Agarwal SK, Kester MB, Debelenko LV, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Human molecular genetics. 1997 Jul;6(7):1169–1175. doi: 10.1093/hmg/6.7.1169. [DOI] [PubMed] [Google Scholar]
- 40.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Human mutation. 2008 Jan;29(1):22–32. doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 41.Malanga D, De Gisi S, Riccardi M, et al. Functional characterization of a rare germline mutation in the gene encoding the cyclin-dependent kinase inhibitor p27Kip1 (CDKN1B) in a Spanish patient with multiple endocrine neoplasia-like phenotype. Eur J Endocrinol. 2012 Mar;166(3):551–560. doi: 10.1530/EJE-11-0929. [DOI] [PubMed] [Google Scholar]
- 42.Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proceedings of the National Academy of Sciences of the United States of America. 2006 Oct 17;103(42):15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tonelli F, Giudici F, Giusti F, et al. A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome. Eur J Endocrinol. 2014 Aug;171(2):K7–K17. doi: 10.1530/EJE-14-0080. [DOI] [PubMed] [Google Scholar]
- 44.Occhi G, Regazzo D, Trivellin G, et al. A novel mutation in the upstream open reading frame of the CDKN1B gene causes a MEN4 phenotype. PLoS genetics. 2013 Mar;9(3):e1003350. doi: 10.1371/journal.pgen.1003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sambugaro S, Di Ruvo M, Ambrosio MR, et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5'UTR region. Endocrine. 2015 May;49(1):58–64. doi: 10.1007/s12020-015-0540-y. [DOI] [PubMed] [Google Scholar]
- 46.Schernthaner-Reiter MH, Trivellin G, Stratakis CA. MEN1, MEN4, and Carney Complex: Pathology and Molecular Genetics. Neuroendocrinology. 2016;103(1):18–31. doi: 10.1159/000371819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991 Dec 12;325(24):1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 48.Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014 Jun;99(6):1955–1969. doi: 10.1210/jc.2013-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vortmeyer AO, Glasker S, Mehta GU, et al. Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2012 Jul;97(7):2404–2413. doi: 10.1210/jc.2012-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xekouki P, Szarek E, Bullova P, et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J Clin Endocrinol Metab. 2015 May;100(5):E710–719. doi: 10.1210/jc.2014-4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xekouki P, Pacak K, Almeida M, et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: a new association for SDH? J Clin Endocrinol Metab. 2012 Mar;97(3):E357–366. doi: 10.1210/jc.2011-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Denes J, Swords F, Rattenberry E, et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab. 2015 Mar;100(3):E531–541. doi: 10.1210/jc.2014-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1): diagnosis and management. Handbook of clinical neurology. 2013;115:939–955. doi: 10.1016/B978-0-444-52902-2.00053-9. [DOI] [PubMed] [Google Scholar]
- 54.Wimmer K, Yao S, Claes K, et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes, chromosomes & cancer. 2006 Mar;45(3):265–276. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- 55.Manski TJ, Haworth CS, Duval-Arnould BJ, Rushing EJ. Optic pathway glioma infiltrating into somatostatinergic pathways in a young boy with gigantism. Case report. Journal of neurosurgery. 1994 Oct;81(4):595–600. doi: 10.3171/jns.1994.81.4.0595. [DOI] [PubMed] [Google Scholar]
- 56.Fuqua JS, Berkovitz GD. Growth hormone excess in a child with neurofibromatosis type 1 and optic pathway tumor: a patient report. Clinical pediatrics. 1998 Dec;37(12):749–752. doi: 10.1177/000992289803701207. [DOI] [PubMed] [Google Scholar]
- 57.Josefson J, Listernick R, Fangusaro JR, Charrow J, Habiby R. Growth hormone excess in children with neurofibromatosis type 1-associated and sporadic optic pathway tumors. The Journal of pediatrics. 2011 Mar;158(3):433–436. doi: 10.1016/j.jpeds.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 58.Vesterlund M, Zadjali F, Persson T, et al. The SOCS2 ubiquitin ligase complex regulates growth hormone receptor levels. PloS one. 2011;6(9):e25358. doi: 10.1371/journal.pone.0025358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sovio U, Bennett AJ, Millwood IY, et al. Genetic determinants of height growth assessed longitudinally from infancy to adulthood in the northern Finland birth cohort 1966. PLoS genetics. 2009 Mar;5(3):e1000409. doi: 10.1371/journal.pgen.1000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Faucz FR, Horvath AD, Azevedo MF, et al. Is IGSF1 involved in human pituitary tumor formation? Endocrine-related cancer. 2015 Feb;22(1):47–54. doi: 10.1530/ERC-14-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]