

Abstract
Sandhoff disease (SD) is a lysosomal storage disorder characterized by the absence of hydrolytic enzyme β-N-acetylhexosaminidase (Hex), which results in storage of GM2 ganglioside in neurons and unremitting neurodegeneration. Neuron loss initially affects fine motor skills, but rapidly progresses to loss of all body faculties, a vegetative state, and death by five years of age in humans. A well-established feline model of SD allows characterization of the disease in a large animal model and provides a means to test the safety and efficacy of therapeutic interventions before initiating clinical trials. In this study, we demonstrate a robust central nervous system (CNS) inflammatory response in feline SD, primarily marked by expansion and activation of the microglial cell population. Quantification of major histocompatibility complex II (MHC-II) labeling revealed significant up-regulation throughout the CNS with areas rich in white matter most severely affected. Expression of the leukocyte chemokine macrophage inflammatory protein-1 alpha (MIP-1α) was also up-regulated in the brain. SD cats were treated with intracranial delivery of adeno-associated viral (AAV) vectors expressing feline Hex, with a study endpoint 16 weeks post treatment. AAV-mediated gene delivery repressed the expansion and activation of microglia and normalized MHC-II and MIP-1α levels. These data reiterate the profound inflammatory response in SD and show that neuroinflammation is abrogated after AAV-mediated restoration of enzymatic activity.
Keywords: Lysosomal storage disorder, neurodegenerative disease, neuroinflammation, microglia activation, cytokines/chemokines, gene therapy, adeno-associated virus, hexosaminidase, GM2 ganglioside
Introduction
GM2 gangliosidosis is an inherited metabolic disorder resulting from a mutation in one of the genes encoding β-N-acetylhexosaminidase (Hex, EC 3.2.1.52), a lysosomal hydrolase. Deficiency in Hex activity leads to accumulation of GM2 ganglioside in neurons, which causes unrelenting neurodegeneration and death by approximately 5 years of age. Despite being characterized over 100 years ago (Tay, 1881), GM2 gangliosidosis remains incurable today. The GM2 gangliosidoses include Sandhoff disease (SD) and Tay-Sachs disease (TSD), both the consequence of a deficiency in Hex, an enzyme comprised of two subunits, α and β. TSD results from a mutation in the gene encoding the α subunit (HEXA), while SD is caused by a mutation in the gene encoding the β subunit (HEXB). Though the pathogenic nature of GM2 gangliosidosis has not been fully elucidated, neuron loss is a prominent component. The milieu of neuronal death is complex, involving numerous cell types such as microglia and astrocytes, and a myriad of signaling molecules including cytokines and chemokines. A more complete understanding of neuronal death could provide therapeutic targets and allow for more comprehensive intervention in the disease process.1
The HEXB-/- mouse model has progressive, profound neuropathology that closely recapitulates human SD (Sango et al., 1995). It has been hypothesized in HEXB-/- mice and human TSD and SD patients, that neuronal death results from an apoptotic cascade activated by storage material (Huang et al., 1997). Though abundant in late disease stages, apoptotic neurons were not detectable during the first two months of life in HEXB-/- mice. Because activated microglia were present by 2 months of age in select areas of the central nervous system (CNS), neuroinflammation may precede, and potentially exacerbate, apoptosis (Wada et al., 2000). Several molecules indicative of microglia activation are up-regulated in the HEXB-/- mouse brain including MHC-II, tumor necrosis factor alpha (TNF-α), interleukin 1 beta (IL-1β), transforming growth factor beta 1 (TGFβ1) (Jeyakumar et al., 2003), and sphingosine kinase 1 (Sphk1) (Wu et al., 2008). Subsequent to bone marrow transplantation or substrate reduction therapy, HEXB-/- mice lived significantly longer and had normalized cytokine levels in the brain (Jeyakumar et al., 2003) and knocking down Sphk1 expression in HEXB-/- mice attenuated disease progression and microgliosis (Wu et al., 2008), suggesting that it is possible to treat the inflammatory component of SD. In human TSD and SD patients, cerebral cortical sections also had a significantly increased number of activated microglia (Myerowitz et al., 2002). A 15 month old Tay-Sachs patient with typical signs of neurodegeneration on MRI had elevated TNF-α and interleukin 5 levels in cerebrospinal fluid, while other cytokines and chemokines were within the normal range (Hayase et al., 2010).
It has been shown that blood monocytes contribute to the expansion of the microglia population by migrating through the vascular endothelium and into the brain parenchyma (Wu and Proia, 2004). MIP-1α, a chemokine responsible for monocyte recruitment from the blood, was increased throughout the brain and spinal cord of HEXB-/- mice, even at the neonatal stage (Wu and Proia, 2004, Tsuji et al., 2005b, Kyrkanides et al., 2008). MIP-1α co-localizes with resident glial cells such as astrocytes (Wu and Proia, 2004, Tsuji et al., 2005b) and microglia, but not GM2 containing neurons (Tsuji et al., 2005b). Knocking out MIP-1α in the HEXB-/- mouse resulted in diminished pathology, decreased apoptotic neuronal death, and increased body weight, behavioral function, and survival (Wu and Proia, 2004). This again suggests that treating the inflammatory component of SD may have clinical benefit.
In recent years, great progress has been made in further characterization of the disease process and refinement of therapeutic approaches. However, the challenge remains to treat both the underlying enzymatic deficiency and the neuroinflammatory process. Intracranial injection of HEXB-/- mice with adeno-associated virus serotype 1 (AAV1) vectors encoding human HEXA and HEXB prevented the activation and expansion of microglial cells. Early intervention (4 weeks of age) resulted in significant extension in lifespan; however, delaying treatment until 12 weeks of age was ineffective despite only minor differences in observed neuroinflammatory properties between treatment ages (Cachon-Gonzalez et al., 2014). These studies provide evidence that AAV gene therapy is capable of alleviating the neuroinflammatory response. However, other underlying disease mechanisms may lead to an age-dependent reduction of therapeutic effectiveness of AAV vectors.
The SD cat model, first described in 1977 (Cork et al., 1977), has been well-characterized in the intervening years (Baker et al., 1979, Martin et al., 2004) and presents an unparalleled opportunity to test therapeutic options for translation to human clinical trials. The feline model recapitulates the disease phenotype, neuropathological alterations, and biochemical defects observed in human patients. Furthermore, the complexity and size of the feline brain more closely resemble that of a human and the longer life span allows for more extensive analysis of therapies. Therefore, after success in HEXB-/- mice, the same AAV1 vectors encoding human Hex were tested in the feline model of SD. Two SD cats treated by bilateral thalamic injection lived to 7.0 and 8.2 months of age, compared with an untreated life span of 4.5 ± 0.5 months. The limited therapeutic effect may have been due to robust humoral immune response to the AAV capsid and/or human Hex protein. AAV vectors encoding feline α and β Hex subunits were injected bilaterally into the thalamus of SD cats, which reduced the immune response and increased survival to 10.4 ± 3.7 months of age, or > 2 times that of untreated cats (Bradbury et al., 2013). Further studies were conducted combining thalamic infusion with additional delivery routes such as bilateral injection into the deep cerebellar nuclei (DCN) (McCurdy et al., 2015), or intracerebroventricular (ICV) injection (Rockwell et al., 2015). Restoration of Hex activity, clearance of storage material, and a panel of biomarkers from combined delivery routes have been reported elsewhere (Bradbury et al., 2015, McCurdy et al., 2015, Rockwell et al., 2015). In the current study, we analyze the effect of intracranial gene therapy with thalamus + DCN and thalamus + ICV injection routes on the neuroinflammatory response in SD.
Experimental procedures
Vectors and Surgery
Vectors were produced as previously described (Bradbury et al., 2013). Feline HEX transgene expression was driven by the CBA promoter (including the CMV immediate-early enhancer fused to the chicken β-actin promoter) (Matalon et al., 2003) and the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) was included for enhancement of gene expression.
All animal procedures were approved by the Auburn University Institutional Animal Care and Use Committee. Bilateral injections of the thalamus and DCN were performed according to a previously published protocol (Bradbury et al., 2013). ICV injection was performed under ultrasound guidance to confirm correct placement of the injection needle. After visualization of the left lateral ventricle with an 8-5 MHz Philips HDI 5000 ultrasound probe (Philips Healthcare, Andover, MA), a single entry site was made through the skull with a 20G spinal needle. Vector was delivered using a Hamilton syringe (Harvard Apparatus, Holliston, MA) fitted with a 25G non-coring needle (Harvard Apparatus). A total of 200 μL was delivered in 10-15 μL aliquots at a rate of 3-5 μL/second with approximately 1 minute between aliquots. Total vector doses were as follows: Thalamus, ∼4.5×1011 vg distributed equally between right and left thalami; DCN, ∼1.6×1011 vg distributed equally between right and left DCN; ICV, ∼6.4×1011 vg injected into the left lateral ventricle. Cats were treated at 4-7 weeks of age, prior to symptom onset (Table 1).
Table 1.
| ID | Age at Treatment (weeks) | Age at endpoint (weeks) | Treatment Route | Dose (g.e.) |
|---|---|---|---|---|
| 7-762 | 5.0 | 21.0 | Thal+DCN | 6.1 × 1011* |
| 7-770 | 5.6 | 21.9 | ||
| 7-774 | 4.3 | 20.3 | ||
| 7-957 | 4.7 | 21.1 | Thal+ICV** | 1.1×1012 |
| 7-960 | 7.1 | 23.0 | ||
| 7-972 | 6.9 | 22.7 | ||
| 7-981 | 5.3 | 22.4 |
g.e.: genome equivalent; thal: thalamus, DCN: deep cerebellar nuclei, ICV: intracerebroventricular
Same dose as previously reported (McCurdy et al 2015) but titered by new method also used for Thal+ICV cohort.
Note: Tissues from cat 7-981 were not available for all analyses.
At humane endpoint (4.5 ± 0.5 months in untreated SD cats) or a predetermined endpoint at 16 weeks after treatment, animals were euthanized by pentobarbital overdose (100 mg/kg) in accordance with the American Veterinary Medical Association guidelines for euthanasia. After cats were transcardially perfused with cold, heparinized saline, tissue samples were fixed in neutral buffered formalin, frozen in optimal cutting temperature (OCT) medium, or flash frozen in liquid nitrogen and stored at -80oC for analysis. Only tissue from the thalamus + ICV cohort was available for immunohistochemistry and immunofluorescence analysis. Tissue from the thalamus + DCN cohort was used for other assays.
Immunohistochemistry
Prior to immunolabeling, 6 μm paraffin sections mounted on glass slides were deparaffinized and endogenous peroxidase-like activity was quenched with 0.3% hydrogen peroxide in absolute methanol for 30 minutes. Antigen retrieval was performed by heat treatment in Tris-EDTA buffer (10 mmol/l Tris, 1 mmol/l EDTA, 0.05% Tween 20, pH 9.0) for 20 minutes. Subsequently sections were blocked with 5% normal horse serum and 0.5% BSA in PBS (pH 7.5) for 1 hour followed by incubation with ionized calcium binding protein 1 (Iba1) (1:100; Biocare, Concord, CA) or MHC-II (1:4, AbD Serotec, Oxford, UK) for 1 hour. Sections were then rinsed with PBS + 0.1%BSA and incubated with horse anti-rabbit/mouse IgG conjugated with HRP (pre-diluted; Vector Laboratories, Burlingame, CA) for 1 hour. Color development with the Vectastain RTU ABC reagent was performed as recommended by the manufacturer (Vector Laboratories). For immunofluorescence, primary antibodies included Iba1 (1:100; Biocare), MHC-II (1:4, Serotec), Hex β-subunit (Martin et al., 2004) (1:1000), and microtubule associated protein 2 (MAP2) (1:3000; Neuromics, Minneapolis, MN) incubated for 1 hour. Sections were then rinsed and incubated with 488-conjugated, 594-conjugated, or 647-conjugated secondary antibodies.
Cell quantification
Stained brain sections were scanned on an Aperio ScanScope CS2 (Aperio, Vista, CA) and quantified using Visiopharm Quantitative Digital Pathology software (Visiopharm, Hoersholm, Denmark). The same brain regions were analyzed for all animals. In each section the maximum allotted area was quantified for each brain area of interest, including: internal capsule white matter (WM), cerebral cortex gray matter (GM), and thalamus (Thal) at the level of the thalamus and in cerebellar white matter (WM), cerebellar cortex grey matter (GM), deep cerebellar nuclei (DCN), and brainstem (BS) at the level of the DCN.
Quantitative PCR
Total RNA was isolated from OCT embedded brain tissue using the RNeasy mini kit (Qiagen, Limburg, Netherlands) according to the manufacturer's instruction. RNA was treated with DNAse (Invitrogen, Carlsbad, CA) and complementary DNA synthesis was performed using the qScript cDNA synthesis kit (Quanta Biosciences, Gaithersburg, MD). Quantitative PCR was conducted with 300 ng of complementary DNA using TaqMan Fast Universal PCR Master Mix (Applied Biosystems, Carlsbad, CA) and a C1000 Thermal Cycler (BioRad, Hercules, CA). MIP-1α and IL-1β mRNA expression were normalized to a ribosomal protein housekeeping gene, RPL-17. Primer and probe sequences are as follows: MIP-1α forward primer CCT ATG TCT CCA AGC AGA TTC C; MIP-1α reverse primer GTC AGT CAC ATA TTC CTG GAC C; MIP-1α probe 5′-/56-FAM/TTT GAG ACC AGC AGC CAA TGC TCC AA/31ABkFQ/-3′; RPL-17 forward primer ATC CAC GAG ACC ACC TTC AA; RPL-17 reverse primer CCA GAC AGC ACC GTG TTA; RPL-17 probe/56-FAM/TTA AGA ACA CAC GGG AAA CTG CCC AG/31ABkFQ/-3′.
Statistics
Significance was determined by two-tailed t-test with SAS software (SAS, Cary, NC).
Results
Microgliosis in the SD cat at humane endpoint (4.5 ± 0.5 months) was analyzed with an antibody against Iba1, a microglia/macrophage-specific protein that localizes to both activated and resting microglia. Microglia in the SD cat brain were almost entirely of an amoeboid morphology exemplified by an enlarged cell body and shortened processes (Fig. 1 A), and had markedly increased immunoreactivity for MHC-II, a marker of antigen presentation (Fig. 1 B). Results were consistent throughout the cerebrum (Fig. 1 A-B), cerebellum (not shown), and spinal cord (not shown) of SD cats. In contrast, microglia in age-matched normal cats maintained a ramified morphology designated by a small cell body and elongated, branching processes (Fig. 1 C). Normal cats lacked positive labeling for MHC-II (Fig. 1 D), indicating an absence of activation. The labeling profile was comparable in the cerebellum and spinal cord (not shown). Seven SD cats were treated with monocistronic AAVrh8 vectors encoding feline HEX α and β subunits between 4-7 weeks of age, prior to symptom onset. Two treatment cohorts were based on injection route: bilateral thalamic and DCN injection (n=3) (McCurdy et al., 2015) or bilateral thalamic and unilateral ICV injection (n=4) (Rockwell et al., 2015) (Table 1). Treated SD cats were euthanized 16 weeks after injection, an age comparable to the humane endpoint of untreated SD cats. Previous reports showed that all treated cats had clearly preserved neurologic function and improved disease phenotype, though mild symptoms such as fine tremors and slight hind limb weakness were present. Also, previously published postmortem analysis revealed above-normal levels of Hex activity throughout the brain and spinal cord, with clearance of GM2 ganglioside storage (McCurdy et al., 2015, Rockwell et al., 2015). Microglia from treated cats retained a ramified morphology and lacked expression of MHC-II in regions of cleared storage such as the thalamus (Fig. 1 E-F) and DCN (not shown), comparable to normal controls. Cat 7-960 had an isolated, discrete population of MHC-II positive cells in the reticular thalamic nuclei (Fig. 2 A, indicated by black arrows) and a modest number of sporadic positively stained cells throughout the cerebellum. Interestingly, this cat was the oldest of all cats at the time of treatment, with injection at 7.1 weeks of age (Table 1). Despite an absence of MHC-II signal in the remaining cats, there appeared to be an increased number of microglia as seen by Iba1 labeling, most notably in defined locations in the thalamus (Fig. 2 B, indicated by black arrows) and DCN.
Figure 1. Activation of microglia in feline Sandhoff disease and attenuation with AAV gene therapy.

Representative images of Iba1 and MHC-II expression in the thalamus are shown. In SD cats at humane endpoint (n=5), labeling with Iba1 shows activation of microglia characterized by an amoeboid morphology with a large cell body and shortened processes (A) and immunereactivity of MHC-II (B). Iba1 labeling from a normal, age-matched control cat shows a resting microglia population characterized by a small cell body and long, branching processes (C) and a lack of MHC-II labeling (D). In AAV-treated SD cats sixteen weeks after Thal+ICV injection, resting, ramified microglia are documented as Iba1-positive cells (E) with negligible MHC-II expression (F) (n=3). Scale bar represents 100 μm (A)-(E), 200 μm (F), and all insets 50 μm.
Figure 2. Isolated areas of microglia activation and expansion in AAV-treated SD cats.

Sixteen weeks after Thal+ICV AAV-therapy, one treated cat showed isolated activation of microglia, indicated by MHC-II immunereactivity, in the region of the reticular thalamic nuclei (A). Another treated cat showed discrete expansion of resting microglia, indicated by Iba1 labeling (B) without MHC-II expression in the region (also shown in immunofluorescence imaging in Figure 3). Scale bars represent 3 mm.
Microglia cell activation was quantified by measuring the area of selected MHC-II positive brain regions of SD, normal, and AAV treated SD cats. Measurements were taken in coronal brain sections that included the thalamus (Fig. 3 A) or DCN (Fig. 3 B). In untreated SD cats, MHC-II was significantly up-regulated in the internal capsule (p = 0.0079), cerebral cortex (p = 0.0452), and thalamus (p = 0.0251) when compared to normal, age-matched control cats (Fig. 3 C-E). MHC-II also was significantly increased in cerebellar white matter (p = 0.009), cerebellar cortex (p = 0.0159), and in the DCN (p < 0.0001) (Fig. 3 F-H). The difference in MHC-II positive area was not significant in the brainstem (p = 0.0740) due to considerable variability across animals of the untreated SD cohort (Fig. 3 I). Sixteen weeks after AAV gene therapy, MHC-II was normalized in all areas of the brain analyzed (p > 0.05) (Fig. 3 C-I). Double immunofluorescence labeling of untreated SD cat brain (Fig 4) confirmed that MHC-II expression was found in Iba-1 positive microglia of amoeboid rather than ramified morphology (Fig 4 A), and was absent in GFAP-positive astroglial cells (Fig. 4 B).
Figure 3. Up-regulation of MHC-II in untreated SD cat brain is normalized after AAV gene delivery.

MHC-II positive area was quantified in internal capsule white matter (WM), cerebral cortex gray matter (GM), and thalamus (Thal) at the level of the thalamic injection site (A) and in cerebellar white matter (WM), cerebellar cortex grey matter (GM), deep cerebellar nuclei (DCN), and brainstem (BS) at the level of the DCN injection site (B) of SD (n=5) and normal, age-matched cats (n=6). The area of positive labeling was significantly increased in untreated SD cats in all areas of the brain analyzed (C-H) except for the brainstem (G). Sixteen weeks after Thal+ICV AAV therapy (SD+AAV, n=3), MHC-II was normalized in all areas of the brain analyzed (C-G). * p ≤ 0.05; ** p ≤ 0.01; error bars represent standard deviation. Orienting image from SD cat; scale bars represent 5 mm (A) or 3mm (B).
Figure 4. MHC-II expression associated with microglia but not astrocytes in the untreated SD cat brain.

Double immunofluorescence labeling of untreated SD cat brain with cell type specific antibodies for (A) Iba1 (red), and MHC-II (green) confirms that MHC-II is expressed in microglia, but absent in (B) GFAP-positive (red) astrocytes. Images are a merged stack of 20 layers (6 μm total) from a confocal microscope. Scale bars represent 20 μm.
After treatment, the majority of cells expressing Hex were neurons, as indicated by immunofluoresence with an antibody to the feline Hex β-subunit and MAP2 (Fig. 5 A). High Hex expression near the thalamic injection site caused vesiculated intracytoplasmic inclusions (Fig. 5 A). Triple labeling by the addition of an antibody to Iba-1 demonstrated that most microglia did not express high levels of Hex (Fig. 5 B). While rare Hex-positive labeling could be found in activated microglial cells, it was not apparent in ramified, resting microglia (not shown).
Figure 5. Hex expression in neurons and microglial cells after AAV treatment.

Immunofluorescence with an antibody to the feline Hex β-subunit (orange) demonstrated high levels of the therapeutic protein in granular inclusions in neurons (MAP2, green) near the thalamic injection site (A). Triple label with MAP2 (green), Iba-1 (red) and Hexβ (orange) showed little evidence of Hex in the majority of microglial cells (B). Image B is a merged stack of 15-20 layers (5 μm total) from a confocal microscope. Scale bar represents 20 μm. The images were acquired from Thal+ICV AAV treated cat 7-972 (Table 1).
MIP-1α functions in recruitment of microglial precursor cells from the blood and thus plays a role in expansion of the microglia population in the brain. We therefore measured MIP-1α expression in the brains of SD, normal, and AAV treated SD cats by quantitative reverse transcription PCR, normalizing expression to a ribosomal protein, RPL-17. As anticipated, a significant up-regulation of MIP-1α was seen in the brain of SD cats (p = 0.01, n = 6) at humane endpoint when compared to normal age-matched cats (n=6) (Fig. 6). Furthermore, 16 weeks after treatment with AAV gene therapy, levels of MIP-1α were not significantly different from normal but were significantly different from untreated SD cats (p = 0.01, n=3 for Thal+DCN cohort; p = 0.004, n=4 for Thal+ICV cohort). There was no statistical difference in MIP-1α expression between the 2 treatment cohorts (p = 0.67). Unfortunately, protein analysis for feline MIP-1α was not possible due to the lack of a cross-reactive antibody. TNF-α expression in untreated SD cats was variable, with elevated expression only emerging in the oldest cat analyzed (4.9 months of age). Sixteen weeks after AAV gene therapy, TNF-α expression was at or below normal levels. Immunostaining corroborated these findings and revealed little to no labeling of TNF-α in untreated SD cats at the mean humane endpoint (4.5 months of age) (data not shown). IL-1β and Sphk1 expression were also analyzed and were not significantly altered with disease or therapy (data not shown).
Figure 6. MIP-1α expression in the SD cat brain after intracranial injection of AAV.
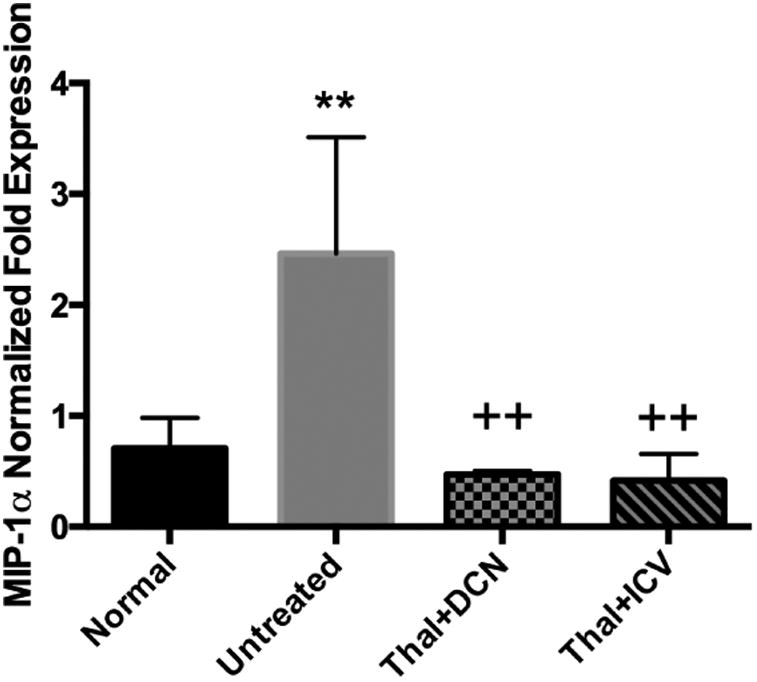
mRNA expression of MIP-1α was significantly elevated in the brain of SD cats (p = 0.01, n=6) at humane endpoint when compared to a normal age-matched cats (n=6). Sixteen weeks after AAV gene therapy in either cohort, MIP-1α expression was not different from normal age-matched cats but was significantly decreased compared to untreated SD cats (p=0.01 for Thal+DCN, n=3; p=0.004 for Thal+ICV, n=4) (A). Error bars represent standard deviation. ** p ≤ 0.01 as compared to normal; ++ p ≤ 0.01 as compared to untreated SD.
Discussion
In human patients (Myerowitz et al., 2002) and HEXB-/- mice (Wada et al., 2000), CNS degeneration is accompanied by marked neuroinflammation, with the expansion and activation of microglia as the most prominent contributor. The feline model of SD was established over 30 years ago (Cork et al., 1977) and it has been an important model in understanding the human disease, but the inflammatory component of CNS pathology had not been explored thoroughly in SD cats. In normal, age-matched control cats, the microglia population is almost entirely in a resting state, with a ramified morphology and lack of MHC-II expression. In contrast, nearly all microglia in the SD brain were activated, expressing the antigen presenting molecule MHC-II and exhibiting an amoeboid morphology (i.e., an enlarged cell body and shortened processes). The amoeboid morphology is associated with an actively phagocytic state in which microglia ingest pathogens or cellular debris for antigen presentation. MHC-II was significantly elevated in all brain areas analyzed except for the brainstem, in which labeling variability was high across untreated SD cats. As previously shown in HEXB-/- mice (Cachon-Gonzalez et al., 2014), brain regions rich in white matter were most severely affected. Also, microglia activation was greatest in the cerebellum, which is consistent with the prominent cerebellar phenotype of feline SD in which whole-body tremors are the most debilitating feature.
As reported previously, sixteen weeks after intracranial injection of AAVrh8-fHEX, all SD cats expressed Hex levels above normal throughout the brain. Treatment dramatically ameliorated clinical signs of disease, with cats demonstrating slight hind limb weakness and whole-body tremors that were mild (Thal+ICV) (Rockwell et al., 2015) or nonexistent (Thal+DCN) (McCurdy et al., 2015). The current report extends previous findings by showing that SD cats treated with AAV had marked attenuation of microglia activation, with a ramified morphology and lack of MHC-II expression, in areas of GM2 ganglioside clearance. When quantified, levels of MHC-II were normalized in all areas of the brain analyzed, demonstrating widespread therapeutic impact. These results show that pre-symptomatic AAV gene therapy precludes the neuroinflammatory process in SD cats for at least 16 weeks. The cerebellum of some treated cats did show increased Iba1 labeling in the absence of MHC-II expression. Hex activity was ≥ 5-fold normal throughout the cerebellum after Thal+ICV injection (Rockwell et al., 2015); therefore, it is unlikely that the Iba1 staining is attributable to low levels or poor distribution of the therapeutic enzyme. It is probable that pathology had initiated in the cerebellum prior to the age of treatment. Many of the early disease signs, such as tremors, are attributed to pathology in the cerebellum suggesting that disease may present first in this brain region. Since gene therapy is capable of preventing but not reversing pathology, it is possible that subclinical cerebellar pathology is present by 4-7 weeks of age while pathology in the cerebrum is less advanced. Of 7 AAV-treated cats reported in this study, a single cat that was the oldest at the time of treatment (∼7 weeks) did have a discrete population of MHC-II positive cells restricted to the reticular thalamic nuclei, perhaps underscoring the importance of treatment age, as documented in SD mice (Cachon-Gonzalez et al., 2014).
In lysosomal diseases, microglia respond to dying cells laden with storage material and remove the cellular debris by phagocytosis; however, SD microglia are Hex-deficient and thus are unable to hydrolyze GM2 ganglioside. In response, it is thought that leukocyte precursors of microglia are recruited to the CNS via MIP-1α, furthering the expansion of activated microglia (Wada et al., 2000). AAV-treated cats in the current study had normalized levels of MIP-1α, suggesting diminished recruitment of microglia precursors from the blood after gene therapy. Since only background levels of HexA activity were found in peripheral blood mononuclear cells from treated cats (data not shown), reduction of microglial recruitment/activation cannot be attributed to an infiltrating population of enzyme-positive cells, as in SD mice treated by bone marrow transplantation (Wada et al., 2000).
Knockout of MIP-1α or CCR2 (a chemokine receptor involved in PBMC infiltration) led to modestly improved survival in SD mice (Wu and Proia, 2004, Kyrkanides et al., 2008), suggesting that targeted inhibition of pro-inflammatory cytokines could influence microglia recruitment and improve outcomes. More recent characterization of SD microglial cell lines identified potential signaling pathways that modulate expression of MIP-1α, providing promising targets for inhibition (Kawashita et al., 2009). Though pre-symptomatic gene therapy prevented microglial activation and MIP-1α elevation in SD cats, most human patients will be treated after the onset of clinical signs when microglial activation is already underway. In such cases, reducing neuroinflammation early in the disease process, perhaps by targeting MIP-1α or its receptors, could be a critical component of a comprehensive treatment regimen.
When Hex was selectively restored in neurons but not microglia of HEXB-/-mice, microglial activation was not attenuated but the animals retained normal rotarod performance to at least 7 weeks beyond the humane endpoint of untreated animals. A subset of these mice were followed to > 1 year of age, at which time they continued to behave normally (Kyrkanides et al., 2012). That neuronal Hex production is insufficient to attenuate neuroinflammation raises the question of whether the enzyme must be restored in microglia for optimal therapeutic effect. Our treatment strategy for SD cats was intracranial injection of AAVrh8, which transduces neurons for a long-lasting source of Hex. However, AAVrh8 is not known to transduce microglia, as shown for other serotypes such as AAV2 and AAV5 (Cucchiarini et al., 2003). In AAVrh8-treated SD cats, Hex expression was apparent in neurons at high levels. Hex also was noted in discrete microglia surrounding transduced neurons, but it is unclear whether microglial Hex was derived from vector transduction, phagocytosis, or cross-correction (the process whereby functional lysosomal enzymes can be endocytosed by diseased cells). It is possible that cross-correction of microglia was achieved by the high-level of Hex expression in SD cats, shown to be above normal throughout the brain and up to 45-fold normal at the injection sites. The secretion and recapture ability of Hex has previously been reported in primary microglial cells from the neonatal brains of Sandhoff disease mice (Tsuji et al., 2005a). Tsuji et al established that Hex was taken up via cell-surface glycoreceptors, primarily the mannose receptor, of SD microglia that were then capable of degrading the accumulated GM2 and GlcNAc-oligosaccharides (Tsuji et al., 2005a). Further study is needed to determine whether enzyme restoration in microglia, or other glial cells, is required for long-term therapy and how best to achieve it.
In conclusion, CNS-wide Hex expression and profound amelioration of clinical disease after intracranial gene therapy (McCurdy et al., 2015, Rockwell et al., 2015) correlated with widespread attenuation of neuroinflammation in feline SD. Such unparalleled short-term therapeutic benefit in a large animal model of SD merits long-term studies with this approach, which are ongoing. In conjunction with the success of AAV therapy in SD mice, our data encourage consideration of human clinical trials. Furthermore, this approach may provide benefit to other monogenic neurodegenerative diseases beyond the scope of lysosomal storage disorders.
Highlights.
Neuroinflammation is a prominent pathological feature of feline Sandhoff disease.
A robust infiltration and activation of microglia was evident in CNS of SD cats.
MHCII and MIP-1α were up-regulated in the brain of SD cats.
Intracranial AAV gene therapy attenuated inflammation, normalizing MHCII and MIP-1α.
Acknowledgments
This work was funded by National Institutes of Health grant U01NS064096 and the National Tay-Sachs and Allied Diseases Association. Many thanks to Dr. Gary Swain for his assistance in acquiring confocal IF images.
Footnotes
Abbreviations: Sandhoff disease (SD); p-N-acetylhexosaminidase (Hex); major histocompatibility complex II (MHC-II); macrophage inflammatory protein-1 alpha (MIP-1α); adeno-associated virus (AAV); Tay-Sachs disease (TSD); central nervous system (CNS); tumor necrosis factor alpha (TNF-α); interleukin 1 beta (IL-1β); transforming growth factor beta 1 (TGFβ1); glial fibrillary acidic protein (GFAP); ionized calcium binding protein 1 (Iba1); sphingosine kinase 1 (Sphk1); peripheral blood mononuclear cell (PBMC); deep cerebellar nuclei (DCN); intracerebroventricular (ICV); woodchuck hepatitis virus post-transcriptional regulatory element (WPRE); α-mannosidase (Mann); β-galactosidase (βgal); granulocyte macrophage colony-stimulating factor (GM-CSF); interferon gamma (IFNγ); interleukin 4 (IL-4); regulated on activation, normal T cell expressed (RANTES); stem cell factor (SCF); monocyte chemotactic protein (MCP-1).
Conflict of Interest: On behalf of all authors, the corresponding author states that there is no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker HJ, Reynolds GD, Walkley SU, Cox NR, Baker GH. The gangliosidoses: comparative features and research applications. Vet Pathol. 1979;16:635–649. doi: 10.1177/030098587901600602. [DOI] [PubMed] [Google Scholar]
- Bradbury AM, Cochran JN, McCurdy VJ, Johnson AK, Brunson BL, Gray-Edwards H, Leroy SG, Hwang M, Randle AN, Jackson LS, Morrison NE, Baek RC, Seyfried TN, Cheng SH, Cox NR, Baker HJ, Cachon-Gonzalez MB, Cox TM, Sena-Esteves M, Martin DR. Therapeutic response in feline sandhoff disease despite immunity to intracranial gene therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:1306–1315. doi: 10.1038/mt.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury AM, Gray-Edwards HL, Shirley JL, McCurdy VJ, Colaco AN, Randle AN, Christopherson PW, Bird AC, Johnson AK, Wilson DU, Hudson JA, De Pompa NL, Sorjonen DC, Brunson BL, Jeyakumar M, Platt FM, Baker HJ, Cox NR, Sena-Esteves M, Martin DR. Biomarkers for disease progression and AAV therapeutic efficacy in feline Sandhoff disease. Experimental neurology. 2015;263:102–112. doi: 10.1016/j.expneurol.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachon-Gonzalez MB, Wang SZ, Ziegler R, Cheng SH, Cox TM. Reversibility of neuropathology in Tay-Sachs-related diseases. Human molecular genetics. 2014;23:730–748. doi: 10.1093/hmg/ddt459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cork LC, Munnell JF, Lorenz MD, Murphy JV, Baker HJ, Rattazzi MC. GM2 ganglioside lysosomal storage disease in cats with beta-hexosaminidase deficiency. Science. 1977;196:1014–1017. doi: 10.1126/science.404709. [DOI] [PubMed] [Google Scholar]
- Cucchiarini M, Ren XL, Perides G, Terwilliger EF. Selective gene expression in brain microglia mediated via adeno-associated virus type 2 and type 5 vectors. Gene Ther. 2003;10:657–667. doi: 10.1038/sj.gt.3301925. [DOI] [PubMed] [Google Scholar]
- Hayase T, Shimizu J, Goto T, Nozaki Y, Mori M, Takahashi N, Namba E, Yamagata T, Momoi MY. Unilaterally and rapidly progressing white matter lesion and elevated cytokines in a patient with Tay-Sachs disease. Brain Dev. 2010;32:244–247. doi: 10.1016/j.braindev.2009.01.007. [DOI] [PubMed] [Google Scholar]
- Huang JQ, Trasler JM, Igdoura S, Michaud J, Hanal N, Gravel RA. Apoptotic cell death in mouse models of GM2 gangliosidosis and observations on human Tay-Sachs and Sandhoff diseases. Human molecular genetics. 1997;6:1879–1885. doi: 10.1093/hmg/6.11.1879. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Thomas R, Elliot-Smith E, Smith DA, van der Spoel AC, d'Azzo A, Perry VH, Butters TD, Dwek RA, Platt FM. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain. 2003;126:974–987. doi: 10.1093/brain/awg089. [DOI] [PubMed] [Google Scholar]
- Kawashita E, Tsuji D, Kawashima N, Nakayama K, Matsuno H, Itoh K. Abnormal production of macrophage inflammatory protein-1alpha by microglial cell lines derived from neonatal brains of Sandhoff disease model mice. Journal of neurochemistry. 2009;109:1215–1224. doi: 10.1111/j.1471-4159.2009.06041.x. [DOI] [PubMed] [Google Scholar]
- Kyrkanides S, Brouxhon SM, Tallents RH, Miller JN, Olschowka JA, O'Banion MK. Conditional expression of human beta-hexosaminidase in the neurons of Sandhoff disease rescues mice from neurodegeneration but not neuroinflammation. J Neuroinflammation. 2012;9:186. doi: 10.1186/1742-2094-9-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrkanides S, Miller AW, Miller JN, Tallents RH, Brouxhon SM, Olschowka ME, O'Banion MK, Olschowka JA. Peripheral blood mononuclear cell infiltration and neuroinflammation in the HexB-/- mouse model of neurodegeneration. J Neuroimmunol. 2008;203:50–57. doi: 10.1016/j.jneuroim.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DR, Krum BK, Varadarajan GS, Hathcock TL, Smith BF, Baker HJ. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant. Experimental neurology. 2004;187:30–37. doi: 10.1016/j.expneurol.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Matalon R, Surendran S, Rady PL, Quast MJ, Campbell GA, Matalon KM, Tyring SK, Wei J, Peden CS, Ezell EL, Muzyczka N, Mandel RJ. Adeno-associated virus-mediated aspartoacylase gene transfer to the brain of knockout mouse for canavan disease. Molecular therapy : the journal of the American Society of Gene Therapy. 2003;7:580–587. doi: 10.1016/s1525-0016(03)00066-2. [DOI] [PubMed] [Google Scholar]
- McCurdy VJ, Rockwell HE, Arthur JR, Bradbury AM, Johnson AK, Randle AN, Brunson BL, Hwang M, Gray-Edwards HL, Morrison NE, Johnson JA, Baker HJ, Cox NR, Seyfried TN, Sena-Esteves M, Martin DR. Widespread correction of central nervous system disease after intracranial gene therapy in a feline model of Sandhoff disease. Gene Ther. 2015;22:181–189. doi: 10.1038/gt.2014.108. [DOI] [PubMed] [Google Scholar]
- Myerowitz R, Lawson D, Mizukami H, Mi Y, Tifft CJ, Proia RL. Molecular pathophysiology in Tay-Sachs and Sandhoff diseases as revealed by gene expression profiling. Human molecular genetics. 2002;11:1343–1350. doi: 10.1093/hmg/11.11.1343. [DOI] [PubMed] [Google Scholar]
- Rockwell HE, McCurdy VJ, Eaton SC, Wilson DU, Johnson AK, Randle AN, Bradbury AM, Gray-Edwards HL, Baker HJ, Hudson JA, Cox NR, Sena-Esteves M, Seyfried TN, Martin DR. AAV-mediated gene delivery in a feline model of Sandhoff disease corrects lysosomal storage in the central nervous system. ASN Neuro. 2015;7 doi: 10.1177/1759091415569908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- Tay W. Symmetrical Changes in the Region of teh Yellow Spot in Each Eye of an Infant. Transactions of the Ophthalmological Society. 1881;1:55–57. [Google Scholar]
- Tsuji D, Kuroki A, Ishibashi Y, Itakura T, Itoh K. Metabolic correction in microglia derived from Sandhoff disease model mice. Journal of neurochemistry. 2005a;94:1631–1638. doi: 10.1111/j.1471-4159.2005.03317.x. [DOI] [PubMed] [Google Scholar]
- Tsuji D, Kuroki A, Ishibashi Y, Itakura T, Kuwahara J, Yamanaka S, Itoh K. Specific induction of macrophage inflammatory protein 1-alpha in glial cells of Sandhoff disease model mice associated with accumulation of N-acetylhexosaminyl glycoconjugates. Journal of neurochemistry. 2005b;92:1497–1507. doi: 10.1111/j.1471-4159.2005.02986.x. [DOI] [PubMed] [Google Scholar]
- Wada R, Tifft CJ, Proia RL. Microglial activation precedes acute neurodegeneration in Sandhoff disease and is suppressed by bone marrow transplantation. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10954–10959. doi: 10.1073/pnas.97.20.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YP, Mizugishi K, Bektas M, Sandhoff R, Proia RL. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Human molecular genetics. 2008;17:2257–2264. doi: 10.1093/hmg/ddn126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YP, Proia RL. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8425–8430. doi: 10.1073/pnas.0400625101. [DOI] [PMC free article] [PubMed] [Google Scholar]