

Abstract
We present two cases of early-onset Alzheimer's disease due to a novel N135Y mutation in PSEN1. The proband presented with memory and other cognitive symptoms at age 32. Detailed clinical characterization revealed initial deficits in memory with associated dysarthria, progressing to involve executive dysfunction, spastic gait, and episodic confusion with polyspike discharges on long-term electroencephalography. Amyloid- and FDG-PET scans showed typical results of Alzheimer's disease. By history, the proband's father had developed cognitive symptoms at age 42 and died at age 48. Neuropathological evaluation confirmed Alzheimer's disease, with moderate to severe amyloid angiopathy. Skeletal muscle showed type 2 fiber– predominant atrophy with pale central clearing. Genetic testing of the proband revealed an N135Y missense mutation in PSEN1. This mutation was predicted to be pathogenic by in silico analysis. Biochemical analysis confirmed that the mutation caused an increased Aβ42/Aβ40 ratio, consistent with other PSEN1 mutations and with a loss of presenilin function.
Keywords: Alzheimer's disease, Presenilin 1
1. Introduction
Mutations of the presenilin 1 (PSEN1) gene on chromosome 14 account for 76.4% of the known cases of autosomal dominant Alzheimer's disease (ADAD) (Shea et al., 2015; Ryman et al., 2014). They cause a more aggressive form of ADAD than PSEN2 mutations and APP mutations/duplications, with earlier age of onset (43.3 ± 8 years) and possibly a shorter duration until death (Shea et al., 2015). Clinically, those with PSEN1 mutations may exhibit more prominent aphasia, myoclonus, seizures, spasticity, or cerebellar dysfunction (Miller and Boeve, 2009; Shea et al., 2015).
The PSEN1 gene is located on chromosome 14 and consists of 13 exons encoding multiple protein isoforms through alternative mRNA splicing (Miller and Boeve, 2009). The resulting PS1 protein has 467 amino acids with nine transmembrane domains. A systematic review of ADAD suggests a distinction between PSEN1 mutations before and after codon 200 (Shea et al., 2015). Patients with PSEN1 mutations on the amino side of codon 200 have slightly earlier onset and are more likely to be affected by seizures and myoclonus than those with mutations on the carboxy side of codon 200. Patients with PSEN1 mutations after codon 200 are more likely to be affected by visuospatial impairment and spastic paraparesis (Shea et al., 2015). However, this distinction has not been observed among participants in the Dominantly Inherited Alzheimer's Network (DIAN) (Tang et al., in press). Pathologically, amyloid plaques, neurofibrillary tangles, and amyloid angiopathy are common (Miller and Boeve, 2009).
The PS1 protein is a critical component of γ-secretase, a protease required for β-amyloid formation from amyloid precursor protein (APP) (De Strooper, 2003). PS1 forms a protein complex with three other proteins within neuronal membranes: nicastrin, anterior pharynx-defective 1 (Aph1) and presenilin enhancer 2 (Pen2). The complex has aspartyl protease activity and cleaves APP within the transmembrane domain, liberating the C-terminal fragment of APP. It is the final enzymatic step in the production of Aβ40 and Aβ42 (Miller and Boeve, 2009). In vitro studies suggest that pathogenic mutations within PSEN1 increase the ratio of Aβ42, which is more amyloidogenic, to Aβ40 (Duff et al., 1996; Scheuner et al., 1996; Kumar-Singh et al., 2006). More than 220 mutations in the PSEN1 gene have been identified (Shea et al., 2015). Here, we describe the features of ADAD caused by a PSEN1 mutation that has not been previously reported.
2. Materials and Methods
2.1 Genetic Testing
Genetic testing was performed by Athena Diagnostics (Worcester, MA), with screening for sequence variants in APP, PSEN1, PSEN2, and for duplication of APP.
2.2 MRI
As part of the Dominantly Inherited Alzheimer's Network (DIAN) (NIA # U19 AG032438, R. J. Bateman, PI) the participant received a high-resolution (1.1 × 1.1 × 1.2 mm voxel) T1-weighted MPRAGE series acquired in the sagittal direction on a 3T scanner. This structural MRI acquisition was performed using the Alzheimer's Disease Neuroimaging Initiative (ADNI) protocol (Jack et al., 2008; Jack et al., 2010). Images were screened for artifacts and protocol compliance by the ADNI imaging core prior to analysis. Volumetric segmentation and cortical surface reconstruction was performed using FreeSurfer 5.1 (Fischl, 2012). Automatic segmentation of subcortical and cortical structures was accomplished using a probabilistic atlas to assign each voxel an anatomical label (Fischl et al., 2002; Desikan et al., 2006). A trained rater in the DIAN imaging core visually verified segmentations and surfaces for accuracy. Structural MRI volumes were adjusted for differences in head size using a regression approach. A regression was fitted with intracranial volume (ICV) as the independent variable and the region of interest (ROI) as the dependent variable to obtain “B weight”. Then a normalization procedure was performed by subtracting the mean ICV of 28 controls from the subject's ICV, multiplying by the “B weight,” and subtracting this total from the raw regional volume.
2.3 PET
All DIAN sites undergo evaluation to ensure their PET scans are in compliance with common ADNI protocols. All PET images were initially quality controlled by the ADNI Quality Control team. Amyloid imaging was performed with a bolus injection of approximately 15 mCi of [11C] PiB. Dynamic imaging acquisition started either at injection for 70 minutes or 40 minutes post-injection for 30 minutes. The PiB PET data 40 to 70 minutes post-injection was used for analyses. Metabolic imaging with [18F] FDG-PET was performed with a dynamic acquisition that began 30 minutes after a bolus injection of approximately 5 mCi of FDG and lasted for 30 minutes. The last 25 minutes of the FDG scans were used for analyses.
Each PET scan was motion corrected and registered to the same individual's MRI (Eisenstein et al., 2012; Rowland et al., 2005). PET data were analyzed using a region-of-interest (ROI) approach (Su et al., 2015; Su et al., 2013) and corrected for partial volume effects using a regional spread function method (Rousset et al., 1998; Su et al., 2015). Regional values were converted to standardized uptake value ratios (SUVRs) relative to the cerebellar cortex.
2.4 Neuropsychological Assessments
As part of the Dominantly Inherited Alzheimer Network (DIAN), three domain-specific cognitive composites and a global cognitive composite were calculated by averaging z scores from a battery of 13 standard paper-and-pencil neuropsychological tests. Episodic memory testing included Logical Memory Immediate Recall and Delayed Recall and Word List Immediate Recall and Delayed Recall. Language function testing included: Letter Fluency for the letters “F”, “A”, and “S,” Category Fluency for animals and vegetables, and the 30-item version of the Boston Naming Test. The working memory portion of testing included Digit Span (Forwards and Backwards), the Trail Making Test Parts A and B, and the Digit Symbol Coding test.
2.5 Biochemistry
7PA2 Chinese hamster ovarian (CHO) cells (Walsh et al., 2005) stably over-expressing human APP were rapidly thawed from storage in liquid nitrogen using pre-warmed media. Residual freezing media was exchanged to remove dimethyl sulfoxide after the cells had been allowed 1-2 hours to adhere to flasks. Colonies were then expanded, trypsinized, and re-seeded evenly on tissue culture-treated 12-well plates at 1 × 105 cells/well. At approximately 75% confluency, the cell colonies were transiently transfected with wild-type PSEN1 (negative control), N135D PSEN1 (positive control), and N135Y PSEN1 (experimental) using serum-free media and FuGENE® HD transfection reagent at a 3:1 ratio of reagent:DNA. PSEN1 constructs were expressed from an OmicsLink™ CMV promoter-based overexpression vector called pReceiver-M02 (©GeneCopoeia). 1 μg of plasmid per well was sufficient to achieve roughly ∼70% transfection efficiency at 48 hours post-transfection (as estimated by GFP signal at 485 nm excitation). Approximately 48 hours post-transfection, the media was replaced with a reduced volume of serum-free culture medium to limit extraneous protein accumulation and to slightly concentrate the secreted proteins. The 7PA2-conditioned media was collected the following morning and frozen at −80°C with 1:100 HALT™ protease inhibitor. Cells were maintained in Dulbecco's Modified Eagle's Medium supplemented with G418 selection antibiotic, penicillin/streptomycin, L-glutamine, and fetal bovine serum at 37°C in humidified 5% CO2.
Conditioned media harvested from the transfected cells was collected and assayed for the relative ratio of Aβ42:40 using a two-site sandwich ELISA to measure Aβ1-42 and Aβ1-40 levels (Millipore EZHS-SET).
3. Results
3.1 Case 1: Clinical and Imaging Characterization
A 36-year-old right-handed white woman presented to the Memory Disorders Clinic at the University of Alabama at Birmingham with the chief complaint of forgetfulness. Her husband provided the history and stated that he had noticed the insidious onset of memory problems about four years earlier. At that time, he felt that she was forgetting “little things,” or would retrieve only parts of memories for recent events, but performed better when her memory was supported with cues. There was gradual progression and at the time of presentation he stated that she no longer seemed to benefit from cues and had become more repetitive. It would not be unusual for them to have the same conversation three times in a row or for her to write herself more than one identical reminder note. She did not have word-finding difficulty or problems with single-word comprehension, but he had begun to notice slurred speech. On a few occasions she had had difficulty finding her way home when driving. She was still managing some of her instrumental activities of daily living, but he had taken over the household finances 18 months earlier. She was continuing to drive a car and had not been involved in any accidents, nor had she received any tickets. However, she was driving alone less often than before. She was not getting lost in familiar settings and was continuing to cook, but he was concerned about her ability to handle meat with sanitary technique. There was no evidence of hallucinations or delusions, but she was quieter and sometimes tearful.
Past medical history was significant for B12 deficiency secondary to GI surgery.
Family history revealed that her father had died at the age of 48 from dementia and a brain autopsy had revealed Alzheimer disease (AD) (see Case 2). There were no other known cases in the family, although the pedigree was fairly small.
She had a high school education and was employed in the service industry, although she was having some more difficulty with her work duties. She rarely consumed alcohol and never drank to excess, nor did she use any tobacco products or illicit drugs.
The general examination was unremarkable. She scored 16/30 on the Mini-Mental State Examination. On the orientation items, she gave only the state correctly. She missed three points on spelling WORLD backwards and two points on delayed recall. On fluency testing, she generated lists of 12 animals and 10 F-words in one minute each with no repetitions. She was able to draw a clock fairly well, placing all the numbers in the correct positions and arranging the hands correctly to indicate ten minutes past eleven. She performed well with simple monetary calculations except for stating that ten nickels make one dollar. The cranial nerves were intact except that speech was slow and mildly dysarthric. Strength, muscle tone and bulk, coordination, and gait were normal. Sensation was largely unimpaired, except for apparent difficulty detecting small excursions of the fingers and toes during joint position sense testing. Reflexes were 3+ throughout. Clonus was noted at both ankles. A slight jaw jerk and right palmomental sign were elicited.
Previous laboratory studies included a B12 level of 271 pg/ml (normal range 211–911) and thyroid stimulating hormone level of 0.2 ng/ml (normal range 0.3–5.6). An EEG showed mild slowing in the theta range. Blood was drawn for a complete blood count, which revealed mild anemia with a normal mean corpuscular volume and red blood cell distribution width. An MRI scan of the brain performed three years before presentation to our clinic revealed minimal cerebral atrophy and minimal deep white matter ischemic changes, but there were bilateral dilated perivascular spaces in the temporal stems. She and her husband were in agreement that they preferred for her not to take an acetylcholinesterase inhibitor at that time. An MRI scan of the cervical spine was done and was normal.
The patient underwent a battery of neuropsychological tests, with the conclusion that there was a possible cognitive disorder but a concern regarding symptom validity due to variability in performance on the tests. She appeared “lethargic/tired throughout the assessment” and “made self-disparaging remarks regarding her performance.” She had significant deficits in semantic fluency, verbal memory, and executive function (all < 1st percentile).
The patient returned to the clinic three months later with her husband, who stated that she continued to repeat questions, but remained independent with her basic ADLs. He stated that she no longer had the capability to put a complete meal together and had forgotten how to cook some things. She was still driving “occasionally” but her husband had mostly taken it over. Her husband reported that her B12 level, checked by her primary care physician, had increased to a satisfactory level but the exact level was not documented. She had lost seven pounds but her general exam was otherwise unchanged. She scored 18/30 on the MMSE, missing eight orientation items, all three recall items, and the drawing of pentagons. Her clock drawing was more disorganized than at her original visit and the hands pointed to the wrong numbers. A repeat MRI was requested and compared to the initial scan, with the observation that during the intervening three years there had been progression of atrophy in the parietal lobes and hippocampal nuclei. A commercial test for genetic causes of familial AD was ordered. At that time, there was a high clinical suspicion for early onset Alzheimer's disease in the setting of her clinical picture and family history.
Genetic testing revealed a novel g.73173630A>T [chr14, hg38] substitution, corresponding to coding nucleotide 403 in transcript variant 1 of PSEN1(NM_000021.3), causing an asparagine (N) to tyrosine (Y) substitution at amino acid 135. There were no duplications in APP or abnormal DNA sequence variants in APP or PSEN2.
After receiving the results of the commercial genetic test, the patient enrolled in the Dominantly Inherited Alzheimer Network (DIAN) study through which she had a uniform clinical assessment, neuropsychological testing (Table 1) and quantitative structural, functional, and amyloid imaging. Structural brain MRI (Fig. 1) showed decreased volume in the bilateral precuneus, hippocampus, entorhinal, parahippocampal, lateral occipital, amygdala, supramarginal gyrus, and right bank of the superior temporal sulcus with left superior temporal and superior frontal volume loss (Supplementary Table 1). FDG-PET showed decreased metabolism in the bilateral precuneus, posterior cingulate, and lateral orbitofrontal cortices (Fig. 2). On quantitative analysis, there was unilateral decreased metabolism in the left inferior temporal, right precentral, right middle temporal, and right supramarginal cortical areas (Supplementary Table 2). There was a striking degree of hypermetabolism in the left hippocampus (Supplementary Table 2). A Pittsburgh Compound B (PiB) amyloid PET (Fig. 3) showed increased PiB standardized uptake value ratio (SUVR) in the brainstem, striatum, bilateral banks of the superior temporal sulci, inferior parietal lobes, inferior temporal lobes, lateral occipital lobes, posterior cingulate gyri, precunei, superior frontal lobes, and superior parietal lobes (Supplementary Table 3). There were unilateral increases in the left fusiform gyrus, pericalcarine gyrus, superior temporal lobe, and supramarginal gyrus. PiB uptake was corrected using measurements from the cerebellum due to atypically high brainstem PiB uptake (SUVR 2.193).
Table 1. Comparison of Case 1's DIAN neuropsychological test scores to 31 matched mutation-free women.
| Control Group | Case 1 | ||
|---|---|---|---|
|
| |||
| Mean | SD | Z | |
| Boston Naming Test | 27.86 | 1.81 | 0.08 |
| Animal fluency | 23.29 | 5.63 | -2.00 |
| Vegetable fluency | 16.90 | 3.82 | -2.60 |
| WAIS Digit-Symbol Sequencing | 65.68 | 10.37 | -5.08 |
| Digit span backwards (# correct sequences) | 7.58 | 2.28 | -1.57 |
| Digit span backwards (longest correct sequence) | 5.32 | 1.30 | -1.79 |
| Digit span forwards (# correct sequences) | 9.39 | 1.82 | -1.31 |
| Digit span forwards (longest correct sequence) | 7.06 | 1.00 | -1.07 |
| A-words | 10.61 | 3.37 | -0.77 |
| F-words | 13.42 | 4.26 | -0.80 |
| S-words | 15.10 | 4.26 | -1.67 |
| Correct memory units from logical memory | 13.13 | 4.96 | -2.65 |
| Logical memory story A | 14.23 | 4.39 | -3.01 |
| Mini-mental state exam | 29.29 | 1.24 | -9.88 |
| Trails A errors | 0.19 | 0.40 | -0.48 |
| Trails A (time in seconds) | 20.32 | 5.10 | 10.33 |
| Trails B errors | 0.32 | 0.83 | 105.36 |
| Trails B (time in seconds) | 51.13 | 18.64 | NC |
| Delayed word list recall | 3.48 | 1.75 | -1.99 |
| Immediate recall of word list | 5.97 | 1.68 | -2.36 |
| Letter Fluency total | 39.13 | 10.05 | -1.31 |
Mean and standard deviation (SD) are for a group of age-matched controls in the DIAN study. Z = z-score for Case 1; NC = task not completed. Z-scores with absolute value of at least 2.0, indicating tests where the proband's performance was at least two SD's from the mean, are in bold font.
Fig. 1.
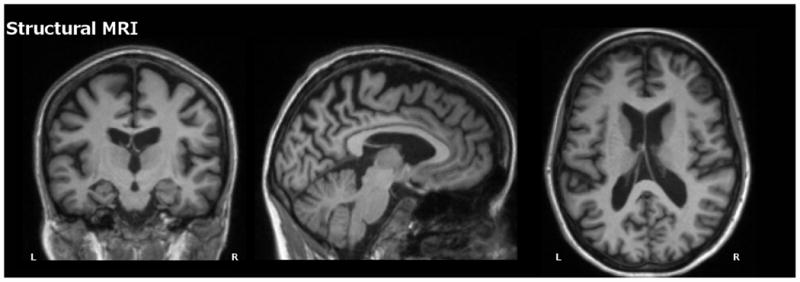
Structural MRI for Case 1 depicting substantial atrophy throughout cortical and subcortical regions.
Fig. 2.
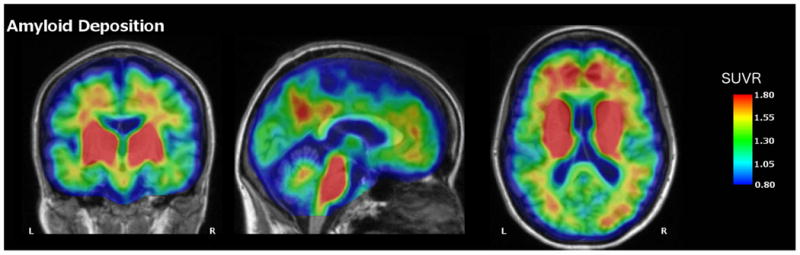
PiB-PET image for Case 1 depicting elevated amyloid deposition in the precuneus, medial prefrontal cortex, bilateral parietal cortex, as well as the basal ganglia and brainstem. Values represent SUVRs relative to cerebellar grey
Fig. 3.
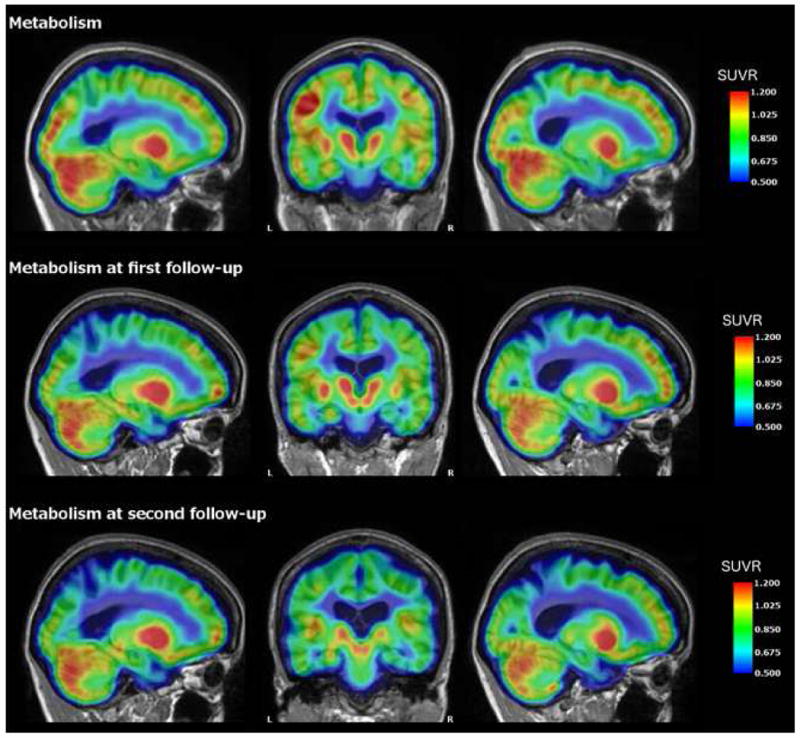
FDG-PET for Case 1 depicting metabolism across the cortex at baseline and annual follow-up one and two years later. There is progressive hypometabolism in the precuneus, which is typical for ADAD mutation carriers as the disease progresses. Of particular note is elevated metabolism in the left hippocampus relative to the right throughout the course. Values represent SUVRs relative to cerebellar gray.
Due to episodic increases in confusion and the hippocampal hypermetabolism on FDG-PET, the patient underwent long-term video electroencephalogram (EEG) monitoring. She had mild generalized slowing suggestive of a mild, nonspecific encephalopathy. She also had frontal spike/polyspike and wave discharges of fluctuating predominance on the right and left. No seizures were seen, but frontal lobe epilepsy was suspected. An anti-thyroperoxidase antibody was checked given her history of thyroid cancer and was negative. She was started on levetiracetam, but was sleepy and more confused on this anticonvulsant and was transitioned to lamotrigine after two months. Lamotrigine did not produce significant drowsiness or confusion and she had no clinically evident seizures upon follow up five months later.
By age 39, she had spasticity in bilateral upper extremities, with brisk deep tendon reflexes throughout, sustained clonus at bilateral ankles, and wide-based and scissoring gait. The motor symptoms had developed progressively since initial evaluation despite repletion of vitamin B12 deficiency and a normal MRI of the cervical spine. Brief bedside cognitive testing continued to decline as did her independence performing her basic activities of daily living.
3.2 Case 2: Neuropathological Characterization
According to his autopsy report, the proband's father had onset of cognitive symptoms at age 42, with rapid onset of a dementia syndrome accompanied by visual and auditory hallucinations. Although a detailed clinical characterization is not available, he was clinically diagnosed as AD. He died at the age of 48 years after being in a severe stage of dementia for the 3 years prior to his death (described as “vegetative” in the autopsy report). He had a history of a seizure disorder as a child, hypertension, and diabetes mellitus. He had suffered from aspiration pneumonia on 3 separate occasions leading up to his death.
Per the autopsy report, macroscopic evaluation of the brain revealed striking cerebral atrophy with a brain weight of 1150 g, frontal and temporal lobe predominant, with some parietal lobe involvement. There was relative sparing of the superior gyrus of the temporal lobes. There was striking atrophy of the hippocampus and amygdala while the basal ganglia, subthalamic nucleus, red nucleus, and substantia nigra were relatively well-preserved. There was moderate reduction of white matter volume, most prominent in the anterior corpus callosum and there were mild to moderate dilations of perivascular spaces in the basal ganglia. In the frontal, temporal, parietal lobes and cingulate gyrus cortex, the number of neuritic plaques ranged from “moderate” to “frequent” using the Consortium to Establish a Registry for Alzheimer Disease (CERAD) semi-quantitative terminology (Mirra et al., 1991). This resulted in a “C” age-related plaque score and in the clinical setting the neuropathologic probabilistic statement of “definite Alzheimer disease” was based on the numerous neurofibrillary tangles in the hippocampal formation and the neocortex. The calcarine cortex also showed scattered tangles and plaques and, in aggregate, a Braak and Braak stage of V/VI was assigned (Braak and Braak, 1991). No Lewy bodies were noted in the brainstem, limbic lobe, amygdala, or neocortex.
The patient had mild, patchy atherosclerosis in scattered vessels with a maximum stenosis of 5–10%. Mild to moderate arteriosclerosis of cerebral arteries and dilation of perivascular spaces in the basal ganglia and thalamus, as is seen in the setting of hypertension, were identified. There was moderate to severe amyloid deposition within subarachnoid and cerebral cortical arteries which was diagnostic of moderate to severe congophilic amyloid angiopathy using the Vonsattel grading scheme (Vonsattel et al., 1991). Mild hypoxic changes were noted in neurons in various cortical, hippocampal, and cerebellar sections, consistent with terminal hypoxia. There was no cerebral dysplasia or heterotopia or evidence of previous meningitis that would explain the patient's history of childhood seizures.
The skeletal muscle was symmetrically atrophied. The right quadriceps muscle was biopsied and showed type 2 fiber predominance greater than is usual for this muscle. No fiber type grouping or grouped atrophy was noted. His type 2 atrophy was consistent with his history of disuse. There were very thin aggregates of granular material noted on H&E and trichrome staining that resembled rimmed vacuoles lacking the vacuolated appearance in the center. The centers of these aggregates were pale and lacked inflammation as is seen in inclusion body myositis. Peripheral nerves showed very mild reduction in the number of myelinated axons, consistent with that seen commonly in adults.
3.3 Biochemical Characterization of N135Y
The N135Y variant in PSEN1 has not been reported in published studies or in ClinVar. However, N135 is homologous to N141 in PS2, the site of the classical PSEN2 mutation identified in Volga German kindreds (Levy-Lahad et al., 1995). N135D and N135S mutations at the same residue in PSEN1 mutation have been identified in other AD kindreds (Crook et al., 1997; Rudzinski et al., 2008). The nucleotide variant underlying the N135Y mutation has a CADD (v1.3) score of 27.7, which places its damage ranking in the top 0.2% of all possible variants in the genome (Kircher et al., 2014). Other tools, such as PolyPhen-2 (Adzhubei et al., 2010) and PROVEAN (Choi et al., 2012), also suggested a high likelihood that the N135Y mutation would be pathogenic.
To determine the effects of N135Y on PS1 function, we expressed this mutant in cultured cells stably expressing human APP. AD-associated mutations in PSEN1 cause a loss of function that selectively affects production of Aβ40 relative to Aβ42, resulting in an increase in Aβ42/Aβ40 ratio (Chávez-Gutiérrez et al., 2012, De Strooper, 2007, Xia et al., 2015). We observed this expected effect with N135D PS1, which had lower Aβ40 production and a higher Aβ42/Aβ40 ratio than wild-type presenilin (Fig. 4). Consistent with its predicted pathogenicity, N135Y had indistinguishable effects from N135D (Fig. 4). We evaluated this mutation according to a decision tree developed for assessing pathogenicity of mutations hypothesized to cause AD (Guerreiro, 2010). Based on this decision tree, we conclude that N135Y is definitely pathogenic, citing (1) our evidence of early-onset AD in two related individuals, (2) previous reports of early-onset AD due to two other mutations at N135, (3) conservation of N135 between PSEN1 and PSEN2, with pathogenic mutations described at the homologous site in PSEN2, (4) the observation that N135 lines up with the loci of other pathogenic mutations in the second transmembrane domain of presenilin (Hardy and Crook, 2001), and (5) the effect of N135Y on Aβ42/Aβ40 ratio, which is similar to that seen in other PSEN1 mutations.
Fig 4.
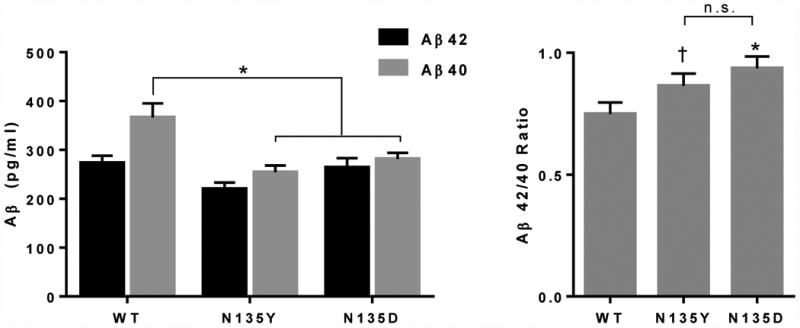
Biochemical characterization of N135Y PS1. Wild-type, N135Y, and N135D PS1 were expressed in cells stably transfected with human APP, and Aβ42 and Aβ40 were measured by ELISA in cell culture supernatants. A. Compared to wild-type PS1, both the known mutation N135D and the novel mutation N135Y suppressed Aβ40 production selectively (ANOVA, p < 0.05; N = 3; * indicates p < 0.05 on post-hoc tests). B. As a result, both mutations produced a higher Aβ42/Aβ40 ratio (ANOVA, p < 0.05;. N = 3; * indicates p < 0.05 and † indicates p = 0.06 on post-hoc tests).
4. Discussion
This is the first report of the clinical syndrome associated with a N135Y mutation in the PSEN1 gene. The cases we identified developed AD in their 30s–40s associated with spasticity, hippocampal hyperexcitability, and epileptiform discharges. Differences in age at onset of 10 years among individuals with the same mutation are not common, but do occur (Ryman et al., 2014), and may result from the contribution of other genes to overall dementia risk (Lalli et al., 2015). Due to the limited pedigree and the proband's co-morbidities, it is not possible to say with certainty which of her clinical and imaging features are due to a direct effect of the mutation.
The proband developed progressive spasticity, with early dysarthria and eventual paraparesis and gait disorder. Although her history of gastric bypass and B12 deficiency could suggest an alternative explanation for her spasticity, continued progression of the spastic gait despite adequate repletion of B12 suggests that the mutation is a contributing factor. Spasticity has been described in many kindreds with PSEN1 mutations and seems to be a unique feature as it is rare in patients with PSEN2 mutations, APP mutations or duplications, or sporadic AD (Shea et al., 2015). The cause of spasticity may be due to age of onset, mutation effect, distribution of pathology (e.g., atypically high deposition of amyloid in brainstem), or other factors. Although a higher prevalence of spasticity among individuals with mutations after codon 200 has been reported in the literature, a more recent study of the DIAN cohort suggests little difference in spasticity prevalence between mutations pre and post codon 200 (Tang et al., in press).
Hyperexcitability is another interesting feature of the clinical case. The proband had very strong hyperactivity of the left hippocampus on FDG-PET (the left hippocampus was more atrophic on structural imaging). She also had fluctuating confusion that, along with the imaging finding, prompted video-EEG telemetry monitoring. This demonstrated epileptiform activity in both frontal lobes. Hyperexcitability has been described in both sporadic and autosomal dominantly inherited AD, more commonly in the latter (Palop and Mucke, 2009). The observation of epileptiform activity without overt seizures raises the question of whether routine EEG screening may have value in patients with EOAD and whether hyperexcitability or seizures are more common with some mutations than others. In the case we describe, it is not clear whether use of anticonvulsants benefited the patient in terms of cognition or rate of functional decline, but these questions may be addressed in future clinical trials.
The patient's father's abnormal skeletal muscle findings suggest a possible variant of inclusion body myositis (IBM) without the typical inflammatory changes. A PSEN1 mutation mouse model also developed histopathological features in skeletal muscle resembling IBM including centric nuclei, intracellular accumulation of Aβ peptide, and enhanced inflammation around affected muscle fibers (Kitazawa et al., 2006). Muscle pathology in AD has been understudied and represents a future research need.
AD-causing mutations in PSEN1 are spread throughout the protein, particularly in regions that code for the transmembrane domains of the protein. N135 is in the second transmembrane (TM2) domain of PS1, near the interface with the extracellular/luminal side of the membrane. Many other PSEN1 mutations map to TM2, including common mutations at M146, identified in the original reports of PSEN1 mutations (Sherrington et al., 1995), and other mutations at PSEN1 N135, including N135D and N135S (Crook et al., 1997; Rudzinski et al., 2008). Of note, N135D represents a transition from a polar asparagine residue to a charged (acidic) aspartate residue. Such changes between amino acid categories are often pathogenic. However, the two other mutations described at this site (N135Y and N135S), both substitute other polar residues for the asparagine. This finding indicates that changes within amino acid categories remain pathogenic, and suggests that the asparagine itself may be critical to normal presenilin-1 function. N135 is homologous to N141 in PS2, which is the site of the mutation identified in the original Volga Germans with PSEN2 mutations (Levy-Lahad et al., 1995). Our biochemical analysis indicates that N135Y has effects on Aβ production consistent with those previously described for other pathogenic AD-causing mutations.
Supplementary Material
We report a father and daughter with a previously undescribed presenilin-1 mutation
The proband had a memory disorder with spastic gait and polyspike discharges on EEG
The father had autopsy-confirmed Alzheimer's disease
Biochemical analysis confirmed an increase in Aβ42/Aβ40 ratio due to the mutation
Acknowledgments
This research was supported by the National Institutes of Health (R01NS075487, U19AG032438, and U-01-AG042791), the National Institute of Aging (U19 AG032438) and the Veterans Administration (E6553W).
Footnotes
Disclosure statement: The authors have no conflicts of interest to disclose related to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Chávez-Gutiérrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, Wiltfang J, Serneels L, Karran E, Gijsen H, Schymkowitz J, Rousseau F, Broersen K, De Strooper B. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012 May 16;31(10):2261–74. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook R, Ellis R, Shanks M, Thal LJ, Perez-Tur J, Baker M, Hutton M, Haltia T, Hardy J, Galasko D. Early-onset Alzheimer's disease with a presenilin-1 mutation at the site corresponding to the Volga German presenilin-2 mutation. Ann Neurol. 1997;42:124–128. doi: 10.1002/ana.410420121. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007 Feb;8(2):141–6. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Segonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, Albert MS, Killiany RJ. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Eisenstein SA, Koller JM, Piccirillo M, Kim A, Antenor-Dorsey JA, Videen TO, Snyder AZ, Karimi M, Moerlein SM, Black KJ, Perlmutter JS, Hershey T. Characterization of extrastriatal D2 in vivo specific binding of [ (1)(8)F] (N-methyl)benperidol using PET. Synapse. 2012;66:770–780. doi: 10.1002/syn.21566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B. FreeSurfer. Neuroimage. 2012;62:774–781. doi: 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Guerreiro RJ, Baquero M, Blesa R, Boada M, Brás JM, Bullido MJ, Calado A, Crook R, Ferreira C, Frank A, Gómez-Isla T, Hernández I, Lleó A, Machado A, Martínez-Lage P, Masdeu J, Molina-Porce lL, Molinuevo JL, Pastor P, Pérez-Tur J, Relvas R, Oliveira CR, Ribeiro MH, Rogaeva E, Sa A, Samaranch L, Sánchez-Valle R, Santana I, Tàrraga L, Valdivieso F, Singleton A, Hardy J, Clarimón J. Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging. 2010 May;31(5):725–31. doi: 10.1016/j.neurobiolaging.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Crook R. Presenilin mutations line up along transmembrane alpha-helices. Neurosci Lett. 2001 Jun 29;306(3):203–5. doi: 10.1016/s0304-3940(01)01910-3. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, Dale AM, Carmichael OW, Tosun D, Weiner MW, Alzheimer's Disease Neuroimaging, I Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2010;6:212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, J LW, Ward C, Dale AM, Felmlee JP, Gunter JL, Hill DL, Killiany R, Schuff N, Fox-Bosetti S, Lin C, Studholme C, DeCarli CS, Krueger G, Ward HA, Metzger GJ, Scott KT, Mallozzi R, Blezek D, Levy J, Debbins JP, Fleisher AS, Albert M, Green R, Bartzokis G, Glover G, Mugler J, Weiner MW. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Green KN, Caccamo A, LaFerla FM. Genetically Augmenting Aβ42 Levels in Skeletal Muscle Exacerbates Inclusion Body Myositis-Like Pathology and Motor Deficits in Transgenic Mice. Am J Pathol. 2006;168:1986–1997. doi: 10.2353/ajpath.2006.051232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Singh S, Theuns J, Van Broeck B, Pirici D, Vennekens K, Corsmit E, Cruts M, Dermaut B, Wang R, Van Broeckhoven C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat. 2006;27:686–695. doi: 10.1002/humu.20336. [DOI] [PubMed] [Google Scholar]
- Lalli MA, Bettcher BM, Arcila ML, Garcia G, Guzman C, Madrigal L, Ramirez L, Acosta-Uribe J, Baena A, Wojta KJ, Coppola G, Fitch R, de Both MD, Huentelman MJ, Reiman EM, Brunkow ME, Glusman G, Roach JC, Kao AW, Lopera F, Kosik KS. Whole-genome sequencing suggests a chemokine gene cluster that modifies age at onset in familial Alzheimer's disease. Mol Psychiatry. 2015 Nov;20(11):1294–300. doi: 10.1038/mp.2015.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Guenette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Miller BL, Boeve BF. The behavioral neurology of dementia. Cambridge University Press; Cambridge, UK; New York: 2009. [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol. 2009;66:435–440. doi: 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med. 1998;39:904–911. [PubMed] [Google Scholar]
- Rowland DJ, Garbow JR, Laforest R, Snyder AZ. Registration of [18F]FDG microPET and small-animal MRI. Nucl Med Biol. 2005;32:567–572. doi: 10.1016/j.nucmedbio.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Rudzinski LA, Fletcher RM, Dickson DW, Crook R, Hutton ML, Adamson J, Graff-Radford NR. Early onset familial Alzheimer Disease with spastic paraparesis, dysarthria, and seizures and N135S mutation in PSEN1. Alzheimer Dis Assoc Disord. 2008;22:299–307. doi: 10.1097/WAD.0b013e3181732399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JB, Lopera F, Martins R, Masters CL, Mayeux RP, McDade E, Moreno S, Reiman EM, Ringman JM, Salloway S, Schofield PR, Sperling R, Tariot PN, Xiong C, Morris JC, Bateman RJ, Dominantly Inherited Alzheimer, N Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Shea YF, Chu LW, Chan AO, Ha J, Li Y, Song YQ. A systematic review of familial Alzheimer's disease: Differences in presentation of clinical features among three mutated genes and potential ethnic differences. J Formos Med Assoc. 2015 doi: 10.1016/j.jfma.2015.08.004. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HAR, Haines JL, Pericak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, Bateman RJ, Cairns NJ, Aldea P, Cash L, Christensen JJ, Friedrichsen K, Hornbeck RC, Farrar AM, Owen CJ, Mayeux R, Brickman AM, Klunk W, Price JC, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Schofield PR, Buckles V, Morris JC, Benzinger TL, Dominantly Inherited Alzheimer, N Partial volume correction in quantitative amyloid imaging. Neuroimage. 2015;107:55–64. doi: 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, D'Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA, Benzinger TL. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One. 2013;8:e73377. doi: 10.1371/journal.pone.0073377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Ryman DC, McDade E, Jasielec MS, Buckles VD, Cairns NJ, Fagan AM, Goate A, Marcus DS, Xiong C, Allegri RF, Chhatwal JP, Danek A, Farlow MR, Fox N, Ghetti B, Graff-Radford NR, Laske C, Martins RN, Masters CL, Mayeux RP, Ringman JM, Rossor MN, Salloway SP, Schofield PR, Morris JC, Bateman RJ. the Dominantly Inherited Alzheimer Network. Neurological Manifestations of Autosomal Dominant Alzheimer's Disease from the DIAN cohort and a meta-analysis. Lancet Neurology. doi: 10.1016/S1474-4422(16)30229-0. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP., Jr Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol. 1991;30:637–649. doi: 10.1002/ana.410300503. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Shankar GM, Townsend M, Fadeeva JV, Betts V, Podlisny MB, Cleary JP, Ashe KH, Rowan MJ, Selkoe DJ. The role of cell-derived oligomers of Abeta in Alzheimer's disease and avenues for therapeutic intervention. Biochem Soc Trans. 2005;33:1087–1090. doi: 10.1042/BST20051087. [DOI] [PubMed] [Google Scholar]
- Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ., 3rd Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. Neuron. 2015;85:967–981. doi: 10.1016/j.neuron.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.