

Abstract
Neuropathic pain impacts approximately 3–4.5% of the global population and remains an unresolved health problem. The management of neuropathic pain has two distinct goals—prevention of development and control of established neuropathic pain. We examined the impact of both prophylactic and therapeutic treatments with the tricyclic antidepressant desipramine on the development and maintenance of toxic neuropathic pain induced by the chemotherapeutic agent paclitaxel. We also investigated the involvement of endogenous analgesic (i.e., endogenous opioid and endocannabinoid) systems in the antinociceptive actions of desipramine in these two distinct phases of neuropathic pain. Chronic subcutaneous infusion of desipramine via osmotic pumps suppressed both the development and maintenance of paclitaxel-induced neuropathic pain. However, only prophylactic desipramine treatment blocked the development of neuropathic pain throughout the three month observation interval. The opioid receptor antagonist naloxone blocked the antinociceptive effects of both prophylactic and therapeutic desipramine treatments throughout the entire timecourse of desipramine-induced antinociception. By contrast, cannabinoid CB1 and CB2 receptor antagonists partially attenuated the antinociceptive actions of desipramine in a manner that was restricted to the development phase of paclitaxel-induced neuropathic pain only. Paclitaxel decreased cell viability in TMD231 tumor cells in an MTT assay in vitro. Notably, desipramine (1 nM to 1 μM) alone did not alter tumor cell viability and did not prevent the cytotoxic effects of paclitaxel under identical conditions. The highest concentration of desipramine (10 μM) reduced tumor cell viability alone and enhanced the cytotoxic effects of paclitaxel. Our study identifies a previously unrecognized preemptive analgesic strategy that prevents development of paclitaxel-induced neuropathic pain, and also dissects receptor mechanisms underlying desipramine-induced antinociceptive effects. This information may be applied to improve current therapeutic strategies with the goal of preventing and managing neuropathic pain induced by chemotherapeutic treatment.
Keywords: antidepressant, chemotherapy, neuropathic pain, opioid, tumor cell viability, norepinephrine
Graphical Abstract
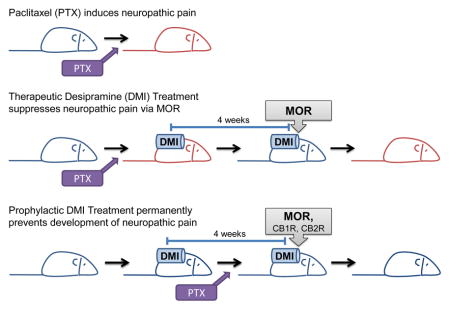
1. Introduction
Neuropathic pain is a global health concern that detrimentally impacts quality of life and produces a high socioeconomic burden (1, 2). Neuropathic pain is caused by damage to the nervous system due to lesions (e.g., abnormality or obvious trauma in diagnostic investigations), disease states (e.g., diabetes mellitus, stroke, multiple sclerosis), or toxic challenges (e.g., chemotherapy) (3, 4). Neuropathic pain treatment involves two distinct goals—prevention of the development of neuropathic pain and control of established neuropathic pain (5). Existing treatments for neuropathic pain are far from satisfactory due to their limited efficacy and adverse side effect profiles, which makes effective management of neuropathic pain an unmet and challenging clinical need (2, 5). Research efforts to date have been largely devoted to identifying novel therapeutic interventions for treating established neuropathic pain (6). However, it is equally important to understand the mechanisms of current approved pain-relieving medications, as this knowledge can be applied to improve such medications and even facilitate the development of novel analgesics for managing toxic neuropathic pain.
Tricyclic antidepressants (TCAs), such as desipramine (Norpramin®), are first-line analgesics used in all clinical neuropathic pain conditions (2, 7, 8), including chemotherapy-induced neuropathic pain (9). The recommended doses for TCAs are 25–150 mg, administered once or via two divided doses daily to patients (2). Common side effects of TCAs include anticholinergic effects, sedation, and adverse cardiac effects (2, 10). The therapeutic ratio between TCAs’ beneficial effects over adverse effects is moderate, similar to most other first-line analgesics (e.g., serotonin-norepinephrine reuptake inhibitors and anticonvulsants) (2). Thus, it is important to elucidate the mechanisms required for TCAs’ analgesic effects if we are to effectively isolate them from the signaling cascades involved in the unwanted side-effects, and achieve a more favorable therapeutic ratio between desirable and undesirable effects.
Therapeutic efficacy of desipramine, a tricyclic antidepressant, is attributed to its ability to inhibit the reuptake of the catecholamine neurotransmitter norepineprine (11–14). Although the noradrenergic mechanism of antinociceptive action of desipramine is well-documented (11–14), blockade of norepineprine reuptake is immediate but therapeutic efficacy is not apparent for several weeks following drug administration. The disconnection between the timecourse of norepinephrine reuptake and therapeutic efficacy suggests that mechanisms downstream of norepinephrine reuptake may contribute to the anti-allodynic efficacy of this agent. We thus evaluated potential contributions of downstream recruitment of endogenous analgesic systems to the anti-allodynic efficacy of desipramine in a rat model of paclitaxel-induced neuropathic pain. The discovery of endogenous analgesic systems has advanced our understanding of pain modulation and fostered development of novel pharmacotherapies (15–18). The endogenous opioid (19) and endogenous cannabinoid (endocannabinoid) (20) systems are amongst the best studied endogenous analgesic systems. Regulation of endogenous ligands (e.g., endorphins and endocannabinoids) and their receptors modulate pain states (21–25). Crosstalk between the endogenous analgesic signaling systems and signaling pathways associated with conventional therapeutic agents have been described (26–32). In the present study, we explored whether the TCA desipramine would engage endogenous analgesic systems to suppress neuropathic pain for two reasons. Firstly, desipramine interacts with endogenous opioid system in both in vitro assays (33–36) and in vivo studies (31, 35, 37–40). Secondly, both the endogenous opioid (41–43) and endocannabinoid (44–47) systems interacts with noradrenergic signaling. A role for the endogenous analgesic system in desipramine-produced antinociception effects on chemotherapy-induced allodynia has never been evaluated. We, therefore, used desipramine to explore the impact and potential contributions of endogenous analgesic mechanisms mechanisms likely to be downstream of blockade of norepinephrine reuptake to desipramine-produced antinociception.
In the present study, we tested the hypothesis that chronic administration of desipramine recruits endogenous analgesic systems to suppress chemotherapy-induced peripheral neuropathic pain induced by paclitaxel, a taxane-based chemotherapeutic agent. We varied the timing of desipramine administration to examine the efficacy of chronic treatment in preventing the development of paclitaxel-induced neuropathic pain and in suppressing the maintenance of established neuropathic pain. We also explored the contribution of endogenous analgesic systems to the antinociceptive action of desipramine using both prophylactic and therapeutic dosing schedules. Finally, we examined the impact of desipramine on tumor cell viability in the presence and absence of paclitaxel in vitro to ascertain whether desipramine could interfere with the anti-tumor efficacy of paclitaxel.
2. Material and Methods
2.1. Subjects
One hundred and fourteen adult male Sprague-Dawley rats (Harlan, Indianapolis, IN, USA), weighing 275 to 350 g, were used in these experiments. All animals were single-housed in a temperature-controlled facility, provided with food and water ad libitum. Animals were maintained on a regular 12 h light/ 12 h dark cycle (lights on at 7 am). All experiments were performed during the light cycle. All experimental procedures were approved by the Bloomington Institutional Animal Care and Use Committee of Indiana University and followed the guidelines for the treatment of animals of the International Association for the Study of Pain (48).
2.2. Drugs and Chemicals
Paclitaxel was purchased from Tecoland Corporation (Edison, NJ, USA) and was dissolved in a cremophor-containing vehicle (1:1:4 ratio of cremophor® EL/ ethanol/ saline) and delivered to rats intraperitoneally (i.p.). Desipramine (DMI), naloxone hydrochloride dehydrate, dimethyl sulfoxide (DMSO), cremophor® EL, polyethylene glycol 400 (PEG400), and acetone were purchased from Sigma-Aldrich (St. Louis, MO, USA). AM251 and AM630 were synthesized by the Makriyannis laboratory (Boston, MA, USA). Saline was obtained from Aqualite System (Lake Forest, IL, USA). Desipramine was dissolved in autoclaved distilled H2O as previously described (49). Naloxone was dissolved in saline. (−)-Naloxone hydrochloride (Sigma Aldrich N7758) used in this study is a μ-opioid receptor (MOR) antagonist that does not inhibit monoamine uptake (50) and is distinct from (+)-naloxone that blocks toll-like receptor 4 (TLR4) but does not bind to μ-opioid receptors (51, 52). AM251 and AM630 were dissolved in a 1:1 mixture of PEG400 and DMSO. Paclitaxel-treated animals receiving distilled H2O (vehicle in lieu of desipramine), saline (vehicle in lieu of naloxone), or 1:1 mixture of PEG400 and DMSO (vehicle in lieu of AM251 and AM630) showed no significant difference in mechanical and cold responsiveness or locomotor activity (P > 0.21) consistent with our previous publication (53), and were thus pooled together as a paclitaxel-vehicle group. Desipramine, vehicle, and all antagonists were delivered subcutaneously to rats in separate osmotic pumps. Rats were allowed six days to fully recover after implantation surgery prior to behavioral testing.
The following reagents were used in the in vitro studies: L-glutamine, Fetal Bovine Serum (FBS), penicillin streptomycin, and Roswell Park Memorial Institute medium (RPMI-1640), Dulbecco's Modified Eagle Medium (DMEM) were obtained from GIBCO (Grand Island, NY, USA). DMSO and MEM non-essential amino acids (NEAA), were obtained from Sigma-Aldrich (St. Louis, MO, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl-2H-tetrazolium bromide (MTT) was obtained from Promega (Madison, Wisconsin, USA). Breast adenocarcinoma TMD-231 cells were a gift from Dr. Harikrishna Nakshatri (Indiana University School of Medicine).
2.3. Osmotic pumps implantation
Osmotic pumps (model 2ML4) were purchased from Alzet (Cupertino, CA, USA) and implanted into rats subcutaneously (s.c.) following the manufacturer’s instruction, as described in our previously published work (54). After filling osmotic pumps with compounds and inserting the flow moderator according to the manufacturer’s instruction, the outer surfaces of the pumps were sterilized using Pro Advantage Alcohol Prep Pads (National Distribution & Contracting, Nashville, TN, USA). Animals were placed on a thermoregulation device and anesthetized using isoflurane. The incision site between the animal's scapulae was shaved; the exposed skin was cleaned with 70% ethanol, povidone, and 70% ethanol in a sequential order. A horizontal incision was performed on the skin between the scapulae and following blunt dissection, osmotic pumps were implanted subcutaneously with the flow moderator facing toward the sacral region of the spinal cord pointing away from the incision. Incisions were then sutured with 4-0 silk (Ethicon, Somerville, NJ, USA). Animals were monitored after surgery until fully awake and were then monitored daily for 14 days. Sutures were removed on post-surgical day 14. One animal developed seroma after surgery and was euthanized and not used for data collection.
2.4. General experimental protocol
Animals were randomly assigned to experimental groups. The experimenter was blinded to the experimental condition and osmotic pump contents in all studies. The treatment groups were separated to multiple cohorts and each cohort contained multiple groups and testing of the different cohorts overlapped. Paclitaxel was used to produce chemotherapy-induced neuropathic pain. Paclitaxel (2 mg/kg i.p.) was administered to rats four times on alternate days (day 0, 2, 4, 6) in a volume of 1 ml/kg (cumulative dose: 8 mg/kg i.p.) as previously described (55). Control rats received an equal volume of cremophor-containing vehicle. Mechanical paw withdrawal thresholds (to assess mechanical allodynia) were assessed every two days whereas the frequency of withdrawal to acetone (to assess cold allodynia) was assessed every four to six days. The timecourse of paclitaxel-induced allodynia (Fig. 1) was divided into two phases, i.e., the development phase (from day 1 to day 17) and the maintenance phase (from day 17 to day 86) of neuropathic pain. The development phase was further divided into early and late development phases to differentiate the intervals marked by active paclitaxel treatments (early development, day 1–6) and that occurring after cessation of active paclitaxel treatments (late development, day 7–17).
Figure 1. Chemotherapy-induced peripheral neuropathic pain.

Timecourse of paclitaxel-induced (A) mechanical and (B) cold allodynia. Animals all received osmotic pump containing sterile H2O (the vehicle of desipramine). Data are expressed as mean ± SEM (n = 6–9 per group). Arrows show timing of injections of paclitaxel (PTX) or cremophor-vehicle (CR). *P < 0.05 vs. cremophor vehicle.
In Experiment #1, the effects of prophylactic treatment (see Fig. 2A) with desipramine on the development and maintenance of paclitaxel-induced allodynia were examined. Pharmacological manipulations were delivered to rats via osmotic pumps for 28 days, beginning six days prior to initial paclitaxel/cremophor vehicle injection (i.e., day -6) and terminating upon pump removal on day 22 following initiation of paclitaxel/cremophor vehicle dosing. Animals received chronic infusion of vehicle, desipramine (10 mg/kg/day s.c.) (35) alone or co-administered (in a separate pump) with the opioid receptor antagonist naloxone (12 mg/kg/day s.c.) (56), the CB1 antagonist AM251 (3 mg/kg/day s.c.) (54), or the CB2 antagonist AM630 (3 mg/kg/day s.c.) (54). Separate groups received naloxone (12 mg/kg/day s.c.), AM251 (3 mg/kg/day s.c.), or AM630 (3 mg/kg/day s.c.) alone. Mechanical thresholds and cold withdrawal frequencies were measured prior to pharmacological manipulations (day -8 and day -7), during the chronic infusion of compounds (day 0–21), and after the pumps were removed (day 26–86). Locomotor activity was assessed in all groups during chronic infusion of compounds (day 19) and 9 days after the pumps were removed (day 31).
Figure 2. Prophylactic treatment with desipramine permanently blocked the development and maintenance of paclitaxel-induced allodynia.

(A) Testing scheme used to evaluate impact of prophylactic infusion of desipramine for 4 weeks on the development and maintenance of paclitaxel-induced allodynia. Chronic infusions were initiated one week prior to the onset of paclitaxel dosing and continued for 3 additional weeks. The presence of residual analgesia following cessation of drug delivery (i.e., osmotic pumps were removed) was also evaluated. (B–E) Chronic infusion of desipramine (10 mg/kg/day s.c., delivered from day −6 to day 22), initiated prior to paclitaxel dosing, suppressed the development and maintenance of paclitaxel-induced (B) mechanical and (C) cold allodynia, but did not alter responsiveness to (D) mechanical or (E) cold stimulation in animals treated with cremophor-vehicle. Arrows show timing of injections of paclitaxel (PTX) or cremophor-vehicle (CR). Dotted lines show timing of implantation and removal of the osmotic pumps containing desipramine (DMI) or vehicle. Data are expressed as mean ± SEM (n = 5–6 per group). *P < 0.05 vs. vehicle.
In Experiment #2, the effects of therapeutic treatment (see Fig. 5A) with desipramine on the maintenance phase of paclitaxel-induced allodynia were assessed. After paclitaxel-induced allodynia was fully established on day 18, rats received pharmacological manipulations delivered by osmotic pumps from day 18 to day 46 following initiation of paclitaxel/cremophor vehicle dosing. Animals received chronic infusions of vehicle, desipramine (10 mg/kg/day s.c.) alone or co-administered (in a separate pump) with the opioid receptor antagonist naloxone (12 mg/kg/day s.c.), the CB1 antagonist AM251 (3 mg/kg/day s.c.), or the CB2 antagonist AM630 (3 mg/kg/day s.c.). Separate groups received naloxone (12 mg/kg/day s.c.), AM251 (3 mg/kg/day s.c.), or AM630 (3 mg/kg/day s.c.) alone. Mechanical thresholds and cold withdrawal frequencies were measured prior to pharmacological manipulations (on day 0–17), during the chronic infusion of compounds (day 22–45), or after the pumps were removed (day 50–75). Locomotor activity was assessed in all groups during the chronic infusion of compounds (day 43) and after the pumps were removed (day 55).
Figure 5. Therapeutic treatment with desipramine suppressed the maintenance of paclitaxel-induced allodynia.

(A) Testing scheme used to evaluate impact of chronic infusion of desipramine for 4 weeks in a therapeutic dosing paradigm on the maintenance of paclitaxel-induced allodynia. Infusions were initiated after establishment of paclitaxel-induced allodynia. The presence of residual analgesia following cessation of drug delivery (i.e. osmotic pumps were removed) was also evaluated. (B-D) Chronic infusion of desipramine (10 mg/kg/day s.c., delivered from day 18 to day 46), delivered after the establishment of paclitaxel-induced allodynia, suppressed paclitaxel-induced (B) mechanical and (C) cold allodynia, but did not alter responsiveness to (D) mechanical or (E) cold stimulation in animals treated with cremophor-vehicle. Arrows show timing of injections of paclitaxel (PTX) or cremophor-vehicle (CR). Dotted lines show timing of implantation and removal of the osmotic pumps containing desipramine (DMI) or vehicle. Data are expressed as mean ± SEM (n = 6–10 per group). *P < 0.05 vs. vehicle.
In Experiment #3, the impacts of desipramine on paclitaxel-produced cytotoxic effects in tumor cells were assessed using MTT assay in vitro. First, we determined the effects of serial log10 dilutions of paclitaxel (0.1 nM–1 μM) on breast adenocarcinoma TMD-231 cells. Then, we evaluated the impact of desipramine on paclitaxel-produced cytotoxicity by testing serial log10 dilutions of desipramine (1 nM 10 μM) alone or in combination of paclitaxel (20 nM) on the cell viability of TMD-231 cells.
2.5. Assessement of mechanical allodynia in rats
Withdrawal thresholds to mechanical stimulation were measured in duplicate for each paw using an electronic von Frey anesthesiometer equipped with a 800-g range probe (IITC Life Science Inc., Woodland Hills, CA) as described previously (57). Mechanical paw withdrawal thresholds were measured in duplicate for each paw and were reported as the mean of duplicate determinations averaged across paws.
2.6. Assessment of cold allodynia in rats
Paw withdrawal frequencies to cold stimulation were measured using the acetone method as previously described (57). Rats were placed underneath transparent plastic cages on an elevated metal mesh table. After habituation, an acetone bubble that formed at the end of a blunt 1-ml syringe was gently presented to the plantar surface of the hind paw. Animals were observed for 20 seconds after acetone application. Acetone was applied to each paw of the animal 5 times alternately with a 3-min interval between applications. Paw withdrawal on a given trial was deemed present if animals showed one or more forms of unilateral nocifensive behavior (i.e., withdrawing, raising, licking, shaking, or repetitive stepping on the stimulated paw), whereas trials on which an animal did not show any unilateral behavior were counted as zero. Paw withdrawal frequencies were recorded as the percentage of the total number of occurrences of paw withdrawal out of 10 trials per animal (i.e., five trials per paw).
2.7. Assessment of Locomotor Activity
Locomotor activity in rats was measured using an activity meter (Coulbourn Instruments, Whitehall, PA) and calculated by TruScan 2.03 (Coulbourn) following manufacturer’s instruction as previously described (54). Animals were placed in the center of a 40.64 x 40.64 x 40.64 cm3 activity chamber in a dark room (i.e., 50 lux illumination) and measured for total distance (cm) traveled during a 15-min observation interval. Animals were not habituated to the open field pre-drug because all drugs were administered via chronic infusion and locomotor activity tests were performed in the same animals used to evaluate anti-allodynic efficacy.
2.8. Cell culture
TMD-231 cells were cultured in DMEM medium with penicillin–streptomycin (100 units/mL 100 μg/mL), 0.1 mM NEAA, 2 mM L-glutamine, and 10 % fetal bovine serum (v/v) at 37 °C under 5 % CO2 in humidified incubator conditions. Cells were passaged every three days and confluent cells were used as the test cell line.
2.9. MTT cytotoxicity assay
TMD-231 cells were plated onto 96-well plates (1 × 103 cells/well) and incubated for 24 h at 37°C. Vehicle (0.1% DMSO), various concentrations of paclitaxel (serial log10 dilutions from 0.1 nM to 1 μM, and 10, 20, 40, 80 nM), and despiramine (serial log10 dilutions from 1 nM to 10 μM) alone or in combination with paclitaxel (20 nM) were added to each well and cells were incubated for 72 h at 37 °C in a humidified incubator containing 5% CO2. Following the incubation period, 10 μL of MTT solution (10 mg/mL) was added into each well followed by further incubation for 4 h in the incubator at 37°C. 100 μL of solubilization solution was added per well. The absorbance was measured using a microplate reader at a wavelength of 570 nm. The absorbance of untreated cells was set as 100% viability and the values of treated cells were calculated as percentage of the absorbance of untreated cells.
2.10. Statistical analyses
Analysis of variance (ANOVA) for repeated measures was used to determine the time course of paclitaxel-induced allodynia and chronic drug effects, followed by Bonferroni post hoc tests for comparisons between groups. The Sphericity-Assumed correction was applied to all repeated factors. One-way ANOVA was used to identify the source of significant interactions at each time point, followed by Bonferroni post hoc tests or student t-tests as appropriate. A 2 x 2 ANOVA was used to compare the impact of desipramine (or vehicle treatment) on locomotor activity in paclitaxel- or cremophor-treated rats at each timepoint. Paired t-tests were additionally used to compare locomotor activity levels during and after chronic desipramine infusion. The dose-response curve and IC50 value for paclitaxel on inhibiting tumor cell viability were determined using GraphPad Prism (CA, USA). One way ANOVA followed by Bonferroni post hoc tests was used to analyze in vitro data to assess potential impact of desipramine on the cytotoxic effects of paclitaxel on tumor cell viability. Statistical analyses were performed using IBM-SPSS Statistics version 21.0 (SPSS Inc., Chicago, IL, USA) and GraphPad Prism version 5.02 for windows software (GraphPad Software, San Diego, CA USA). P < 0.05 was considered statistically significant. Figures were presented with the relevant statistical groups.
3. Results
3.1. Paclitaxel induced neuropathic pain in rats
Paclitaxel treatment induced hypersensitivities to mechanical (F1,13 = 77.11, P < 0.0001, Fig. 1A) and cold (F1,13 = 85.66, P < 0.0001, Fig. 1B) stimulation relative to rats treated with the cremophor-vehicle. In paclitaxel-treated animals, mechanical (P < 0.05) and cold (P < 0.01) allodynia emerged on day 4 and day 11, respectively. Paclitaxel-evoked mechanical and cold allodynia were maximal on day 17 and was maintained until at least day 86 post initiation of paclitaxel dosing. To further study the effects of drug manipulations on the time course of paclitaxel-induced allodynia, the time course of drug action was divided into a development phase (day 1 to day 17, where allodynia was gradually developing), and a maintenance phase (day 18 to day 86, where maximal allodynia was already established and stably maintained throughout the remaining observation interval). Paclitaxel accumulation in the dorsal root ganglion (DRG) reaches a peak after all doses of paclitaxel have been administered in the animals (day 7) and the accumulation is largely reduced on day 16 (58). Thus, we further divided the development phase into an early (i.e., from day 1 to day 6, during which paclitaxel injections were performed) and a late (i.e., from day 7 to day 17, following termination of paclitaxel dosing) development phase. This separation enabled us to further evaluate the temporal resolution of drug effects relative to the timing of both paclitaxel treatments and development of resulting allodynia.
3.2. Prophylactic treatment with desipramine suppressed the development and establishment of paclitaxel-induced neuropathic pain
To study the effects of desipramine treatment on the development and establishment of paclitaxel-induced allodynia, we used a prophylactic dosing paradigm where desipramine was delivered via a 4-week osmotic pump implanted to animals beginning 6 days (day -6) prior to initiation of paclitaxel dosing (Fig. 2A). This chronic infusion of desipramine was sustained during the period of paclitaxel dosing (day 0–6) and covered both the development phase (day 1–17) and the first 5 days of the establishment (day 18–22) of paclitaxel allodynia (Fig. 2A).
Prophylactic treatment with desipramine (10 mg/kg/day s.c.), delivered from day -6 to day 22 following initial paclitaxel dosing, blocked the development of paclitaxel-induced mechanical (F1,9 = 166.07, P < 0.0001, Fig. 2B) and cold allodynia (F1,9 = 60.85, P < 0.0001, Fig. 2C) on all days tested (F31,279 = 5.77, P < 0.0001 mechanical, F17,153 = 2.48, P < 0.01 cold). Effects of desipramine on the development (day 1–17) and maintenance phases of paclitaxel-evoked allodynia [during (day 18–22) and after (day 23–86) chronic infusion] were further analyzed. Prophylactic desipramine treatment suppressed the development (F1,9 = 24.21, P < 0.01 mechanical; F1,9 = 14.07, P < 0.01, day1–17 cold) and prevented the establishment (i.e., maintenance) of paclitaxel-induced mechanical (F1,9 = 176.22, P < 0.0001) and cold (F1,9 = 62.39, P < 0.0001) allodynia relative to the vehicle group during the period of chronic infusion (from day -6 to day 22). Moreover, prophylactic desipramine treatment also produced residual antinociceptive effects, preventing the maintenance of paclitaxel-evoked mechanical (F1,9 = 403.13, P < 0.0001, Fig. 2B) and cold (F1,9 = 62.63, P < 0.0001, Fig. 2C) allodynia compared to the vehicle group from day 26 to day 86 (P < 0.01 mechanical, P < 0.05 cold). Thus, prophylactic infusion of desipramine prevented the development of paclitaxel-induced allodynia and allodynia completely failed to develop even after the drug was no longer administered.
Prophylactic treatment with desipramine (10 mg/kg/day s.c., delivered from day -6 to day 22) did not alter responsiveness to mechanical (F1,10 = 4.09, P = 0.07, Fig. 2D) or cold (F1,10 = 3.64, P = 0.09, Fig. 2E) stimulation in animals treated with cremophor vehicle in lieu of paclitaxel on any day tested (F31,310 = 1.45, P = 0.06 mechanical, F17,170 = 1.53, P = 0.09 cold).
3.3. Prophylactic desipramine treatment-induced prevention of paclitaxel allodynia were fully blocked by opioid receptor antagonist naloxone
Anti-allodynic effects of prophylactic treatment with desipramine (10 mg/kg/day s.c., delivered from day -6 to day 22) on paclitaxel-evoked mechanical (F2,14 = 162.67, P < 0.0001, Fig. 3A) and cold (F2,14 = 54.46, P < 0.0001, Fig. 3B) hypersensitivities were fully blocked by concurrent administration of the opioid receptor antagonist naloxone (12 mg/kg/day s.c.) (P < 0.0001 mechanical, P < 0.0001 cold). Naloxone-induced blockade of desipramine-induced anti-allodynic effects were present throughout both the development (P < 0.0001 mechanical, P < 0.05 cold) and maintenance (P < 0.0001 mechanical, P < 0.0001 cold) phases of paclitaxel-induced allodynia (Fig. 3A–B). Moreover, the inhibitory effects were sustained when the pumps were removed and drugs were no longer being delivered (P < 0.0001 mechanical, P < 0.0001 cold, Fig. 3A–B). Naloxone (12 mg/kg/day s.c.) alone, delivered from day -6 to day 22, did not alter paclitaxel-induced mechanical (F1,7 = 0.09, P > 0.77) or cold (F1,7 = 0.10, P > 0.76) responsiveness compared to vehicle (Fig. 3C–D).
Figure 3. Opioid receptor antagonist naloxone blocked the anti-nociceptive effects of prophylactic desipramine treatment on paclitaxel-induced allodynia.

The anti-allodynic effects of prophylactic treatment with desipramine (10 mg/kg/day s.c., delivered from day −6 to day 22) on paclitaxel-evoked (A) mechanical and (B) cold hypersensitivities were blocked by naloxone (12 mg/kg/day s.c.). Naloxone alone did not alter paclitaxel-induced (C) mechanical or (D) cold allodynia. Data are expressed as mean ± SEM (n = 4–6 per group). Arrows show timing of injections of paclitaxel. Dotted lines show timing of implantation and removal of the osmotic pumps. *P < 0.05 vs. vehicle. xP < 0.05 vs. desipramine.
3.4. Partial involvement of cannabinoid CB1 and CB2 signaling in the anti-allodynic effects of prophylactic desipramine treatment
Anti-allodynic effects of prophylactic desipramine treatment (10 mg/kg/day s.c., delivered from day -6 to day 22) on paclitaxel-evoked mechanical (F2,13 = 26.28, P < 0.0001, Fig. 4A) and cold (F2,13 = 16.30, P < 0.0001, Fig. 4B) hypersensitivities were not altered by the CB1 antagonist AM251 (3 mg/kg/day s.c.) over the entire observation interval but impact of antagonist treatment approached significance (P = 0.08 mechanical, P = 0.05 cold). However, AM251 reliably attenuated desipramine-induced anti-allodynic effects in both modalities in the late development phase (after all doses of paclitaxel were given and before the maximal allodynia was established, day 7–17) (P < 0.05 mechanical, P < 0.01 cold, Fig. 4A–B). AM251 also attenuated desipramine-induced suppression of cold (P < 0.05), but not mechanical (P = 0.40), allodynia during the maintenance phase of paclitaxel-induced allodynia (Fig. 4A–B). The suppressive effects of AM251 were no longer present when compounds were removed (P = 0.08 mechanical, P = 0.13 cold, Fig. 4A–B).
Figure 4. Partial involvement of cannabinoid receptor signaling in the anti-allodynic effects of prophylactic desipramine treatment.

The CB1 antagonist AM251 (3 mg/kg/day s.c, delivered from day -6 to day 22) partially blocked the anti-allodynic effects of chronic desipramine (10 mg/kg/day s.c., delivered from day -6 to day 22) on (A) mechincal and (B) cold hypersensitivities in the late development phase of pactlitaxel-induced allodynia. The CB2 antagonist AM630 (3 mg/kg/day s.c., delivered from day -6 to day 22) transiently blocked the anti-allodynic effects of chronic desipramine (10 mg/kg/day s.c., delivered from day −6 to day 22) on (C) mechanical, but not (D) cold allodynia, during the late development phase of paclitaxel-induced allodynia. AM251 or AM630 alone did not alter paclitaxel-induced (E) mechanical or (F) cold allodynia. Data are expressed as mean ± SEM (n = 5–6 per group). Arrows show timing of injections of paclitaxel. Dotted lines show timing of implantation and removal of the osmotic pumps. *P < 0.05 vs. vehicle. xP < 0.05 vs. desipramine.
Prophylactic treatment with desipramine suppressed mechanical (F2,14 = 79.15, P < 0.0001, Fig. 4C) and cold (F2,14 = 25.62, P < 0.0001, Fig. 4D) allodynia in a time-dependent manner; anti-allodynic effects were not blocked by the CB2 antagonist AM630 (3 mg/kg/day s.c.) (P = 0.17 mechanical, P = 0.11 cold). However, AM630 reliably, though transiently, blocked desipramine-induced suppression of mechanical allodynia when analysis was restricted to the late development phase of paclitaxel-induced allodynia (P < 0.01 mechanical) (Fig. 4C–D).
Neither AM251 (3 mg/kg/day s.c.) nor AM630 (3 mg/kg/day s.c.) alone, delivered from day -6 to day 22, altered paclitaxel-induced hypersensitivities to either mechanical (F2,14 = 3.72, P > 0.05) or cold (F2,14 = 0.41, P > 0.68) stimulation compared to the vehicle group (Fig. 4E–F).
3.5. Therapeutic treatment with desipramine suppressed the maintenance of paclitaxel-induced allodynia
To study the effects of desipramine treatment on the maintenance of paclitaxel-induced allodynia, we designed a therapeutic dosing paradigm where the 4-week chronic infusion of desipramine was restricted to timepoints when paclitaxel allodynia was maximal and fully established (i.e., chronic infusion beginning on day 18, Fig. 5A).
Therapeutic treatment with desipramine (10 mg/kg/day s.c.), delivered from day 18 to day 46 following initial paclitaxel dosing, reversed established mechanical (F1,14 = 24.21, P < 0.0001, Fig. 5B) and cold (F1,14 = 55.66, P < 0.0001, Fig. 5C) allodynia in a time-dependent manner (F33,462 = 15.33, P < 0.0001 mechanical, F13,182 = 14.13, P < 0.0001 cold). Desipramine suppressed mechanical (P < 0.0001) and cold (P < 0.0001) allodynia relative to the vehicle group during the time period associated with chronic infusion (from day 18 to day 46). Moreover, desipramine produced residual antinociceptive effects on mechanical (P < 0.01) and cold (P < 0.0001) allodynia compared to the vehicle group (i.e., from day 46 to day 58 following initiation of paclitaxel dosing) after the pumps were removed and drugs were no longer being infused. However, neuropathic pain eventually returned (Fig. 5B–C).
By contrast, desipramine, infused during the same interval, did not alter responsiveness to mechanical (F1,10 = 1.44, P = 0.26, Fig. 5D) or cold (F1,10 = 0.86, P = 0.38, Fig. 5E) stimulation in animals treated with cremophor-vehicle in lieu of paclitaxel.
3.6. Anti-allodynic effects of therapeutic desipramine treatment on the maintenance of paclitaxel-induced allodynia was fully blocked by naloxone
Anti-allodynic effects of therapeutic treatment with desipramine (10 mg/kg/day s.c., delivered from day 18 to day 46) on paclitaxel-evoked mechanical (F2,19 = 11.79, P < 0.0001, Fig. 6A) and cold (F2,19 = 18.12, P < 0.0001, Fig. 6B) hypersensitivities were fully blocked by the opioid receptor antagonist naloxone (12 mg/kg/day s.c.). This blockade was observed both during the chronic infusion interval (day 18 to day 46, P < 0.0001 mechanical, P < 0.0001 cold) and after termination of chronic infusion (i.e., from day 47 to 58 following initiation of paclitaxel dosing), when residual anti-allodynic effects of therapeutic desipramine treatment (P < 0.01 mechanical, P < 0.0001 cold) were apparent. Naloxone (12 mg/kg/day s.c.) infusion alone was associated with a modest attenuation of paclitaxel-induced mechanical allodynia that was restricted to the interval following removal of the osmotic pump (F1,13 = 4.69, P < 0.05 mechanical, Fig. 6C). However, naloxone (12 mg/kg/day s.c.) alone, delivered from day 18 to day 46, did not alter paclitaxel-induced hypersensitivity to cold stimulation (F1,13 = 3.03, P = 0.11 cold, Fig. 6D).
Figure 6. Opioid receptor antagonist naloxone blocked the anti-nociceptive effects of therapeutic desipramine treatment on paclitaxel-induced allodynia.

The anti-allodynic effects of therapeutic treatment with desipramine (10 mg/kg/day s.c., delivered from day 18 to day 46) on paclitaxel-evoked (A) mechancal and (B) cold hypersensitivities were blocked by naloxone (12 mg/kg/day s.c.), but in each case allodynia eventually returned. Naloxone alone produced a modest attenuation of paclitaxel-induced (C) mechanical but not (D) cold allodynia following removal of the osmotic pump. Data are expressed as mean ± SEM (n = 5–10 per group). Arrows show timing of injections of paclitaxel. Dotted lines show timing of implantation and removal of the osmotic pumps. *P < 0.05 vs. vehicle. xP < 0.05 vs. desipramine.
3.7. Therapeutic desipramine treatment suppressed the maintenance of paclitaxel- induced allodynia independent of cannabinoid signaling
Therapeutic treatment with desipramine (10 mg/kg/day s.c., delivered from day 18 to day 46) suppressed established paclitaxel-evoked mechanical (F2,19 = 25.02, P < 0.0001, Fig. 7A) and cold (F2,19 = 37.68, P < 0.0001, Fig. 7B) hypersensitivities, but these effects were not blocked by the CB1 antagonist AM251 (3 mg/kg/day s.c.) (P = 1.00 mechanical, P = 1.00 cold). Similarly, desipramine-induced suppressions of established mechanical (F2,19 = 22.78, P < 0.0001, Fig. 7C) and cold (F2,19 = 42.27, P < 0.0001, Fig. 7D) allodynia were not blocked by the CB2 antagonist AM630 (3 mg/kg/day s.c.) (P = 1.00 mechanical, P = 1.00 cold). AM251 (3 mg/kg/day s.c.) or AM630 (3 mg/kg/day s.c.) alone, delivered from day 18 to day 46, did not alter nociceptive responsiveness to either mechanical (F2,18 = 1.81, P = 0.19 mechanical, Fig. 7E) or cold stimulation (F2,18 = 0.29, P = 0.76 cold, Fig. 7F).
Figure 7. Therapeutic treatment with desipramine suppressed the maintenance of paclitaxel-induced allodynia independent of cannabinoid receptor signaling.

(A, B) CB1 antagonist AM251 (3 mg/kg/day s.c.) or (C, D) CB2 antagonist AM630 (3 mg/kg/day s.c.) did not block the anti-allodynic effects of chronic desipramine (10 mg/kg/day s.c., delivered from day 18 to day 46) on paclitaxel-evoked (A, C) mechanical and (B, D) cold hypersensitivities. AM251 or AM630 alone did not alter paclitaxel induced (E) mechanical or (F) cold allodynia. Data are expressed as mean ± SEM (n = 5–10 per group). Arrows show timing of injections of paclitaxel. Dotted lines show timing of implantation and removal of the osmotic pumps. *P < 0.05 vs. vehicle.
3.8. Effects of prophylactic desipramine on locomotor activity in paclitaxel-treated rats
Paclitaxel treatment did not alter locomotor activity compared to cremophor-vehicle treatment either during (day 19, P = 0.38) or after (day 43, which is 9 days following pump removal, P = 0.78) prophylactic infusion (i.e., day -6 to day 22) and no significant interaction was observed between infusion pump content (desipramine vs. vehicle) and chemotherapy status (i.e., paclitaxel vs. cremophor-vehicle) on either during (day 19, P = 0.875) or after (day 43, P = 0.223) chronic infusion. Locomotor activity was, however, modestly impacted by osmotic pump content during (day 19, F1,23 = 16.14, P < 0.001) but not after the period of chronic infusion (P = 0.96) (Fig. 8A). Locomotor activity levels did not differ before (day 19) or after (day 31) cessation of desipramine infusions in either paclitaxel-treated (P = 0.95, paired t-test) or cremophor vehicle-treated (P > 0.06, paired t-test) groups. Naloxone and cannabinoid antagonists failed to restore the desipramine-induced changes in locomotor activity in paclitaxel-treated animals (P > 0.80, Fig. 8B).
Figure 8. Impact of chronic infusion of desipramine or vehicle via prophylactic or therapeutic dosing regimens on locomotor activity in rats.

Effects of (A) prophylactic and (C) therapeutic dosing with desipramine on locomotor activity during (day 19 or day 43, respectively) and after (day 31 or day 55, respectively) chronic infusion. Antagonists did not alter desipramine-induced changes on locomotor activity during either the (B) prophylactic (day 19) or (D) therapeutic (day 43) treatment. Data are mean ± SEM (n = 5–10 per group). Abbreviations: CR, cremophor; PTX, paclitaxel; DMI, desipramine. *P < 0.05 vs. vehicle.
3.9. Effects of therapeutic desipramine on locomotor activity in paclitaxel-treated rats
Paclitaxel treatment did not alter locomotor activity compared to cremophor-vehicle treatment either during (day 43, P = 0.21) or after (day 55, which is 9 days following pump removal, P = 0.32) the interval of therapeutic infusion (i.e., day 18 to day 46) (Fig. 8C). Moreover, no significant interaction was observed between infusion pump content (desipramine vs. vehicle) and chemotherapy status (i.e., paclitaxel vs. cremophor-vehicle) at either during (day 43, P = 0.78) or after (day 55, P = 0.22) chronic infusion. Locomotor activity was, however, modestly attenuated by desipramine infusion at the early (day 43, F1,23 = 19.6, P < 0.001) but not at the late (day 55, P = 0.14) time point (Fig. 8C). Moreover, locomotor activity levels did not differ in the presence (day 43) or absence (day 55) of desipramine infusion in either paclitaxel-treated (P = 0.68, paired t-test) or cremophor vehicle-treated (P = 0.86, paired t-test) groups (Fig. 8C). Naloxone and cannabinoid antagonists failed to restore the desipramine-associated reduction in locomotor activity in paclitaxel-treated animals (P > 0.61, Fig. 8D).
3.10. Effects of desipramine and paclitael on tumor cell viability
We used an MTT assay to determine the impact of desipramine on the viability of TMD-231 tumor cells in the presence or absence of paclitaxel. Paclitaxel produced a concentration-dependent reduction in TMD-231 tumor cell viability with an IC50 of 2.5 nm (Fig. 9A). Paclitaxel (20 nM) inhibited TMD-231 cell viability (F11,100 = 99.38, P < 0.0001) relative to vehicle treatment (P < 0.0001). Desipramine (1 nM – 1 μM) alone did not alter TMD-231 cell viability relative to vehicle treatment (P = 1.00 for all desipramine concentrations) (Fig. 9B). Moreover, the combination of desipramine (1 nM – 1 μM) with paclitaxel (20 nM) inhibited TMD-231 cell viability compared to vehicle treatment (P < 0.0001) and produced similar cytotoxic effects compared to the paclitaxel (20 nM) alone group (P = 1.00) for all desipramine concentrations tested (Fig. 9B). Only the highest concentration of desipramine (10 μM), administered alone, reduced tumor cell viability in relative to vehicle (P < 0.001) and the combination of desipramine (10 μM) with paclitaxel (20 nM) further reduced tumor cell viability compared to paclitaxel (20 nM) alone (P < 0.01) (Fig. 9B). Thus, desipramine did not impede paclitaxel-induced reductions in tumor cell viability in TMD-231 cells.
Figure 9. Effects of desipramine and paclitaxel on tumor cell viability.

(A) Paclitaxel reduces TMD-231 tumor cell viability in MTT assay with IC50 of 2.5 nM. (B) Despiramine (1 nm-10 μM) did not impede ability of paclitaxel (20 nM) to reduce tumor cell viability in TMD-231 cells. Only the highest concentration of desipramine (10 μM) enhanced the cytotoxic effects of paclitaxel and reduced tumor cell viability relative to vehicle in the absence of paclitaxel. *P < 0.05 vs. vehicle, xP < 0.05 vs. paclitaxel (20 nM). Values are means ± standard deviation of 8–16 replicates per determination. The same findings were obtained from three different experiments.
4. Discussion
Mechanisms associated with the development of neuropathic pain differ from those that serve to perpetuate and maintain it (5). A better understanding of differences in pharmacological sensitivities produced by currently approved therapeutics during the development and maintenance phases of pathological pain, may elucidate different underlying mechanisms and facilitate optimization of therapeutic ratios (e.g., increased efficacy and reduced side-effects) (3). In the present study, we compared the antinociceptive effects of prophylactic and therapeutic treatment with desipramine in a rat model of chemotherapy-induced neuropathic pain to study antinociceptive effects of this agent during either the development or maintenance phases of neuropathic pain. We also evaluated possible contributions of endogenous opioid and endocannabinoid analgesic systems to desipramine’s antinociceptive efficacy.
Prevention of the development of iatrogenic neuropathic pain is an important goal of neuropathic pain management, as is attenuation of established neuropathic pain (5). We explored the anti-allodynic effects of desipramine using both prophylactic (i.e., administered prior to paclitaxel dosing) and therapeutic (i.e., administered after allodynia is fully established) treatment regimens. Desipramine treatment not only suppressed established paclitaxel-induced allodynia, but also prevented the development of neuropathic pain when dosing occurred both before and coincident with paclitaxel administration. Strikingly, the anti-allodynic effects of prophylactic desipramine treatment were present throughout the entire 92-day observation interval that represents a long period of time in the life span of an adult rat. This observation is important for the clinical scenario where regimens of chemotherapeutic treatment are scheduled and predictable, raising the possibility that effective preemptive and prophylactic analgesic strategies may be attainable. More work is necessary to determine whether prophylactic effects of desipramine on chemotherapy-induced neuropathic pain generalize to other tricyclic agents (59) or monoamine re-uptake inhibitors (60), especially given that preemptive administration of tricyclic antidepressant amitriptyline has been shown to be effective on post-herpetic neuralgia in a clinical trial (59).
Interestingly, the pervasive preventative benefit of desipramine was specific to the development of paclitaxel-induced allodynia; when desipramine was administered (for an equivalent 4-week dosing interval) after neuropathic pain was already established, the residual anti-allodynic effects of therapeutic desipramine treatment only lasted for 12 days, and neuropathic pain eventually returned. Differential efficacies between prophylactic and therapeutic desipramine dosing may be attributed to distinct mechanisms underlying the development and maintenance phases of paclitaxel-induced allodynia. Paclitaxel accumulates in the DRG, and produces mitochondrial toxicity and damage in primary afferent fibers (58, 61, 62). Indeed, in this rat model of paclitaxel-induced allodynia, on day 7 (after all doses of paclitaxel), a high level of accumulation of paclitaxel is observed in DRG where paclitaxel-induced mitochondria abnormalities in the sensory neurons occur and the amount of paclitaxel in DRG decreases after allodynia is fully established (58). More work is necessary to determine whether prophylactic treatment with desipramine prevents these early direct changes induced by paclitaxel, and consequently, neuropathic pain fails to develop. On the other hand, the maintenance of paclitaxel-induced allodynia results from long-term influences produced by paclitaxel, such as increased secretions of proinflammatory cytokines and activation of astrocytes (and possibly microglia) that contribute to central sensitization (63, 64). The maintenance phase of neuropathic pain additionally involves changes in the expression level of proteins associated with nociceptive responsiveness (65). Therapeutic desipramine treatment may attenuate the maintenance of neuropathic pain by suppressing the long-term changes produced by paclitaxel. However, such long-term changes once established by paclitaxel can only be suppressed, but cannot be permanently prevented when desipramine (or its residual antinociceptive efficacy) is no longer present in the system; thus, neuropathic pain is postponed, but not permanently eliminated.
Different classes of chemotherapeutic agents target cancer or solid tumor through different mechanisms. For example, taxanes (e.g., paclitaxel) stabilize microtubule polymerization and formation whereas vinca alkaloids (e.g., vincristine) inhibit microtubule formation thus arresting cell division (66). As a result, the neurotoxicity levels and the degree of severity of peripheral neuropathic pain associated with different classes of chemotherapeutic agents also vary (65). Acute desipramine fails to attenuate established vincristine-induced neuropathic pain (67). However, our study showed that chronic dosing with desipramine is sufficient to suppress paclitaxel-induced allodynia but only prophylactic dosing permanently prevented its development. This difference in desipramine antinociceptive efficacy may reflect distinct mechanisms underlying the development and maintenance of neuropathic pain, different dosing paradigms (e.g. preemptive vs. therapeutic) and durations of dosing, or the chemotherapeutic agent used to induce neuropathic pain (65). It is plausible that chronic dosing with desipramine would suppress neuropathic pain induced by other classes of chemotherapeutic agents, including vincristine, as anti-depressant efficacy of TCAs similarly takes weeks to emerge, a fact which may explain why the analgesic effects of TCAs on chemotherapy neuropathic pain in the clinic is only moderate (2).
We also evaluated whether desipramine could alter anti-tumor effects of paclitaxel in vitro. Importantly, paclitaxel reduced tumor cell viability in a metabolism-based colorimetric assay (i.e., the MTT assay), and these actions were not impeded by desipramine treatment. Moreover, desipramine alone did not alter tumor cell viability in the absence of paclitaxel treatment. However, more work is necessary to determine whether desipramine could influence the metabolism, excretion, or access of the chemotherapeutic agent into neurons in vivo. Our studies do not preclude the possibility that desipramine alters the pharmacokinetics of paclitaxel since both share a tricyclic structure.
Desipramine suppresses neuropathic pain induced by diabetes and traumatic nerve injury through mechanisms that involve blockade of norepinephrine re-uptake, activation of descending pathways in the spinal cord that block pain signals from ascending to the brain, and the involvement of β2-adrenoceptors (11–14, 68, 69). However, the mechanism of its antinociceptive action on chemotherapy-induced neuropathic pain remains incompletely characterized. Blockade of norepinephrine reuptake is immediate but efficacy of tricyclic antidepressants in models of depression and pathological pain takes several weeks to develop. Thus, our studies focused on characterizing endogenous analgesic mechanisms downstream of immediate noradrenergic actions in desipramine-produced suppression of chemotherapy-induced neuropathic pain.
Interactions between desipramine and the endogenous opioid system remain incompletely characterized but are nonetheless supported based upon in vitro studies using cell-based assays (33, 34) and in vivo studies in human (70) and animals (35, 37, 38). In vitro studies suggest that desipramine (35, 36) as well as other TCAs (71, 72) bind to opioid receptors. Moreover, chronic desipramine down-regulates opioid receptors in vivo in rat cerebral cortex (39). Desipramine not only modulates morphine-induced analgesia (40), but also potentiates the antinociceptive effects of morphine on tail-flick nociception and hot-plate-induced thermal nociception (31). In our study, anti-allodynic effects of both prophylactic and therapeutic desipramine dosing strategies were blocked by the opioid receptor antagonist naloxone. These observations are in line with previous findings of naloxone-induced blockade of the analgesic effects of desipramine (71) and other TCAs (e.g., clomipramine) (73) on electric shock-induced nociception. The antinociceptive effects of desipramine on the development and maintenance of paclitaxel-induced allodynia is likely attributable to the interactions between endogenous opioid system and noradrenergic function on pain suppression that occur downstream of blockade of norepinephrine reuptake (74). More studies are needed to further elucidate the role of endogenous opioid system in the actions of other TCAs in chemotherapy-induced neuropathic pain.
Interestingly in our study, the transient contribution of cannabinoid receptors in the antinociceptive action of desipramine was restricted to the development phase of paclitaxel-induced allodynia. The endocannabinoid system is implicated in the descending control of pain (75, 76) and interacts with norepinephrinergic and serotonergic systems (32, 44–47, 77). The partial blockade of the antinociceptive effects of prophylactic desipramine by the CB1 antagonist observed in the present study may be due to the CB1-mediated regulation of norepinephrine levels and modulation of descending pain inhibitory pathways (78, 79). A better understanding of the contribution of cannabinoid CB1 and CB2 receptor mechanisms to the therapeutic actions of TCAs are needed. It would also be worth evaluating whether the effective dosing regimen of desipramine can be modified by co-administration with other modulators of the endogenous analgesia system.
Clinical studies have shown that patients treated with TCAs often develop strong sedation (2). In the present study, we observed a modest reduction in locomotor activity in animals receiving chronic desipramine compared to vehicle infusions irrespective of paclitaxel or cremophor-vehicle treatment. Importantly, locomotor activity did not differ during chronic infusion of desipramine or after cessation of drug delivery (i.e., 9 days following osmotic pump removal) in experiments employing either prophylactic or therapeutic dosing strategies. These findings suggest that any observed locomotor effects of desipramine were minor. Locomotor activity also did not differ between the vehicle and desipramine groups after removal of the osmotic pumps. Finally, sedative effects cannot account for the anti-allodynic efficacy of desipramine observed in our study. Naloxone eliminated the anti-allodynic effects of desipramine infusion, but did not alter desipramine-induced changes in locomotor activity. Moreover, a much higher dose of desipramine is required to produce sedation in animals (80), and no obvious sedation was apparent in any animal via visual inspection performed by an investigator blinded to experimental conditions.
In conclusion, chronic infusion of desipramine suppressed the development and maintenance of paclitaxel-induced neuropathic pain, but only prophylactic treatment permanently blocked the development of neuropathic pain. These results are important because desipramine is an US Food and Drug Administration (FDA)-approved medication already widely used in humans that could be fast tracked into clinical trials as a first line preemptive and prophylactic treatment for preventing the development of taxane-induced neuropathic pain. The endogenous opioid system, and to a lesser extent, the endocannabinoid system, is implicated in the anti-allodynic mechanisms of desipramine, although their roles may differ during the development and maintenance phases of paclitaxel-induced neuropathic pain. Importantly, insights gained from the present study could be applied to improve current treatments and identify novel therapeutic targets for preventing and managing toxic neuropathic pain evoked by chemotherapy.
Acknowledgments
The authors are grateful to Dr. Phil Holmes for helpful discussions. This work was supported by the National Institutes of Health [DA041229 (to AGH), DA037673 (to AGH), DA009158 (to AM and AGH), and CA200417 (to AGH)]. AM is a consultant for MAK scientific. All authors state no conflict of interests.
Abbreviation
- ANOVA
analysis of variance
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CR
cremophor
- DMI
desipramine
- DMSO
dimethyl sulfoxide
- DMEM
Dulbecco's Modified Eagle Medium
- DRG
dorsal root ganglion
- FBS
Fetal Bbovine Sserum
- FDA
US Food and Drug Administration
- i.p
intraperitoneal injection
- MOR
μ-opioid receptor
- MTT
3-(4,5-dimethyktguazik-2-yl)-2,5-diphenyltetrazolium bromide
- NEAA
non-essential amino acids
- PTX
paclitaxel
- TCAs
tricyclic antidepressants
- TLR4
toll-like receptor 4
- PEG400
polyethylene glycol 400
- s.c
subcutaneous injection
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Berger A, Dukes EM, Oster G. Clinical characteristics and economic costs of patients with painful neuropathic disorders. The journal of pain : official journal of the American Pain Society. 2004;5:143–149. doi: 10.1016/j.jpain.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. The Lancet Neurology. 2015;14:162–173. doi: 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uhmand WK, Yung JH. Neurologic Complications of Cancer Therapy. Current treatment options in neurology. 1999;1:428–437. doi: 10.1007/s11940-996-0006-x. [DOI] [PubMed] [Google Scholar]
- 4.Merskeyand H, Bogduk N. Classification of Chronic Pain. IASP Press; 1994. Part III: Pain Terms, A Current List with Definitions and Notes on Usage. [Google Scholar]
- 5.Woolfand RJ, Mannion CJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- 6.Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advances and future challenges. The Journal of pharmacology and experimental therapeutics. 2006;319:507–514. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- 7.Baron R. Neuropathic pain: a clinical perspective. Handbook of experimental pharmacology. 2009:3–30. doi: 10.1007/978-3-540-79090-7_1. [DOI] [PubMed] [Google Scholar]
- 8.O'Connorand AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. The American journal of medicine. 2009;122:S22–32. doi: 10.1016/j.amjmed.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 9.Pachman DR, Barton DL, Watson JC, Loprinzi CL. Chemotherapy-induced peripheral neuropathy: prevention and treatment. Clinical pharmacology and therapeutics. 2011;90:377–387. doi: 10.1038/clpt.2011.115. [DOI] [PubMed] [Google Scholar]
- 10.Glassmanand JT, Bigger AH., Jr Cardiovascular effects of therapeutic doses of tricyclic antidepressants. A review. Archives of general psychiatry. 1981;38:815–820. doi: 10.1001/archpsyc.1981.01780320095011. [DOI] [PubMed] [Google Scholar]
- 11.Dharmshaktu P, Tayal V, Kalra BS. Efficacy of antidepressants as analgesics: a review. Journal of clinical pharmacology. 2012;52:6–17. doi: 10.1177/0091270010394852. [DOI] [PubMed] [Google Scholar]
- 12.Max MB, Lynch SA, Muir J, Shoaf SE, Smoller B, Dubner R. Effects of desipramine, amitriptyline, and fluoxetine on pain in diabetic neuropathy. The New England journal of medicine. 1992;326:1250–1256. doi: 10.1056/NEJM199205073261904. [DOI] [PubMed] [Google Scholar]
- 13.Furst S. Transmitters involved in antinociception in the spinal cord. Brain research bulletin. 1999;48:129–141. doi: 10.1016/s0361-9230(98)00159-2. [DOI] [PubMed] [Google Scholar]
- 14.Raisman R, Sette M, Pimoule C, Briley M, Langer SZ. High-affinity [3H]desipramine binding in the peripheral and central nervous system: a specific site associated with the neuronal uptake of noradrenaline. European journal of pharmacology. 1982;78:345–351. doi: 10.1016/0014-2999(82)90036-x. [DOI] [PubMed] [Google Scholar]
- 15.Basbaumand HL, Fields AI. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annual review of neuroscience. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- 16.Watkinsand DJ, Mayer LR. Multiple endogenous opiate and non-opiate analgesia systems: evidence of their existence and clinical implications. Annals of the New York Academy of Sciences. 1986;467:273–299. doi: 10.1111/j.1749-6632.1986.tb14635.x. [DOI] [PubMed] [Google Scholar]
- 17.Terenius L. Endogenous peptides and analgesia. Annual review of pharmacology and toxicology. 1978;18:189–204. doi: 10.1146/annurev.pa.18.040178.001201. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt BL, Hamamoto DT, Simone DA, Wilcox GL. Mechanism of cancer pain. Molecular interventions. 2010;10:164–178. doi: 10.1124/mi.10.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumovaand B, Jakoubek A. Analgesia and impact induced by anticipation stress: involvement of the endogenous opioid peptide system. Brain research. 1989;503:273–280. doi: 10.1016/0006-8993(89)91674-0. [DOI] [PubMed] [Google Scholar]
- 20.Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. Pain modulation by release of the endogenous cannabinoid anandamide. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:12198–12203. doi: 10.1073/pnas.96.21.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Przewlockiand B, Przewlocka R. Opioids in neuropathic pain. Current pharmaceutical design. 2005;11:3013–3025. doi: 10.2174/1381612054865055. [DOI] [PubMed] [Google Scholar]
- 22.Cravattand AH, Lichtman BF. The endogenous cannabinoid system and its role in nociceptive behavior. Journal of neurobiology. 2004;61:149–160. doi: 10.1002/neu.20080. [DOI] [PubMed] [Google Scholar]
- 23.Jacoband K, Ramabadran JJ. Role of opiate receptors and endogenous ligands in nociception. Pharmacology & therapeutics. 1981;14:177–196. doi: 10.1016/0163-7258(81)90060-7. [DOI] [PubMed] [Google Scholar]
- 24.Deng L, Guindon J, Cornett BL, Makriyannis A, Mackie K, Hohmann AG. Chronic cannabinoid receptor 2 activation reverses paclitaxel neuropathy without tolerance or cannabinoid receptor 1-dependent withdrawal. Biological psychiatry. 2015;77:475–487. doi: 10.1016/j.biopsych.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng L, Cornett BL, Mackie K, Hohmann AG. CB1 Knockout Mice Unveil Sustained CB2-Mediated Antiallodynic Effects of the Mixed CB1/CB2 Agonist CP55,940 in a Mouse Model of Paclitaxel-Induced Neuropathic Pain. Molecular pharmacology. 2015;88:64–74. doi: 10.1124/mol.115.098483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melik Parsadaniantz S, Rivat C, Rostene W, Reaux-Le Goazigo A. Opioid and chemokine receptor crosstalk: a promising target for pain therapy? Nature reviews Neuroscience. 2015;16:69–78. doi: 10.1038/nrn3858. [DOI] [PubMed] [Google Scholar]
- 27.Cristino L, Luongo L, Imperatore R, Boccella S, Becker T, Morello G, Piscitelli F, Busetto G, Maione S, Di Marzo V. Orexin-A and Endocannabinoid Activation of the Descending Antinociceptive Pathway Underlies Altered Pain Perception in Leptin Signaling Deficiency. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2016;41:508–520. doi: 10.1038/npp.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luongo L, Maione S, Di Marzo V. Endocannabinoids and neuropathic pain: focus on neuron-glia and endocannabinoid-neurotrophin interactions. The European journal of neuroscience. 2014;39:401–408. doi: 10.1111/ejn.12440. [DOI] [PubMed] [Google Scholar]
- 29.Palazzo E, de Novellis V, Petrosino S, Marabese I, Vita D, Giordano C, Di Marzo V, Mangoni GS, Rossi F, Maione S. Neuropathic pain and the endocannabinoid system in the dorsal raphe: pharmacological treatment and interactions with the serotonergic system. The European journal of neuroscience. 2006;24:2011–2020. doi: 10.1111/j.1460-9568.2006.05086.x. [DOI] [PubMed] [Google Scholar]
- 30.He LF. Involvement of endogenous opioid peptides in acupuncture analgesia. Pain. 1987;31:99–121. doi: 10.1016/0304-3959(87)90011-X. [DOI] [PubMed] [Google Scholar]
- 31.Suhand LL, Tseng HH. Intrathecal administration of thiorphan, bestatin, desipramine and fluoxetine differentially potentiate the antinociceptive effects induced by beta-endorphin and morphine, administered intracerebroventricularly. Neuropharmacology. 1990;29:207–214. doi: 10.1016/0028-3908(90)90003-a. [DOI] [PubMed] [Google Scholar]
- 32.Mallet C, Daulhac L, Bonnefont J, Ledent C, Etienne M, Chapuy E, Libert F, Eschalier A. Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain. 2008;139:190–200. doi: 10.1016/j.pain.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 33.Tocque B, Albouz S, Boutry JM, Le Saux F, Hauw JJ, Bourdon R, Baumann N, Zalc B. Desipramine elicits the expression of opiate receptors and sulfogalactosylceramide synthesis in rat C6 glioma cells. Journal of neurochemistry. 1984;42:1101–1106. doi: 10.1111/j.1471-4159.1984.tb12716.x. [DOI] [PubMed] [Google Scholar]
- 34.Barg J, Belcheva MM, Bem WT, Lambourne B, McLachlan JA, Tolman KC, Johnson FE, Coscia CJ. Desipramine modulation of sigma and opioid peptide receptor expression in glial cells. Peptides. 1991;12:845–849. doi: 10.1016/0196-9781(91)90144-e. [DOI] [PubMed] [Google Scholar]
- 35.Stengaard-Pedersenand M, Schou K. Opioid receptors in the brain of the rat following chronic treatment with desipramine and electroconvulsive shock. Neuropharmacology. 1986;25:1365–1371. doi: 10.1016/0028-3908(86)90110-3. [DOI] [PubMed] [Google Scholar]
- 36.Coddand RF, Walker EE. Mu/delta opioid site selectivity of some antidepressants. NIDA research monograph. 1986;75:351–354. [PubMed] [Google Scholar]
- 37.Devoize JL, Rigal F, Eschalier A, Trolese JF, Renoux M. Influence of naloxone on antidepressant drug effects in the forced swimming test in mice. Psychopharmacology. 1984;84:71–75. doi: 10.1007/BF00432028. [DOI] [PubMed] [Google Scholar]
- 38.Stengaard-Pedersenand M, Schou K. Opioid peptides and receptors in relation to affective illness. Effects of desipramine and lithium on opioid receptors in rat brain. Acta pharmacologica et toxicologica. 1985;56(Suppl 1):170–179. doi: 10.1111/j.1600-0773.1985.tb02509.x. [DOI] [PubMed] [Google Scholar]
- 39.Reisineand P, Soubrie T. Loss of rat cerebral cortical opiate receptors following chronic desimipramine treatment. European journal of pharmacology. 1982;77:39–44. doi: 10.1016/0014-2999(82)90532-5. [DOI] [PubMed] [Google Scholar]
- 40.O'Neilland KA, Valentino D. Chronic desipramine attenuates morphine analgesia. Pharmacology, biochemistry, and behavior. 1986;24:155–158. doi: 10.1016/0091-3057(86)90061-4. [DOI] [PubMed] [Google Scholar]
- 41.Werling LL, Brown SR, Cox BM. Opioid receptor regulation of the release of norepinephrine in brain. Neuropharmacology. 1987;26:987–996. doi: 10.1016/0028-3908(87)90077-3. [DOI] [PubMed] [Google Scholar]
- 42.Quirarte GL, Galvez R, Roozendaal B, McGaugh JL. Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain research. 1998;808:134–140. doi: 10.1016/s0006-8993(98)00795-1. [DOI] [PubMed] [Google Scholar]
- 43.Bohn LM, Xu F, Gainetdinov RR, Caron MG. Potentiated opioid analgesia in norepinephrine transporter knock-out mice. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20:9040–9045. doi: 10.1523/JNEUROSCI.20-24-09040.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atsak P, Hauer D, Campolongo P, Schelling G, McGaugh JL, Roozendaal B. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:3504–3509. doi: 10.1073/pnas.1200742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hill MN, Miller GE, Carrier EJ, Gorzalka BB, Hillard CJ. Circulating endocannabinoids and N-acyl ethanolamines are differentially regulated in major depression and following exposure to social stress. Psychoneuroendocrinology. 2009;34:1257–1262. doi: 10.1016/j.psyneuen.2009.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin WJ, Lai NK, Patrick SL, Tsou K, Walker JM. Antinociceptive actions of cannabinoids following intraventricular administration in rats. Brain research. 1993;629:300–304. doi: 10.1016/0006-8993(93)91334-o. [DOI] [PubMed] [Google Scholar]
- 47.Gregg LC, Jung KM, Spradley JM, Nyilas R, Suplita RL, 2nd, Zimmer A, Watanabe M, Mackie K, Katona I, Piomelli D, Hohmann AG. Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-alpha initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:9457–9468. doi: 10.1523/JNEUROSCI.0013-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 49.Page ME, Detke MJ, Dalvi A, Kirby LG, Lucki I. Serotonergic mediation of the effects of fluoxetine, but not desipramine, in the rat forced swimming test. Psychopharmacology. 1999;147:162–167. doi: 10.1007/s002130051156. [DOI] [PubMed] [Google Scholar]
- 50.Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. The Journal of pharmacology and experimental therapeutics. 1995;274:1263–1270. [PubMed] [Google Scholar]
- 51.Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, Somogyi AA, Yin H, Maier SF, Rice KC, Watkins LR. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, Slivka PF, Coats BD, Rezvani N, Wieseler J, Hughes TS, Landgraf KE, Chan S, Fong S, Phipps S, Falke JJ, Leinwand LA, Maier SF, Yin H, Rice KC, Watkins LR. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain, behavior, and immunity. 2010;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahn EJ, Zvonok AM, Thakur GA, Khanolkar AD, Makriyannis A, Hohmann AG. Selective activation of cannabinoid CB2 receptors suppresses neuropathic nociception induced by treatment with the chemotherapeutic agent paclitaxel in rats. The Journal of pharmacology and experimental therapeutics. 2008;327:584–591. doi: 10.1124/jpet.108.141994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rahn EJ, Deng L, Thakur GA, Vemuri K, Zvonok AM, Lai YY, Makriyannis A, Hohmann AG. Prophylactic cannabinoid administration blocks the development of paclitaxel-induced neuropathic nociception during analgesic treatment and following cessation of drug delivery. Molecular pain. 2014;10:27. doi: 10.1186/1744-8069-10-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94:293–304. doi: 10.1016/S0304-3959(01)00363-3. [DOI] [PubMed] [Google Scholar]
- 56.Alcaraz C, Vargas ML, Fuente T, Milanes MV. Chronic naloxone treatment induces supersensitivity to a mu but not to a kappa agonist at the hypothalamus-pituitary-adrenocortical axis level. The Journal of pharmacology and experimental therapeutics. 1993;266:1602–1606. [PubMed] [Google Scholar]
- 57.Deng L, Guindon J, Vemuri VK, Thakur GA, White FA, Makriyannis A, Hohmann AG. The maintenance of cisplatin- and paclitaxel-induced mechanical and cold allodynia is suppressed by cannabinoid CB(2) receptor activation and independent of CXCR4 signaling in models of chemotherapy-induced peripheral neuropathy. Molecular pain. 2012;8:71. doi: 10.1186/1744-8069-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao WH, Zheng H, Zheng FY, Nuydens R, Meert TF, Bennett GJ. Mitochondrial abnormality in sensory, but not motor, axons in paclitaxel-evoked painful peripheral neuropathy in the rat. Neuroscience. 2011;199:461–469. doi: 10.1016/j.neuroscience.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bowsher D. The effects of pre-emptive treatment of postherpetic neuralgia with amitriptyline: a randomized, double-blind, placebo-controlled trial. Journal of pain and symptom management. 1997;13:327–331. doi: 10.1016/s0885-3924(97)00077-8. [DOI] [PubMed] [Google Scholar]
- 60.Smith EM, Pang H, Cirrincione C, Fleishman S, Paskett ED, Ahles T, Bressler LR, Fadul CE, Knox C, Le-Lindqwister N, Gilman PB, Shapiro CLO. Alliance for Clinical Trials in. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. Jama. 2013;309:1359–1367. doi: 10.1001/jama.2013.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flattersand GJ, Bennett SJ. Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain. 2006;122:245–257. doi: 10.1016/j.pain.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther. 2006;109:57–77. doi: 10.1016/j.pharmthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 63.Burgos E, Gomez-Nicola D, Pascual D, Martin MI, Nieto-Sampedro M, Goicoechea C. Cannabinoid agonist WIN 55,212-2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. Eur J Pharmacol. 2012;682:62–72. doi: 10.1016/j.ejphar.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 64.Naguib M, Xu JJ, Diaz P, Brown DL, Cogdell D, Bie B, Hu J, Craig S, Hittelman WN. Prevention of Paclitaxel-Induced Neuropathy Through Activation of the Central Cannabinoid Type 2 Receptor System. Anesth Analg. 2012 doi: 10.1213/ANE.0b013e31824b0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaggiand N, Singh AS. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012;291:1–9. doi: 10.1016/j.tox.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 66.Verstappen CC, Heimans JJ, Hoekman K, Postma TJ. Neurotoxic complications of chemotherapy in patients with cancer: clinical signs and optimal management. Drugs. 2003;63:1549–1563. doi: 10.2165/00003495-200363150-00003. [DOI] [PubMed] [Google Scholar]
- 67.Lynch JJ, 3rd, Wade CL, Zhong CM, Mikusa JP, Honore P. Attenuation of mechanical allodynia by clinically utilized drugs in a rat chemotherapy-induced neuropathic pain model. Pain. 2004;110:56–63. doi: 10.1016/j.pain.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 68.Yalcin I, Tessier LH, Petit-Demouliere N, Doridot S, Hein L, Freund-Mercier MJ, Barrot M. Beta2-adrenoceptors are essential for desipramine, venlafaxine or reboxetine action in neuropathic pain. Neurobiology of disease. 2009;33:386–394. doi: 10.1016/j.nbd.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 69.Jett MF, McGuirk J, Waligora D, Hunter JC. The effects of mexiletine, desipramine and fluoxetine in rat models involving central sensitization. Pain. 1997;69:161–169. doi: 10.1016/s0304-3959(96)03231-9. [DOI] [PubMed] [Google Scholar]
- 70.Torpy DJ, Grice JE, Hockings GI, Crosbie GV, Walters MM, Jackson RV. The effect of desipramine on basal and naloxone-stimulated cortisol secretion in humans: interaction of two drugs acting on noradrenergic control of adrenocorticotropin secretion. The Journal of clinical endocrinology and metabolism. 1995;80:802–806. doi: 10.1210/jcem.80.3.7883833. [DOI] [PubMed] [Google Scholar]
- 71.Biegonand A, Samuel D. Interaction of tricyclic antidepressants with opiate receptors. Biochemical pharmacology. 1980;29:460–462. doi: 10.1016/0006-2952(80)90531-6. [DOI] [PubMed] [Google Scholar]
- 72.Somoza E, Galindo A, Bazan E, Guillamon A, Valencia A, Fuentes JA. Antidepressants inhibit enkephalin binding to synaptosome-enriched fractions of rat brain. Neuropsychobiology. 1981;7:297–301. doi: 10.1159/000117864. [DOI] [PubMed] [Google Scholar]
- 73.Eschalier A, Montastruc JL, Devoize JL, Rigal F, Gaillard-Plaza G, Pechadre JC. Influence of naloxone and methysergide on the analgesic effect of clomipramine in rats. European journal of pharmacology. 1981;74:1–7. doi: 10.1016/0014-2999(81)90316-2. [DOI] [PubMed] [Google Scholar]
- 74.Jasmin L, Boudah A, Ohara PT. Long-term effects of decreased noradrenergic central nervous system innervation on pain behavior and opioid antinociception. The Journal of comparative neurology. 2003;460:38–55. doi: 10.1002/cne.10633. [DOI] [PubMed] [Google Scholar]
- 75.Mengand ID, Johansen JP. Antinociception and modulation of rostral ventromedial medulla neuronal activity by local microinfusion of a cannabinoid receptor agonist. Neuroscience. 2004;124:685–693. doi: 10.1016/j.neuroscience.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 76.Meng ID, Manning BH, Martin WJ, Fields HL. An analgesia circuit activated by cannabinoids. Nature. 1998;395:381–383. doi: 10.1038/26481. [DOI] [PubMed] [Google Scholar]
- 77.Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- 78.Oropeza VC, Page ME, Van Bockstaele EJ. Systemic administration of WIN 55,212-2 increases norepinephrine release in the rat frontal cortex. Brain research. 2005;1046:45–54. doi: 10.1016/j.brainres.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 79.Hill MN, Ho WS, Sinopoli KJ, Viau V, Hillard CJ, Gorzalka BB. Involvement of the endocannabinoid system in the ability of long-term tricyclic antidepressant treatment to suppress stress-induced activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2006;31:2591–2599. doi: 10.1038/sj.npp.1301092. [DOI] [PubMed] [Google Scholar]
- 80.Ross SB, Ogren SO, Renyi AL. (Z)-dimethylamino-1-(4-bromophenyl)-1-(3-pyridyl) propene (h 102/09), a new selective inhibitor of the neuronal 5-hydroxytryptamine uptake. Acta pharmacologica et toxicologica. 1976;39:152–166. doi: 10.1111/j.1600-0773.1976.tb03166.x. [DOI] [PubMed] [Google Scholar]