

Abstract
G-protein coupled receptors (GPCRs), the largest family of human membrane proteins, mediate cellular signaling and represent primary targets of about one third of currently marketed drugs. GPCRs undergo highly dynamic structural transitions during signal transduction, from binding of extracellular ligands to coupling with intracellular effector proteins. Molecular dynamics (MD) simulations have been utilized to investigate GPCR signaling mechanisms (such as pathways of ligand binding and receptor activation/deactivation) and to design novel small-molecule drug candidates. Future research directions point towards modeling cooperative binding of multiple orthosteric and allosteric ligands to GPCRs, GPCR oligomerization and interactions of GPCRs with different intracellular signaling proteins. Through methodological and supercomputing advances, MD simulations will continue to provide important insights into GPCR signaling mechanisms and further facilitate structure-based drug design.
Keywords: G-protein Coupled Receptors, Cellular Signaling, Molecular Dynamics, Ligand Binding, Activation, Protein-Protein Interactions
Introduction
G-protein coupled receptors (GPCRs) mediate cellular responses to hormones, neurotransmitters, chemokines and the senses of sight, olfaction and taste. They have served as targets of about one third of currently marketed drugs for treating many human diseases, including cancer, diabetes, obesity, heart failure and neurological diseases[1,2]. Because GPCRs are highly dynamic membrane proteins that undergo large-scale conformational changes during signaling, they have presented challenges for experimental structural determination[3].
Molecular dynamics (MD) is a computational technique that simulates the detailed structural dynamics of biomolecules over time based on an atomic force field[4]. In the absence of particular experimental GPCR structures, computational homology modeling and ab initio predictions[5,6] have been used to construct atomic structures of GPCRs and MD simulations have then been performed to study their dynamics[7]. MD simulations of GPCR computational models have been pioneered by Dahl, Weinstein and the others since the 1990s on the dopamine[8], serotonin[9–11] and opioid receptors[12]. Following the groundbreaking X-ray crystal structure of rhodopsin in 2000[13], many MD simulations were performed to investigate the receptor structural changes and receptor-retinal interactions[14,15]. Another surge of MD studies on GPCRs was triggered by the X-ray structural determination of β2AR in 2007[16,17] and a rapidly growing number of high-resolution GPCR structures thereafter[18]. Several excellent reviews on MD simulations of GPCRs have been presented earlier[19–21].
In this short review, we will briefly discuss the early GPCR MD studies, but primarily outline the latest developments of both conventional and enhanced atomistic MD studies of GPCRs and offer our perspectives. The simulation methods of our focus include long-timescale conventional MD; Markov state models (MSM)[22] that extract protein dynamics from many short MD simulations; and enhanced sampling via random acceleration MD (RAMD)[23], steered MD[24], metadynamics[25] and accelerated MD (aMD)[26]. As shown in Figure 1, these studies have provided important insights into GPCR signaling processes (such as ligand binding and unbinding and receptor activation and deactivation conformational transitions), and into the structural basis for small-molecule drug design.
Fig. 1.
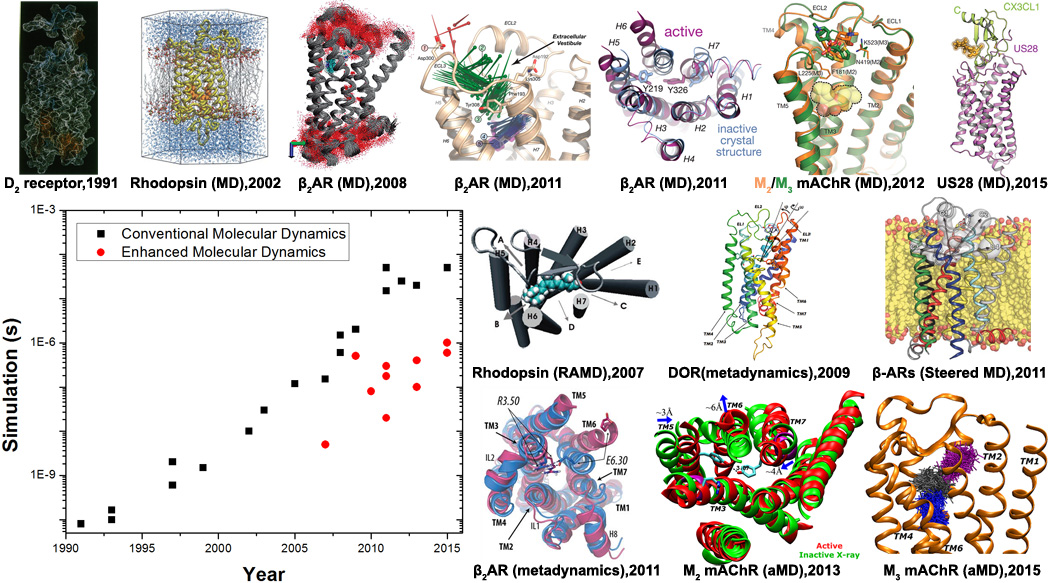
Advances in conventional and enhanced molecular dynamics (MD) simulations of GPCR signaling to date. GPCR structural images are adapted from conventional MD simulations on a homology model of the D2 dopamine receptor in 1991[8], rhodopsin using its X-ray structure in 2002[14,15], flexibility and internal hydration of β2AR following determination of the X-ray structure in 2008[17], drug binding and deactivation processes of β2AR in 2011[32,40], ligand binding to the M2 and M3 mAChRs in 2012[33] and constitutive activity of US28 bound by CX3CL1 chemokine in 2015[49]. Enhanced MD simulations have also been applied to explore structural dynamics of GPCRs. For example, ligand unbinding has been investigated through random acceleration MD (RAMD) simulations on rhodopsin in 2007[27] and β2AR in 2009[28], metadynamics simulations of δ-opioid receptor (DOR) in 2009[31] and steered MD simulations of the b-ARs in 2011[30]. Moreover, GPCR activation processes have been characterized through combined metadynamics and adiabatic biasing MD (ABMD) simulations of rhodopsin in 2010[50] and β2AR in 2011[42], as well as aMD simulations of M2 mAChR in 2013[59] and M3 mAChR in 2015[51]. In addition, ligand binding to the M3 mAChR captured through aMD simulations was reported in 2015[36].
Molecular mechanisms of ligand binding to GPCRs
It is of great pharmaceutical interest to understand the molecular mechanisms of ligand recognition by GPCRs. However, ligand binding takes place over typically microseconds and even longer timescales for ligand unbinding or drug release. Due to limited computing power, these timescales were beyond the reach of conventional MD even in ten years ago. Instead, enhanced MD approaches were first adopted to address this issue. Notably, RAMD was applied to simulate the ligand unbinding from rhodopsin in 2007[27] and β2AR in 2009[28]. For rhodopsin, the retinal was found to exit predominantly from the clefts between the transmembrane (TM) 4/TM5 helices or between the TM5/TM6 helices near the extracellular side. In contrast, in β2AR the extracellular surface opening was the most frequently observed ligand egress point (pathway A). With the salt bridge formed between Asp192 in the extracellular loop 2 (ECL2) and Lys3057.32 (the superscripts denote Ballesteros-Weinstein residue numbering[29]), the dominant ligand exit pathway A was divided into two subpathways, in which one surface opening is formed between ECL2 and TM5/TM6/TM7 helices (subpathway A1) and the other is formed between ECL2 and TM2/TM3/TM7 helices (subpathway A2). These two subpathways appeared to have “practically equal frequency” for ligand exit from the β2AR X-ray structure, although RAMD simulations based on a putative ligand-free conformation of β2AR suggested that subpathway A2 is more favorable for ligand unbinding[28]. In 2011, steered MD was used to simulate ligand dissociation from the β1 and β2 adrenergic receptors (β-ARs)[30]. The simulations produced similar free energy profiles for two exit channels C1 and C2 of β-ARs that correspond to the aforementioned subpathways A1 and A2, respectively. It was thus suggested that “both routes may serve indistinguishably for ligand entry and exit”. Nevertheless, the secondary binding pocket in channel C2 of β2AR that is located at ~15Å from the orthosteric site was found less favorable than in channel C1. Moreover, the conformational change of Phe218ECL2 in β1AR and Phe193ECL2 in β2AR correlated with ligand solvation during unbinding[30]. In addition, metadynamics was applied to investigate ligand binding to the δ-opioid receptor (DOR) using a homology model[31]. The metadynamics simulations also suggested a preferential pathway of antagonist binding to DOR through a cleft formed between ECL2-ECL3 in the extracellular region.
Conventional MD was not applied to study GPCR ligand binding until 2011, when Dror et al. used the specialized supercomputer Anton to simulate binding of drug molecules to the β-ARs[32]. In contrast to the RAMD and SMD studies, the Anton MD simulations showed that the alprenolol antagonist bound to β2AR along a single dominant pathway, in which alprenolol passed between ECL2-ECL3 and then through the crevice between ECL2 and the TM4/TM6/TM7 helices (the subpathway A1[28] or channel C1[30] discussed above) to reach the orthosteric site. In only one out of 12 simulations that captured antagonist binding to β2AR, alprenolol entered between ECL2 and the TM2/TM7 helices (subpathway A2[28] or channel C2[30]). Besides, as alprenolol enters the extracellular vestibule of β2AR, the majority of ligand dehydration occurs, which contributes to the primary observed barrier for ligand binding[32]. In 2012, more Anton MD simulations on the M2 and M3 muscarinic acetylcholine receptors (mAChRs) showed that the large tiotropium (TTP) antagonist could not enter the deep orthosteric site during a simulation, but just bound to the extracellular vestibule[33]. In comparison, the endogenous agonist acetylcholine (ACh) of much smaller size was able to reach the orthosteric site with transient pause in the extracellular vestibule. In 2013, binding of several known negative allosteric modulators (NAMs) to the extracellular vestibule of the M2 receptor was captured in further Anton MD simulations[34]. The modulators typically form cation-π interactions with aromatic residues in the receptor extracellular vestibule. The extracellular allosteric binding mode was confirmed by mutation experiments and later by the X-ray structure of the active M2 receptor that is recognized by a positive allosteric modulator (PAM)[35].
In 2013, we performed aMD simulations to investigate the binding of three ligands to the M3 mAChR: the antagonist TTP, full agonist ACh, and partial agonist arecoline (ARc)[36]. In comparison with the previous Anton MD simulations[33], aMD greatly accelerated the binding of ACh to the receptor orthosteric ligand-binding site (~80 times speedup) and binding of TTP to the extracellular vestibule. Further aMD simulations also captured binding of ARc to the receptor orthosteric site. All three ligands were observed to pause in the extracellular vestibule during binding. The metastable intermediate states identified during ligand binding were in agreement with the Anton MD simulations[33]. Recently, metadynamics simulations have also captured the binding of a PAM to DOR in the presence of an agonist[37]. These studies demonstrate the applicability of both conventional and enhanced MD simulations to ligand binding of GPCRs.
Conformational sampling of GPCR deactivation and activation pathways
With emerging X-ray structures of GPCRs determined in both inactive and active states (e.g., the β2AR [16,38] and M2 mAChR[35,39]), extensive MD simulations have been applied to investigate GPCR activation mechanisms[40–46]. In 2011, Dror et al. observed deactivation of β2AR from the active X-ray structure upon removal of the G-protein or its mimetic nanobody through Anton MD simulations[40]. The MD simulations revealed an intermediate conformation of β2AR during the deactivation process. Vaidehi and co-workers combined a coarse-grained conformational sampling method and all-atom MD simulations to investigate ensemble conformations of the human β2AR bound by different ligands[41,47]. Results showed that the binding of full or partial agonist leads to selection of a subset of conformations including the active and inactive states, while the inverse agonist selects only inactive state conformations. In 2012, Shan et al. observed distinct conformational states and dynamics in the serotonin 2A receptor (5-HT2A) receptor bound by the full agonist, partial agonist and inverse agonist[43]. The three ligands induce distinct conformational changes in several known GPCR activation elements and different responses in the lipid membrane. In 2014, The Google Exacycle cloudcomputing platform was used to simulate β2AR for a total of 2.15 milliseconds by combining short cMD runs[46]. MSMs revealed multiple activation pathways of β2AR and the simulation-derived structures were found to improve molecular docking of ligand molecules. In 2015, Neale et al. investigated the effects of lipid binding on activation of β2AR through long-timescale MD simulations[48]. Recently, Anton MD simulations also provided important insights into the constitutive activity of the US28 receptor that is bound by the CXCL1 chemokine[49].
Metadynamics combined with adiabatic biasing MD (ABMD) simulations was used to investigate activation and free energy landscapes of rhodopsin in 2010[50] and β2AR in 2011[42]. The metadynamics simulations identified important low-energy intermediate states of the two GPCRs along their activation pathways. In the case of β2AR, distinct free energy profiles were obtained when the receptor was bound by the inverse agonist, neutral antagonist and agonist. In 2013, Li et al. also applied metadynamics simulations to the adenosine 2A receptor and suggested that different ligands were able to shift the receptor conformational equilibrium towards the active and inactive states[45].
In 2013, Miao et al. applied aMD simulations on the M2 mAChR and captured its activation at an atomistic level[44]. Starting from the inactive X-ray structure with antagonist removed, the receptor undergoes large-scale structural rearrangements to an active state via two intermediate conformations. The receptor activation involves major conformational transitions in the TM5, TM6 and TM7 helices and changes in the receptor dynamic network. Notably, Tyr2065.58 and Tyr4407.53 relocate their side chains towards each other forming hydrogen-bonding interactions and the cytoplasmic end of TM6 tilts outward by ~6 Å. Subsequent determination of the X-ray structure of the active M2 receptor largely confirmed these predictions[35]. In 2015, we also captured activation of the M3 mAChR and investigated the allosteric effects of sodium ion binding through extensive aMD simulations[51]. Results showed that with the D2.50 residue deprotonated, the M3 receptor was bound by an allosteric sodium ion and confined mostly in the inactive state with remarkably reduced flexibility. In contrast, the D2.50-protonated receptor did not exhibit sodium ion binding to this site and sampled significantly larger conformational space. The receptor activation was observed and characterized by structural rearrangements of the TM helices via dynamic hydrogen bond and salt bridge interactions. Network analysis revealed that the allosteric signaling between residue D2.50 and key residues in the intracellular, extracellular and orthosteric pockets is significantly weakened upon sodium ion binding, which precludes receptor activation.
Computer-aided drug design targeting GPCRs
Due to the immense cost in high-throughput screening of large compound libraries, computer-aided design has become an attractive approach for drug discovery targeting GPCRs[52,53]. Moreover, design of receptor-specific orthosteric drugs has been challenging and mostly unsuccessful in the case of GPCRs, largely because residues in the orthosteric site are highly conserved across different receptor subtypes, e.g., the five mAChRs[33,39,54]. Alternatively, development of allosteric modulators, which bind to GPCR allosteric sites with great subtype specificity, has emerged as a new paradigm for GPCR drug discovery[55–57].
In 2015, Huang et al. demonstrated a computer-aided design approach to design allosteric modulators of two understudied GPCRs, the GPR68 and GPR65, by combining computational homology modeling, molecular docking and experimental assays[58]. First, yeast-based screening against 24 selected GPCRs revealed that the Lorazepam drug acts as a PAM of GPR68. Based on the CXCR4 template, 3307 homology models were constructed and refined to capture Lorazepam binding. With an optimized binding mode chosen, a library of 3.1 million compounds was computationally docked to predict new GPR68 allosteric modulators. Functional assays confirmed that ogerin among many was a potent PAM of GPR68. Application of the same approach also found allosteric agonists and NAMs of GPR65. While homology models appear to generate feasible receptor structures for molecular docking, it was also noted that this protocol demands iterative cycles of modeling and optimization[58].
Since GPCRs are highly flexible membrane proteins, especially in the druggable allosteric sites[34,59], MD simulations have been proposed to construct receptor ensembles for molecular docking in order to account for the receptor flexibility. Recently, Miao et al. have combined aMD simulations and ensemble docking with experimental binding and functional assays to design allosteric modulators of the M2 mAChR[60]. AMD simulations were carried out to construct the receptor ensembles. Compounds obtained from the National Cancer Institute (NCI) Diversity Set (~1,600) were docked to the receptor ensembles to identify novel allosteric modulators. In the first round, 10 top-ranked compounds were selected from the Glide virtual screening workflow (VSW) calculations. Although one compound significantly slowed down the dissociation of an antagonist radioligand, none of them exhibited high enough affinity for the M2 mAChR. In the second round, retrospective docking of known ligands showed that combining aMD simulations with the Glide induced fit docking (IFD) provided much improved docking enrichment factors compared with the VSW. Glide IFD was thus applied in ensemble docking of the M2 mAChR and 38 top-ranked compounds were selected for experimental testing. Results showed that about half of the 38 hits altered the radioligand dissociation rate, a hallmark of allosteric behavior. Through further experimental characterization using competition binding, 12 compounds were identified with ≤30µM affinity. With final in vivo functional experiments on six selected lead compounds, we confirmed four of them as new NAMs and one as PAM of agonist-mediated response at the M2 mAChR. This study also demonstrates a successful structure-based design approach to effectively account for receptor flexibility and identify chemically diverse GPCR allosteric modulators.
Computing and methodological advances will continue to facilitate future simulations and drug discovery of GPCRs
Shown in Figure 2 is an overview of the cellular signaling processes associated with GPCRs. Upon binding of extracellular ligands, GPCRs undergo large-scale conformational changes and interact with intracellular effector proteins (such as the heterotrimeric G protein and arrestin) to activate various cell signaling pathways. Extracellular ligands include small organic molecules, neurotransmitters, hormones, lipids, chemokines, etc. Orthosteric ligands bind to a GPCR site that is recognized by the endogenous ligand. They can act as antagonists, inverse agonists, full and partial agonists and shift GPCRs towards different conformational states for cell signaling. Furthermore, the GPCR signaling can be regulated by allosteric modulators (e.g., organic molecules and polypeptides) that bind to regions topographically distant from the orthosteric site. During the past 25 years, MD studies of GPCRs have seen tremendous advances, from the early simulation of a GPCR homology model for only 80 ps in 1991[8] to the latest 50 µs Anton simulations (6 orders of magnitude increase)[40] and microsecond enhanced MD simulations[36]. MD simulations have greatly helped us to understand the dynamic mechanisms of ligand recognition and activation of GPCRs.
Fig. 2.
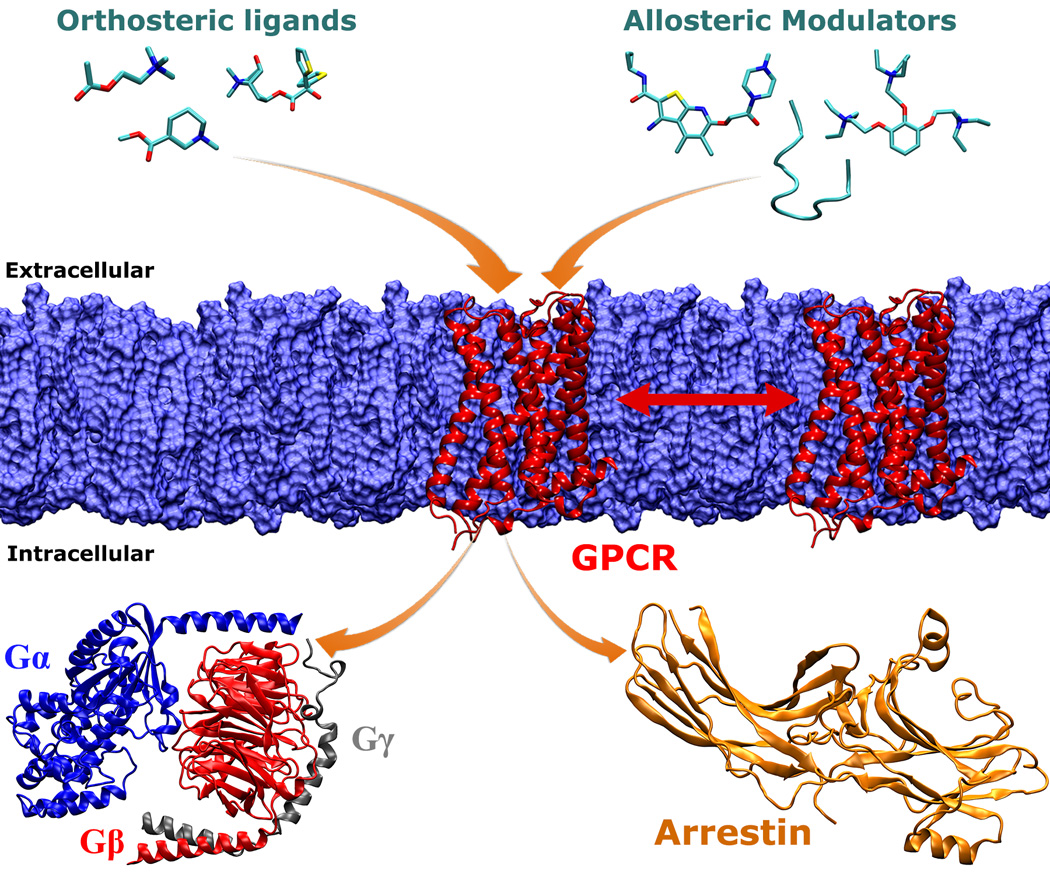
An overview of cellular signaling processes associated with GPCR membrane proteins. Upon stimulation by binding of extracellular ligands (or light in the case of rhodopsin), GPCRs undergo large-scale conformational changes and interact with intracellular effector proteins (such as the heterotrimeric G protein and arrestin) to activate various cell signaling pathways. Extracellular ligands include orthosteric ligands and allosteric modulators. Orthosteric ligands bind to a GPCR site that is recognized by the endogenous ligand. They can act as antagonists, inverse agonists, full and partial agonists and shift GPCRs towards different conformational states for cell signaling. The GPCR dynamics and function can be further regulated by allosteric modulators (e.g., organic molecules and polypeptides) that bind to regions topographically distant from the orthosteric site. On the intracellular side, different subtypes of activated GPCRs are able to bind specific G proteins, such as the Gs, Gi and Gq proteins. In addition to the G protein mediated signaling pathway, GPCRs can also bind to arrestin, leading to internalization of the receptors. Finally, GPCRs might form oligomers during their function in the lipid membrane. MD simulations will continue to deepen our understanding of these GPCR signaling processes.
Despite the above successes, the following aspects of GPCR dynamics and functional mechanisms remain unclear, but MD simulations will continue to deepen our understanding of GPCRs and facilitate related drug design. Particularly, recognition of GPCRs by large lipids and polypeptides (like chemokines) and mechanisms of cooperative binding and allosteric modulation between multiple orthosteric and allosteric ligands of GPCRs require further investigation. Moreover, the interactions of GPCRs with intracellular proteins remain poorly understood, largely due to the increased system size and complexity. Different subtypes of GPCRs are able to bind specific G proteins, such as the Gs, Gi and Gq proteins. The binding specificity between GPCRs-G proteins is subject to future research. In addition, given two milestone crystal structures of the GPCR-G protein[38] and GPCR-arrestin[61] complexes, a detailed understanding of the molecular mechanisms underlying the coupling of GPCRs with the G protein versus arrestin (i.e., biased agonism) is in order. Finally, GPCRs likely form oligomers during their function in the lipid membrane. While coarse-grained MD has been used to study GPCR oligomerization[62,63], it is important to study the effects of oligomerization on the structural dynamics and cell signaling of GPCRs through more detailed MD simulations.
Highlights.
GPCRs mediate cellular signaling and represent important drug targets
A review on molecular dynamics simulations and drug discovery of GPCRs is presented
Remarkable advances will continue to facilitate GPCR simulations and drug discovery
Acknowledgments
This work was supported by NSF (grant MCB1020765), NIH (grant GM31749), Howard Hughes Medical Institute, National Biomedical Computation Resource (NBCR), and the national supercomputer centers (the XSEDE awards TG-MCA93S013 and TG-MCB140011 and the NERSC award project M1925).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
Special interest (·) or outstanding interest (··):
- 1.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson KA. New paradigms in GPCR drug discovery. Biochemical Pharmacology. 2015;98:541–555. doi: 10.1016/j.bcp.2015.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevens RC, Cherezov V, Katritch V, Abagyan R, Kuhn P, Rosen H, Wuthrich K. The GPCR Network: a large-scale collaboration to determine human GPCR structure and function. Nature Reviews Drug Discovery. 2013;12:25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature Structural Biology. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 5.Trabanino RJ, Hall SE, Vaidehi N, Floriano WB, Kam VWT, Goddard WA., 3rd First Principles Predictions of the Structure and Function of G-Protein-Coupled Receptors: Validation for Bovine Rhodopsin. Biophysical Journal. 2004;86:1904–1921. doi: 10.1016/S0006-3495(04)74256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li YY, Hou TJ, Goddard WA. Computational Modeling of Structure-Function of G Protein-Coupled Receptors with Applications for Drug Design. Current Medicinal Chemistry. 2010;17:1167–1180. doi: 10.2174/092986710790827807. [DOI] [PubMed] [Google Scholar]
- 7.Li Y, Zhu F, Vaidehi N, Goddard WA, III, Sheinerman F, Reiling S, Morize I, Mu L, Harris K, Ardati A, et al. Prediction of the 3D structure and dynamics of human DP G-protein coupled receptor bound to an agonist and an antagonist. Journal of the American Chemical Society. 2007;129:10720–10731. doi: 10.1021/ja070865d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dahl SG, Edvardsen O, Sylte I. Molecular-Dynamics of Dopamine at the D2 Receptor. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8111–8115. doi: 10.1073/pnas.88.18.8111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang DQ, Weinstein H. Signal Transduction by a 5-Ht2 Receptor - a Mechanistic Hypothesis from Molecular-Dynamics Simulations of the 3-Dimensional Model of the Receptor Complexed to Ligands. Journal of Medicinal Chemistry. 1993;36:934–938. doi: 10.1021/jm00059a021. [DOI] [PubMed] [Google Scholar]
- 10.Sylte I, Edvardsen O, Dahl SG. Molecular-Dynamics of the 5-Ht(1a) Receptor and Ligands. Protein Engineering. 1993;6:691–700. doi: 10.1093/protein/6.7.691. [DOI] [PubMed] [Google Scholar]
- 11.Garmer DR. MD simulations of a 5-HT2A receptor model in DOPC membranes. Journal of Biomolecular Structure & Dynamics. 1997;14:525–546. doi: 10.1080/07391102.1997.10508154. [DOI] [PubMed] [Google Scholar]
- 12.Strahs D, Weinstein H. Comparative modeling and molecular dynamics studies of the delta, kappa and mu opioid receptors. Protein Engineering. 1997;10:1019–1038. doi: 10.1093/protein/10.9.1019. [DOI] [PubMed] [Google Scholar]
- 13.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 14.Rohrig UF, Guidoni L, Rothlisberger U. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 2002;41:10799–10809. doi: 10.1021/bi026011h. [DOI] [PubMed] [Google Scholar]
- 15.Saam J, Tajkhorshid E, Hayashi S, Schulten K. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophysical Journal. 2002;83:3097–3112. doi: 10.1016/S0006-3495(02)75314-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into beta(2)-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 17.Huber T, Menon S, Sakmar TP. Structural basis for ligand binding and specificity in adrenergic receptors: Implications for GPCR-targeted drug discovery. Biochemistry. 2008;47:11013–11023. doi: 10.1021/bi800891r. [DOI] [PubMed] [Google Scholar]
- 18.Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 19.Grossfield A. Recent progress in the study of G protein-coupled receptors with molecular dynamics computer simulations. Biochimica Et Biophysica Acta- Biomembranes. 2011;1808:1868–1878. doi: 10.1016/j.bbamem.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Johnston JM, Filizola M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Current Opinion in Structural Biology. 2011;21:552–558. doi: 10.1016/j.sbi.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanni S, Rothlisberger U. A Closer Look into G Protein Coupled Receptor Activation: X-Ray Crystallography and Long-Scale Molecular Dynamics Simulations. Current Medicinal Chemistry. 2012;19:1135–1145. doi: 10.2174/092986712799320493. [DOI] [PubMed] [Google Scholar]
- 22.Pande VS, Beauchamp K, Bowman GR. Everything you wanted to know about Markov State Models but were afraid to ask. Methods. 2010;52:99–105. doi: 10.1016/j.ymeth.2010.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ludemann SK, Lounnas V, Wade RC. How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. Journal of Molecular Biology. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 24.Grubmuller H, Heymann B, Tavan P. Ligand binding: Molecular mechanics calculation of the streptavidin biotin rupture force. Science. 1996;271:997–999. doi: 10.1126/science.271.5251.997. [DOI] [PubMed] [Google Scholar]
- 25.Laio A, Parrinello M. Escaping free-energy minima. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: A promising and efficient simulation method for biomolecules. Journal of Chemical Physics. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 27.Wang T, Duan Y. Chromophore channeling in the G-protein coupled receptor rhodopsin. Journal of the American Chemical Society. 2007;129:6970–6971. doi: 10.1021/ja0691977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang T, Duan Y. Ligand Entry and Exit Pathways in the beta(2)-Adrenergic Receptor. Journal of Molecular Biology. 2009;392:1102–1115. doi: 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. In: Stuart CS, editor. Methods in Neurosciences. Vol. 25. Academic Press; 1995. pp. 366–428. [Google Scholar]
- 30.Gonzalez A, Perez-Acle T, Pardo L, Deupi X. Molecular Basis of Ligand Dissociation in beta-Adrenergic Receptors. Plos One. 2011;6 doi: 10.1371/journal.pone.0023815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Provasi D, Bortolato A, Filizola M. Exploring Molecular Mechanisms of Ligand Recognition by Opioid Receptors with Metadynamics. Biochemistry. 2009;48:10020–10029. doi: 10.1021/bi901494n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci U S A. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. Landmark microsecond-timescale molecular dynamics simulations that elucidated drug binding pathway to G-protein coupled receptors.
- 33.Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, et al. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dror RO, Green HF, Valant C, Borhani DW, Valcourt JR, Pan AC, Arlow DH, Canals M, Lane JR, Rahmani R, et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature. 2013;503:295–299. doi: 10.1038/nature12595. Highly detailed view of allosteric ligand binding to a G-protein coupled receptor explored through extensive molecular dynamics simulations and validated through mutation experiments.
- 35.Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, et al. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kappel K, Miao Y, McCammon JA. Accelerated Molecular Dynamics Simulations of Ligand Binding to a Muscarinic G-protein Coupled Receptor. Quarterly Reviews of Biophysics. 2015;48:479–487. doi: 10.1017/S0033583515000153. The longest single all-atom enhanced molecular dynamics simulation published to date on ligand binding to a G-protein coupled receptor.
- 37.Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M. Mechanistic Insights into the Allosteric Modulation of Opioid Receptors by Sodium Ions. Biochemistry. 2014;53:5140–5149. doi: 10.1021/bi5006915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasmussen SGF, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the [bgr]2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, et al. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE. Activation mechanism of the β2-adrenergic receptor. Proc Natl Acad Sci U S A. 2011;108:18684–18689. doi: 10.1073/pnas.1110499108. The longest single all-atom conventional molecular dynamics simulation published to date on deactivation of a G-protein coupled receptor.
- 41.Niesen MJM, Bhattacharya S, Vaidehi N. The Role of Conformational Ensembles in Ligand Recognition in G-Protein Coupled Receptors. Journal of the American Chemical Society. 2011;133:13197–13204. doi: 10.1021/ja205313h. [DOI] [PubMed] [Google Scholar]
- 42.Provasi D, Artacho MC, Negri A, Mobarec JC, Filizola M. Ligand-Induced Modulation of the Free-Energy Landscape of G Protein-Coupled Receptors Explored by Adaptive Biasing Techniques. Plos Computational Biology. 2011;7:e1002193. doi: 10.1371/journal.pcbi.1002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shan JF, Khelashvili G, Mondal S, Mehler EL, Weinstein H. Ligand-Dependent Conformations and Dynamics of the Serotonin 5-HT2A Receptor Determine Its Activation and Membrane-Driven Oligomerization Properties. Plos Computational Biology. 2012;8:e1002473. doi: 10.1371/journal.pcbi.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miao Y, Nichols SE, Gasper PM, Metzger VT, McCammon JA. Activation and dynamic network of the M2 muscarinic receptor. Proc Natl Acad Sci U S A. 2013;110:10982–10987. doi: 10.1073/pnas.1309755110. Activation of a G-protein coupled receptor through accelerated molecular dynamics simulation.
- 45.Li J, Jonsson AL, Beuming T, Shelley JC, Voth GA. Ligand-Dependent Activation and Deactivation of the Human Adenosine A2A Receptor. Journal of the American Chemical Society. 2013;135:8749–8759. doi: 10.1021/ja404391q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohlhoff KJ, Shukla D, Lawrenz M, Bowman GR, Konerding DE, Belov D, Altman RB, Pande VS. Cloud-based simulations on Google Exacycle reveal ligand modulation of GPCR activation pathways. Nature Chemistry. 2014;6:15–21. doi: 10.1038/nchem.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vaidehi N, Kenakin T. The role of conformational ensembles of seven transmembrane receptors in functional selectivity. Current Opinion in Pharmacology. 2010;10:775–781. doi: 10.1016/j.coph.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 48.Neale C, Herce HD, Pomes R, Garcia AE. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor? Biophysical Journal. 2015;109:1652–1662. doi: 10.1016/j.bpj.2015.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, Angelini A, Waghray D, Dror RO, Ploegh HL, et al. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science. 2015;347:1113–1117. doi: 10.1126/science.aaa5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Provasi D, Filizola M. Putative Active States of a Prototypic G-Protein-Coupled Receptor from Biased Molecular Dynamics. Biophysical Journal. 2010;98:2347–2355. doi: 10.1016/j.bpj.2010.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miao Y, Caliman AD, McCammon JA. Allosteric Effects of Sodium Ion Binding on Activation of the M3 Muscarinic G-Protein Coupled Receptor. Biophysical Journal. 2015;108:1796–1806. doi: 10.1016/j.bpj.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shoichet BK, Kobilka BK. Structure-based drug screening for G-protein-coupled receptors. Trends in Pharmacological Sciences. 2012;33:268–272. doi: 10.1016/j.tips.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kruse AC, Weiss DR, Rossi M, Hu JX, Hu K, Eitel K, Gmeiner P, Wess J, Kobilka BK, Shoichet BK. Muscarinic Receptors as Model Targets and Antitargets for Structure-Based Ligand Discovery. Molecular Pharmacology. 2013;84:528–540. doi: 10.1124/mol.113.087551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thal DM, Sun BF, Feng D, Nawaratne V, Leach K, Felder CC, Bures MG, Evans DA, Weis WI, Bachhawat P, et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature. 2016;531:335–340. doi: 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christopoulos A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nature Reviews Drug Discovery. 2002;1:198–210. doi: 10.1038/nrd746. [DOI] [PubMed] [Google Scholar]
- 56.Jeffrey Conn P, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wootten D, Christopoulos A, Sexton PM. Emerging paradigms in GPCR allostery: implications for drug discovery. Nat Rev Drug Discov. 2013;12:630–644. doi: 10.1038/nrd4052. [DOI] [PubMed] [Google Scholar]
- 58. Huang XP, Karpiak J, Kroeze WK, Zhu H, Chen X, Moy SS, Saddoris KA, Nikolova VD, Farrell MS, Wang S, et al. Allosteric ligands for the pharmacologically dark receptors GPR68 and GPR65. Nature. 2015;527:477–483. doi: 10.1038/nature15699. An outstanding study combining computational homology modeling, molecular docking and experimental assays to design allosteric drugs of understudied G-protein coupled receptors.
- 59.Miao Y, Nichols SE, McCammon JA. Mapping of Allosteric Druggable Sites in Activation-Associated Conformers of the M2 Muscarinic Receptor. Chemical Biology & Drug Design. 2013;83:237–246. doi: 10.1111/cbdd.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miao Y, Goldfeld D, Moo EV, Sexton PM, Christopoulos A, McCammon JA, Valant C. Structure-based design of chemically diverse allosteric modulators of a muscarinic G protein-coupled receptor. In Preparation. 2016 doi: 10.1073/pnas.1612353113. Structure-based design of allosteric drugs of a G-protein coupled receptor combining accelerated molecular dynamics simulations, ensemble docking and experimental assays.
- 61.Kang YY, Zhou XE, Gao X, He YZ, Liu W, Ishchenko A, Barty A, Sathish D, Yefanov O, Han GW, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnston JM, Wang H, Provasi D, Filizola M. Assessing the Relative Stability of Dimer Interfaces in G Protein-Coupled Receptors. Plos Computational Biology. 2012;8 doi: 10.1371/journal.pcbi.1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Periole X, Knepp AM, Sakmar TP, Marrink SJ, Huber T. Structural Determinants of the Supramolecular Organization of G Protein-Coupled Receptors in Bilayers. Journal of the American Chemical Society. 2012;134:10959–10965. doi: 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]