

Abstract
A single genome encodes a large number of phosphoryl hydrolases for the purposes of phosphate recycling, primary and secondary metabolism, signal transduction and regulation, and protection from xenobiotics. Phosphate monoester hydrolysis faces a high kinetic barrier, yet there are multiple solutions to the problem both in terms of catalytic mechanisms and three-dimensional structure of the hydrolases. Recent structural and mechanistic findings highlight the trigonal-bipyramidal nature of the transition state for enzyme promoted phosphate monoester hydrolysis and the evolution and role of inserted loops/domains in governing substrate specificity and promiscuity. Important questions remain as to how electrostatics modulate water networks and critical proton-transfer events. How substrate targeting and catalysis is achieved by the independently evolved catalytic platforms is compared and contrasted in this article.
Graphical Abstract
Introduction
Group transfer reactions comprise a substantial fraction (ca. 21%) of enzyme-catalyzed reactions (PANTHER Database; URL: http://www.pantherdb.org/genes/index.jsp). Phosphoryl group transfer involved in monoester cleavage is unique among group transfer processes because the phosphate group is negatively charged, which impairs attack by a nucleophile at the phosphorus center. The phosphate monoester hydrolases (phosphatases) or transferases (Enzyme Commission (E.C.) groups 2 or 3) are numerous and ubiquitous. A single genome encodes a large number of phosphatases with 44 in Escherichia coli, 50 in Pseudomonas aeruginosa, 1,103 in homo sapiens, 770 in Zea mays, and 110 in Saccharomyces cerevisiae (SCOP Database; URL: http://scop.mrc-lmb.cam.ac.uk/scop/). These enzymes remove phosphoryl (PO32−) moieties for the purposes of phosphate recycling, primary and secondary metabolism, signal transduction, and regulation and chemical defense (protection from xenobiotics). Substrates for phosphatases include phospholipids, phosphoglycans, phosphoproteins, phosphonucleic acids, and organophosphate metabolites. Although phosphatases generally catalyze hydrolytic P-O bond cleavage in organophosphate monoesters and anhydrides, natural variants include catalysis of hydrolytic P-C bond cleavage in activated organophosphonates (phosphoacetaldehyde and phosphonoacetate hydrolases) and P-N bonds in organophosphoamides (e.g., histidinol-phosphate phosphatase). Other P-O bond hydrolysis reactions that might be found represented in a phosphatase family include nucleoside triphosphate hydrolysis (e.g. sarcoplasmic reticulum H+, Ca2+ ATPase of the haloalkanoate dehalogenase (HAD) superfamily[1]), pyrophosphate hydrolysis, or nucleoside phosphodiester hydrolysis (e.g, Xanthomonas axonopodis nucleotide pyrophosphatase/phosphodiesterase of the alkaline phosphatase (AP) superfamily [2]).
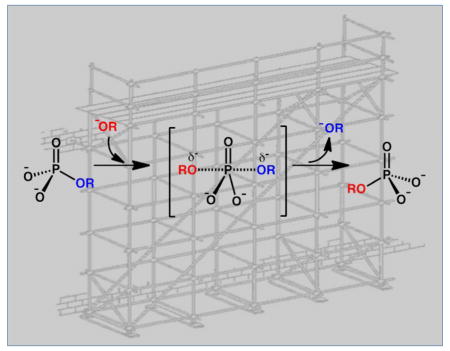
Phosphatases catalyze hydrolysis reactions of phosphate monoesters to form inorganic phosphate and alcohol. (Figure 1A). In this reaction, the phosphorus center undergoes nucleophilic substitution of the “OR” substituent by a hydroxyl group (“OH”) derived from water. The chemical mechanism and energy landscape reflect the timing between the P-OR bond cleavage and HO-P bond formation. The “concerted” reaction proceeds via a single trigonal bipyramidal transition state, whereas the “non-concerted” reaction (addition-elimination or dissociative) proceeds via two trigonal bipyramidal transition states, both of which have the same stereochemical requirements and outcomes (Figure 1A). X-ray crystal structures of transition-state analogs have clearly demonstrated the trigonal bypyramidal geometry and a recent study in alkaline phosphatase shows that inhibition is dependent on the extent to which phosphoryl analogs can mimic this form [3]. Depending on nuances of the active site, the transition state will differ even among orthologous enzymes [4]
Figure 1.
(A) General phosphoryl transfer reaction, with bond lengths and full versus partial charges dependent on the extent of bond making and breaking in the intermediate/transition state but with common trigonal bipyramidal geometry. (B) Stabilization of trigonal bipyramidal geometry/electrostatics by use of general base, GB, general acid, GA, and residues to provide favorable electrostatics, E.
Phosphate monoester hydrolysis faces a high kinetic barrier with uncatalyzed reaction rates of the dianionic form, which is dominant at pH 7.4, of k = 2 × 10−20 s−1 at 25 °C [5]. Nonetheless rates for phosphatase-catalyzed reactions are quite large with k = ca. 1 s−1 to 15,000 s−1 (for discussion see [5,6]). How catalysis is achieved by the independently evolved catalytic platforms supported by the respective folds of the phosphatase families is of considerable interest and the focus of this article.
The mechanism of phosphate monoester hydrolysis in solution is well characterized. The rate of the uncatalyzed process is pH dependent [7,8], where the monoanionic species is more reactive than the dianionic species. This outcome is a consequence of the reduced electrostatic repulsion of the nucleophle as well as the advantage conferred by intramolecular proton transfer from the phosphoryl −OH to the leaving group. In solution and in enzyme-mediated phosphate monoester hydrolysis reactions, selected metal ions may also facilitate the process [9–11]. In mechanisms for metal-ion-dependent hydrolysis, cations such as Fe2+/3+, Zn2+, or Mn2+ are proposed to act by increasing the nucleophilicity of water or by stabilizing a formal hydroxide species [12–14]. Metal ions, for example Mg2+, can also enhance electrophilicity at the phosphorus center, by neutralizing the negative charge on the high-energy intermediate/transition state, and can also serve to co-position the nucleophile and electrophile and increase the acidity of the leaving group.
Overall, the essential elements of phosphoryl group transfer catalysis include 1) steric and electronic activation of nucleophile and electrophile, 2) “shielding” of the nucleophile and phosphorus center, 3) geometric and electronic stabilization of the trigonal bipyramidal transition state and 4) stabilization of the leaving group. The arsenal of phosphate binding and catalytic groups present in proteins, to achieve these goals is limited and, thus, we anticipate a great deal of overlap in catalytic groups (electropositive metal, ion pairs) utilized in Nature to promote this reaction (Figure 1B).
Considering the limited choice of catalytic groups available for phosphatases, relative positioning of amino acids and cofactors becomes important. Therefore, the scaffold must play a critical role as it will dictate how catalysis will be achieved. Ultimately the question becomes, are some folds better adapted to support architectures suitable for phosphoryl group transfer?
Interplay of Scaffold and Phosphoryl-Group Transfer Chemistry
Considerable variation exists in the fold types supporting phosphatase function. A search of the SCOP database (SCOP Database; URL: http://scop.mrc-lmb.cam.ac.uk/scop/) yields 20 superfamilies that include phosphoryl group transfer enzymes, with six superfamilies related to one another by topological permutations (Table S1). Classifying the superfamilies families based on chemical mechanism leads to three distinct reaction types. Of the 20 summarized here, eight superfamilies catalyze phosphoryl group transfer via covalent catalysis with an enzyme residue followed by attack by HO(H), five via metal ion-mediated activation of a HO(H) nucleophile and five via protein-mediated activation of a HO(H) nucleophile (two superfamilies are not well characterized mechanistically). The fold types include Rossmann-like, β sandwich, β-propeller, and five α-helical bundle. Notably, no single fold or mechanism dominates among the families and, thus, catalysis of phosphoryl-group transfer has multiple solutions in terms of both mechanism and protein fold. However, the number of phosphatase members within each family may reflect the evolvability or ease of modifying the existing fold to support new substrate specificities (see section below). In that regard, the superfamilies with the most members are the HAD, the phosphotyrosine protein phosphatase (PTP) I and II superfamilies, and the metallo-dependent phosphatase (MDP) superfamily.
Intricate relationships between function, fold and mechanism exist. Initially, the fold is recruited because it has a certain activity that confers a selective advantage. However, this may not be the primary function, and ultimately the function will choose the scaffold (see [15–17]). The capabilities of the chosen scaffold dictate the mechanism arrived at through evolution. Throughout the evolution process, the evolvability of the scaffold will depend on the stability of the fold to mutations and insertions (see section on substrate specificity). Under selective pressure the proficiency of the new catalyst and the ability to take on new specificities and metabolic niches will impact both its biological spread (number of species) and its occurrence within a family (number of members within a species). For instance, in the HAD superfamily the phosphatases comprise ca. 99% of the superfamily, while in the DNAse1-like superfamily, the acid phosphatase/vanadium-dependent haloperoxidase, and even the AP superfamily, phosphoryl transferase activity is limited to a branch of the superfamily. Subsequent elaboration of the active site under selective pressure to increase catalytic efficiency is constrained by the fold, both in terms of geometry and the original mechanism.
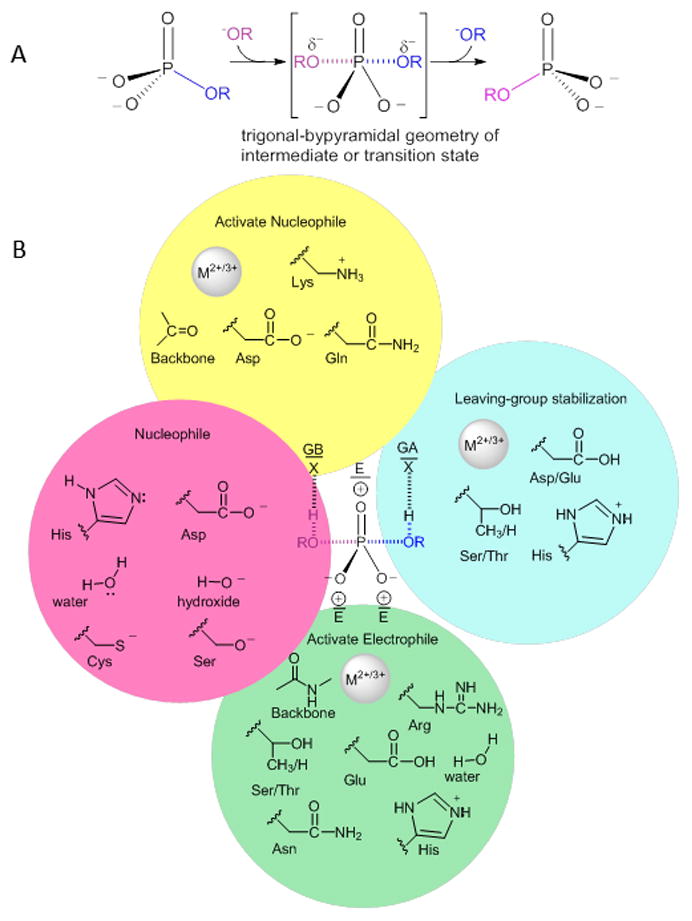
In the case of phosphoryl group transfer via covalent catalysis by an enzyme residue and subsequent attack by a HO(H), the active site nucleophile sets the requirements for supporting residues (Figure 1B). The data in Table S1 show that among these enzymes, the nucleophiles utilized are the side chains of Asp (1 superfamily), Cys (4 superfamilies), Ser (1 superfamily), and His (2 superfamilies). The selection of the nucleophiles predisposes the choice of additional catalytic residues. In the case of Cys and Ser nucleophiles, a general base catalyst allows activation of the Cys/Ser – alternatively, pKa perturbation by the surrounding dielectric can afford ionization of the side chain. The charge on Cys/Ser must then be shielded from the electrophilic center by metal ions or side-chain functionality. Shielding is also necessitated for the carboxylate of Asp nucleophiles. In contrast, the imidazole of His does not require deprotonation (activation), but upon phosphorylation the positive charge must be stabilized, which can be conveniently accomplished through a charge-relay system. Stabilizing elements are summarized in Table S1.
In metal ion-dependent hydrolytic mechanisms metal ions form coordinate bonds to the phosphate oxygen(s) forming an enzyme adduct that has a lifetime that depends on the nature of the metal ion(s) and its ligand-exchange rate. This represents a variation on covalent nucleophilic catalysis wherein the metal ion activates the HO(H) nucleophile. If a metal ion-bound hydroxide is utilized, then ligand exchange between water and phosphate is necessary to enable product dissociation. When a metal ion-bound water is utilized, the situation mirrors the use of an enzyme-activated water nucleophile with scaffold-dependent three-dimensional position and identity of the water activating group.
The placement of the active site in the enzyme scaffold is also significant. In several superfamilies, the active site is located at a topological break in the beta sheet where intervening residues cause two adjacent strands to be distant in the primary sequence. This type of placement is observed in the AP, HAD, His phosphatase, and CheY superfamilies (Figure 2A). Active sites also occur at interfaces between domains (in PTP I and II, MDP family, Rhodenese, carbohydrate phosphatase, DNase I-like, and YopH), locations that can be rationalized in terms of both chemistry and protein dynamics. The network of hydrogen bonds to the nucleophile, electrophilic center, and leaving group varies with fold. However, the scaffold serves to immobilize the nucleophile and electrophile and their supporting residues. For example hotspots of conservation have been observed in the His phosphatase superfamily in the second layer beyond the catalytic site [18]). In contrast, mobile portions of the enzyme in the form of substrate-binding loops or domains are associated with the substrate leaving group, as illustrated in the HAD superfamily, where the dynamics of the cap and core allow substrate discrimination [19,20]. Likewise loop dynamics enable regulation in the MDP superfamily [21].
Figure 2.
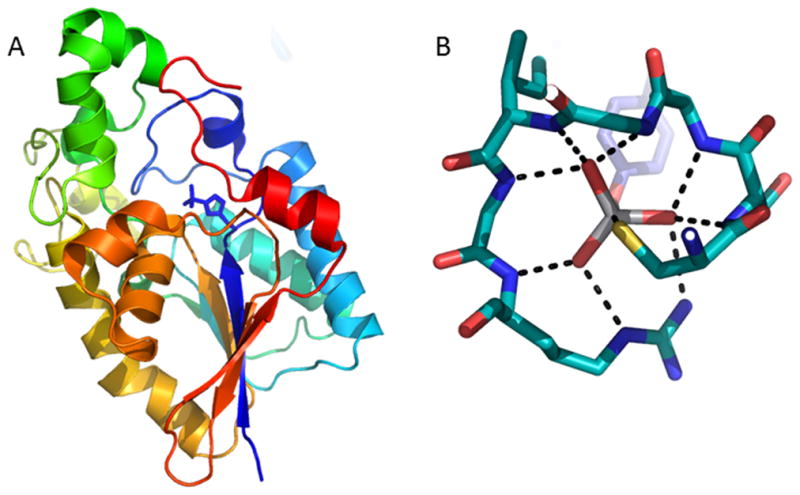
Ribbon diagram of E. coli cofactor-dependent phosphoglycerate mutase (PDB 1E58) with active site residue His10 present as a phosphohistidine (shown as sticks), highlights the active site location in the sheet between strands distant in primary structure (protein color ramped N- to C-terminus, blue to red). (B) Phosphate cradle (residues 215CSAGIGR221) in Cys dependent human PTP IB liganded to the transition-state mimic peptide/VO3− complex (PDB ID 3I7Z). Only Tyr of the peptide is depicted. Hydrogen bonds and trajectory to Cys shown as dashed lines.
Role of the Scaffold in Proton Transfer
With the exception of leaving groups with pKa < ca. 4, a general acid is utilized to protonate the leaving group (usually an alkoxide) and in those enzyme which promote reaction via an EP intermediate, the same residue can act a general base to deprotonate water in the second half reaction. Notably, in six phosphatase superfamilies the carboxylic acid side chain of Asp (less often Glu (one family)) is used as a preferred acid/base. Although exactly how this residue functions as a general acid/base given its pKa is a question that requires future study, it may be that such a residue would participate as part of a water network rather than by directly transferring a proton to/from the bridging substrate oxygen or nucleophilic water. Notably, Asp has lower side chain entropy than Glu [22] and this may be a driving force for its selection as the participating acid/base, to attain greater catalytic efficiency. For His, an acid/base function at neutral pH does not require perturbation of pKa by the active site and indeed this is the second most common acid/base residue used (in three superfamilies).
Role of the Scaffold in -PO32− binding
The phosphoryl groups of the substrate, product and trigonal-bipyramidal transition state have similar electrostatic and geometric requirements for binding and stabilization although subtle but critical variations do exist. Additionally, hydrogen-bond donor groups near the equatorial P-O oxygens in the transition state enhance the electrophilicity of the phosphorus center. Not surprisingly, a common strategy for binding is the use of residues with positively charged side chains. This usage is less common in mechanisms that involve metal ion-mediated activation of a HO(H) nucleophile, presumably because the phosphoryl group is always in the coordination sphere of the metal ion cofactor. In the superfamilies, two utilize Lys for -PO32− binding, four utilize His and 11 utilize Arg. Among the other residues, the polar side chain of Ser is the most prevalent, being utilized in three superfamilies. Use of these residues requires conservation or conservative replacement through evolution in order to retain the core chemistry of the family. In contrast, the use of main-chain interactions requires that the fold itself be retained and be stable to insertions that add specificity (see section on specificity).
Despite necessitating the retention of phi/psi angles around the phosphoryl group binding site, backbone interactions with the transferring phosphate are observed in eleven families from many fold types. A striking example of this is seen in the PTP I superfamily of phosphatases where the conserved HCXXGXXRS/T motif residues form a “cradle” (termed the P-loop) for the phosphoryl group with the nucleophilic Cys positioned for in-line attack at phosphorus (Figure 2B) (note that the P-loop includes three bonds from the Arg side chain). Although the folds of the superfamilies catalyzing phosphoryl transfer differ, a recurring theme is the location of the binding pocket at the N-terminus of an α helix. It is known that the helical dipole provides an electrostatically favorable environment for anion binding at the N-terminus[23], but that 80% of the effect is provided by the first turn of the helix [24]. Optimal ligand positioning involves direct hydrogen bonds to the backbone amide groups [25] (and see [26] for a recent overview of ion-dipole interactions). Examination of the liganded structures shows that in half of the phosphoryl-group transfer superfamilies the active site is positioned such that the transferred group makes direct hydrogen bonds to the N-terminus of an α-helix (Table S1). This is the case even in the metal-ion-dependent alkaline phosphatase and carbohydrate phosphatase superfamilies, where metal ion coordination might contribute considerably to binding. Overall, in terms of evolution, it may be that catalytic scaffolds that provide helices at binding hot-spots and or at sites where dynamics are favorable have been selected to gain greater catalytic proficiency.
Role of the Scaffold in Substrate Specificity
Overall, examination of the reactions and folds of the superfamilies reflect evolution guided by retention of the chemistry of phosphoryl group transfer with elaborations to accept new substrates rather than conservation of a substrate binding mode with modification to allow new chemistry. The evolved enzymes are “modular” containing key elements for binding and catalysis of the phosphoryl group/transition state conserved in a “core” catalytic domain and additional loops or domains inserted to bind substrates (see [27] for a recent review on functional innovation from changes in protein domains). Illustrative examples of how segments or domains are included to enable evolution of new specificities for substrates having a range of physicochemical properties are found in the HAD and His-dependent phosphatase superfamilies (Figure 3)[28,29], as well as in the actin superfamily [30], PP2C[31], and carbohydrate-dependent phosphatases [32]. One might imagine that such substrate-binding segments or domains may have been inserted as a module possessing an existing binding motif or have arisen from co-evolution with the core domain. Recently, it was shown in the HAD superfamily that core catalytic domains with the same specificity domain architecture share greater similarity in sequence and structure. This observation suggests that intramolecular coevolution occurs where the fold diverges differentially in the context of an accessory domain [33]. An ancillary effect of elaboration of the core with a substrate-binding segment/domain is the creation of a bulk solvent-exclusive environment with perturbation of the ionization states of active-site residues and ligands [34].
Figure 3.

Enzymes from the His-dependent phosphatase (A) (PDB ID 1UJC, 1QHF, 1BIF, 1RPA) and HAD (B) (PDB ID 1U7P, 3QU2, 1N9K, 1SU4) superfamilies highlighting inserts (yellow) that change specificity from macromolecules (SixA and MDP) to small molecules. Panel A adapted from Hamada et al. [28].
In the absence of inserted segments/domains the same catalytic core domain, can allow an open site with access to macromolecular ligands (Figure 3A and B). Examples of this phenomenon are observed in the HAD Superfamily (e.g. T4-polynucleotide kinase/phosphatase [35], MDP-1 [36], Scp-1 [37]), His-dependent phosphatase family (SixA [28]), the AP superfamily (PLAP [38]) and the MDP family (calcineurin [39]). As is the case with calcineurin binding to Ca2+ and calmodulin, additional domains may be appended to provide protein-protein docking sites (i.e. specificity) and/or modulate activity [40,41] (Figure 4A). An extreme example of this open active-site architecture, but with a twist that might be considered the “ultimate in substrate specificity”, is found in the chemotaxis phosphatase CheZ superfamily, represented by the phosphatase CheZ and the corresponding phosphoprotein substrate CheY. In this case, the “phosphatase” CheZ active-site contains only a conserved Asp that forms a salt-bridge to CheY and a conserved catalytic Gln that serves to deprotonate the water nucleophile (located on a helix with Asp and Gln separated by a single helical turn). The phosphoprotein substrate CheY carries with it the remainder of the catalytic machinery, including the cofactor Mg2+ and residues that bind and stabilize phosphate and any reaction intermediates [42,43] (Figure 4B). No other phosphoprotein substrate can provide this necessary machinery. Indeed, a small molecule can act as phosphoryl group donor or acceptor, showing that CheY bears the core catalytic machinery [44,45]. As may be expected, CheY possesses auto-phosphatase activity and indeed the fold is a circular permutation of that of the HAD superfamily, a relationship recently probed mechanistically [46].
Figure 4.
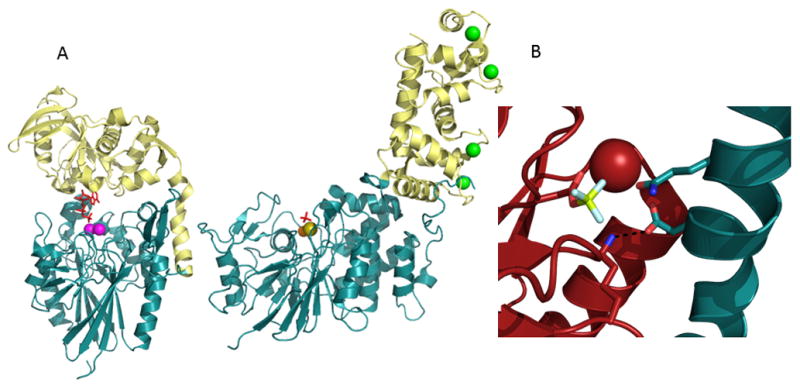
(A) Modes of specificity. In 5′-nucleotidase (PDB ID 1HPU) the inserted domain (yellow) provides specificity toward the nucleotide substrate (red sticks) while in the Ser/Thr phosphatase calcineurin (PDB ID 2PGB) the insert (yellow) provides a Ca2+ (green spheres; phosphate in red sticks) binding domain where occupancy results in partial stimulation of calcineurin activity and allows binding to calmodulin in a Ca2+ dependent manner. (B) In the CheZ family the phosphatase CheZ (blue) bears only two conserved residues (one binding Glu and one catalytic Asn) and CheY (red) supplies the required active site and Mg2+ cofactor (red sphere, phosphoryl group mimic BeF3 shown in yellow and white sticks) (PDB ID 1KMI).
On the opposite end of specificity lies substrate ambiguity, which is exemplified by phosphatases that have broad substrate ranges. Just as the scaffold itself varies, the role of the scaffold in accommodating a broad range of substrates differs amongst superfamilies (recently discussed [47]). It has been pointed out that the large volume, polar solvent accessible surface area and varied electrostatic surface of the active site of the AP superfamily enables binding to diverse substrates [48]. In contrast, in the HAD superfamily, the insertion of active site loops/domains increases the number of possible interactions with different substrates [49,50]. In both cases, the unifying theme is the availability of multiple or physicochemically flexible interaction sites.
Conclusions
The wealth of structural and mechanistic information now available for phosphatases allows special insight into the role played by the protein scaffold in driving mechanisms participating in phosphoryl group transfer. It is obvious that the problem of catalyzing phosphoryl group transfer, though considerable, has many solutions in terms of mechanism and supporting scaffolds. Questions still remain about the mechanisms of pKa modulation and the role of electrostatics and water networks in critical proton-transfer steps in these processes. Finally, the modular nature of the phosphatases provides opportunities for protein engineering.
Supplementary Material
Highlights.
Phosphoryl transfer is supported in 20 superfamilies with varied structural folds
The three basic catalytic mechanisms proceed though trigonal bipyramidal geometry
Inserted loops/domains in scaffolds govern substrate specificity and ambiguity
Important questions remain for critical proton-transfer events
Acknowledgments
Funding: This work is supported by NIH GM098760 to KNA and DD-M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2. 6 A resolution. Nature. 2000;405:647–655. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 2.Zalatan JG, Fenn TD, Brunger AT, Herschlag D. Structural and functional comparisons of nucleotide pyrophosphatase/phosphodiesterase and alkaline phosphatase: implications for mechanism and evolution. Biochemistry. 2006;45:9788–9803. doi: 10.1021/bi060847t. [DOI] [PubMed] [Google Scholar]
- 3•.Peck A, Sunden F, Andrews LD, Pande VS, Herschlag D. Tungstate as a Transition State Analog for Catalysis by Alkaline Phosphatase. Journal of Molecular Biology. 2016;428:2758–2768. doi: 10.1016/j.jmb.2016.05.007. The authors show for the first time that tungstate, commonly used as an inhibitor for phosphatases, acts as a transition state analog in alkaline phosphatase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor Ringia EA, Tyler PC, Evans GB, Furneaux RH, Murkin AS, Schramm VL. Transition state analogue discrimination by related purine nucleoside phosphorylases. J Am Chem Soc. 2006;128:7126–7127. doi: 10.1021/ja061403n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lad C, Williams NH, Wolfenden R. The rate of hydrolysis of phosphomonoester dianions and the exceptional catalytic proficiencies of protein and inositol phosphatases. Proc Natl Acad Sci U S A. 2003;100:5607–5610. doi: 10.1073/pnas.0631607100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandao TA, Hengge AC, Johnson SJ. Insights into the reaction of protein-tyrosine phosphatase 1B: crystal structures for transition state analogs of both catalytic steps. J Biol Chem. 2010;285:15874–15883. doi: 10.1074/jbc.M109.066951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunton CA, Llewellyn DR, Oldham KG, Vernon CA. The reactions of organic phosphates. Part I. The hydrolysis of methyl dihydrogen phosphate. Journal of the Chemical Society (Resumed) 1958:3574–3587. doi: 10.1039/JR9580003574. 716. [DOI] [Google Scholar]
- 8.Kumamoto J, Westheimer FH. The Hydrolysis of Mono- and Dibenzyl Phosphates. Journal of the American Chemical Society. 1955;77:2515–2518. [Google Scholar]
- 9.Cassano AG, Anderson VE, Harris ME. Analysis of solvent nucleophile isotope effects: evidence for concerted mechanisms and nucleophilic activation by metal coordination in nonenzymatic and ribozyme-catalyzed phosphodiester hydrolysis. Biochemistry. 2004;43:10547–10559. doi: 10.1021/bi049188f. [DOI] [PubMed] [Google Scholar]
- 10.Sträter N, Lipscomb WN, Klabunde T, Krebs B. Two-Metal Ion Catalysis in Enzymatic Acyl- and Phosphoryl-Transfer Reactions. Angewandte Chemie International Edition in English. 1996;35:2024–2055. [Google Scholar]
- 11.Herschlag D, Jencks WP. The effect of divalent metal ions on the rate and transition-state structure of phosphoryl-transfer reactions. Journal of the American Chemical Society. 1987;109:4665–4674. [Google Scholar]
- 12.Rodrigues JR, Fernandez A, Canales J, Cabezas A, Ribeiro JM, Costas MJ, Cameselle JC. Characterization of Danio rerio Mn2+-dependent ADP-ribose/CDP-alcohol diphosphatase, the structural prototype of the ADPRibase-Mn-like protein family. PLoS One. 2012;7:e42249. doi: 10.1371/journal.pone.0042249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schenk G, Elliott TW, Leung E, Carrington LE, Mitic N, Gahan LR, Guddat LW. Crystal structures of a purple acid phosphatase, representing different steps of this enzyme’s catalytic cycle. BMC Struct Biol. 2008;8:6. doi: 10.1186/1472-6807-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitić N, Noble CJ, Gahan LR, Hanson GR, Schenk G. Metal-Ion Mutagenesis: Conversion of a Purple Acid Phosphatase from Sweet Potato to a Neutral Phosphatase with the Formation of an Unprecedented Catalytically Competent MnIIMnII Active Site. Journal of the American Chemical Society. 2009;131:8173–8179. doi: 10.1021/ja900797u. [DOI] [PubMed] [Google Scholar]
- 15.Khersonsky O, Tawfik DS. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annual Review of Biochemistry. 2010;79:471–505. doi: 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- 16.O’Brien PJ, Herschlag D. Catalytic promiscuity and the evolution of new enzymatic activities. Chemistry & Biology. 1999;6:R91–R105. doi: 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- 17.Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. doi: 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- 18.Rigden DJ. The histidine phosphatase superfamily: structure and function. Biochem J. 2008;409:333–348. doi: 10.1042/BJ20071097. [DOI] [PubMed] [Google Scholar]
- 19.Dai J, Finci L, Zhang C, Lahiri S, Zhang G, Peisach E, Allen KN, Dunaway-Mariano D. Analysis of the structural determinants underlying discrimination between substrate and solvent in beta-phosphoglucomutase catalysis. Biochemistry. 2009;48:1984–1995. doi: 10.1021/bi801653r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H, Patskovsky Y, Toro R, Farelli JD, Pandya C, Almo SC, Allen KN, Dunaway-Mariano D. Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry. 2011;50:8937–8949. doi: 10.1021/bi201181q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sträter N, Jasper B, Scholte M, Krebs B, Duff AP, Langley DB, Han R, Averill BA, Freeman HC, Guss JM. Crystal Structures of Recombinant Human Purple Acid Phosphatase With and Without an Inhibitory Conformation of the Repression Loop. Journal of Molecular Biology. 2005;351:233–246. doi: 10.1016/j.jmb.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 22.Pickett SD, Sternberg MJ. Empirical scale of side-chain conformational entropy in protein folding. J Mol Biol. 1993;231:825–839. doi: 10.1006/jmbi.1993.1329. [DOI] [PubMed] [Google Scholar]
- 23.Wada A. The alpha-helix as an electric macro-dipole. Adv Biophys. 1976:1–63. [PubMed] [Google Scholar]
- 24.Aqvist J, Luecke H, Quiocho FA, Warshel A. Dipoles localized at helix termini of proteins stabilize charges. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:2026–2030. doi: 10.1073/pnas.88.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He JJ, Quiocho FA. Dominant role of local dipoles in stabilizing uncompensated charges on a sulfate sequestered in a periplasmic active transport protein. Protein Science: A Publication of the Protein Society. 1993;2:1643–1647. doi: 10.1002/pro.5560021010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sippel KH, Quiocho FA. Ion–dipole interactions and their functions in proteins. Protein Science. 2015;24:1040–1046. doi: 10.1002/pro.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27•.Lees JG, Dawson NL, Sillitoe I, Orengo CA. Functional innovation from changes in protein domains and their combinations. Current Opinion in Structural Biology. 2016;38:44–52. doi: 10.1016/j.sbi.2016.05.016. Highlights the innovations in activity and specificity made via insertions and deletions into conserved protein folds. [DOI] [PubMed] [Google Scholar]
- 28.Hamada K, Kato M, Shimizu T, Ihara K, Mizuno T, Hakoshima T. Crystal structure of the protein histidine phosphatase SixA in the multistep His-Asp phosphorelay. Genes Cells. 2005;10:1–11. doi: 10.1111/j.1365-2443.2005.00817.x. [DOI] [PubMed] [Google Scholar]
- 29.Lahiri SD, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. Analysis of the substrate specificity loop of the HAD superfamily cap domain. Biochemistry. 2004;43:2812–2820. doi: 10.1021/bi0356810. [DOI] [PubMed] [Google Scholar]
- 30.Alvarado J, Ghosh A, Janovitz T, Jauregui A, Hasson MS, Sanders DA. Origin of exopolyphosphatase processivity: Fusion of an ASKHA phosphotransferase and a cyclic nucleotide phosphodiesterase homolog. Structure. 2006;14:1263–1272. doi: 10.1016/j.str.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 31••.Wei X, Guo J, Li M, Liu Z. Structural Mechanism Underlying the Specific Recognition between the Arabidopsis State-Transition Phosphatase TAP38/PPH1 and Phosphorylated Light-Harvesting Complex Protein Lhcb1. Plant Cell. 2015;27:1113–1127. doi: 10.1105/tpc.15.00102. The structure of a key regulatory serine/threonine protein phosphatase with substrate peptide demostrates the structural recognition elements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown AK, Meng G, Ghadbane H, Scott DJ, Dover LG, Nigou J, Besra GS, Fütterer K. Dimerization of inositol monophosphatase Mycobacterium tuberculosis SuhB is not constitutive, but induced by binding of the activator Mg(2+) BMC Structural Biology. 2007;7:55–55. doi: 10.1186/1472-6807-7-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pandya C, Brown S, Pieper U, Sali A, Dunaway-Mariano D, Babbitt PC, Xia Y, Allen KN. Consequences of domain insertion on sequence-structure divergence in a superfold. Proc Natl Acad Sci U S A. 2013;110:E3381–3387. doi: 10.1073/pnas.1305519110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Isom DG, Castañeda CA, Cannon BR, García-Moreno EB. Large shifts in pKa values of lysine residues buried inside a protein. Proceedings of the National Academy of Sciences. 2011;108:5260–5265. doi: 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galburt EA, Pelletier J, Wilson G, Stoddard BL. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure. 2002;10:1249–1260. doi: 10.1016/s0969-2126(02)00835-3. [DOI] [PubMed] [Google Scholar]
- 36.Peisach E, Selengut JD, Dunaway-Mariano D, Allen KN. X-ray crystal structure of the hypothetical phosphotyrosine phosphatase MDP-1 of the haloacid dehalogenase superfamily. Biochemistry. 2004;43:12770–12779. doi: 10.1021/bi0490688. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Determinants for Dephosphorylation of the RNA Polymerase II C-Terminal Domain by Scp1. Molecular cell. 2006;24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Du MH, Stigbrand T, Taussig MJ, Ménez A, Stura EA. Crystal Structure of Alkaline Phosphatase from Human Placenta at 1. 8 Å Resolution: IMPLICATION FOR A SUBSTRATE SPECIFICITY. Journal of Biological Chemistry. 2001;276:9158–9165. doi: 10.1074/jbc.M009250200. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends in Cell Biology. 21:91–103. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knöfel T, Sträter N. Mechanism of hydrolysis of phosphate esters by the dimetal center of 5′-nucleotidase based on crystal structures1. Journal of Molecular Biology. 2001;309:239–254. doi: 10.1006/jmbi.2001.4656. [DOI] [PubMed] [Google Scholar]
- 41.Li H, Zhang L, Rao A, Harrison SC, Hogan PG. Structure of calcineurin in complex with PVIVIT peptide: portrait of a low-affinity signalling interaction. J Mol Biol. 2007;369:1296–1306. doi: 10.1016/j.jmb.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 42.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E. coli chemotaxis phosphatase CheZ. Nat Struct Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 43.Pazy Y, Motaleb MA, Guarnieri MT, Charon NW, Zhao R, Silversmith RE. Identical phosphatase mechanisms achieved through distinct modes of binding phosphoprotein substrate. Proc Natl Acad Sci U S A. 2010;107:1924–1929. doi: 10.1073/pnas.0911185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukat GS, McCleary WR, Stock AM, Stock JB. Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors. Proc Natl Acad Sci U S A. 1992;89:718–722. doi: 10.1073/pnas.89.2.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Page SC, Silversmith RE, Collins EJ, Bourret RB. Imidazole as a Small Molecule Analogue in Two-Component Signal Transduction. Biochemistry. 2015;54:7248–7260. doi: 10.1021/acs.biochem.5b01082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Immormino RM, Starbird CA, Silversmith RE, Bourret RB. Probing Mechanistic Similarities between Response Regulator Signaling Proteins and Haloacid Dehalogenase Phosphatases. Biochemistry. 2015;54:3514–3527. doi: 10.1021/acs.biochem.5b00286. The authors demonstrate enhanced phosphatase activity in a response regulator by introducing the homologous general acid/base residue from the HAD family. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Pabis A, Duarte F, Kamerlin SC. Promiscuity in the Enzymatic Catalysis of Phosphate and Sulfate Transfer. Biochemistry. 2016;55:3061–3081. doi: 10.1021/acs.biochem.6b00297. Gives insight into structural and mechanistic basis of promiscuity in enzymes that catalyze for phosphatase and sulfatase activities in a single active site. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.Barrozo A, Duarte F, Bauer P, Carvalho AT, Kamerlin SC. Cooperative Electrostatic Interactions Drive Functional Evolution in the Alkaline Phosphatase Superfamily. J Am Chem Soc. 2015;137:9061–9076. doi: 10.1021/jacs.5b03945. Computational study showing that the promiscuity of alkaline phosphatase family enzymes arises from cooperative electrostatic interactions, allowing the enzyme to catalytically accomodate to different substrates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pandya C, Farelli JD, Dunaway-Mariano D, Allen KN. Enzyme promiscuity: engine of evolutionary innovation. J Biol Chem. 2014;289:30229–30236. doi: 10.1074/jbc.R114.572990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50••.Huang H, Pandya C, Liu C, Al-Obaidi NF, Wang M, Zheng L, Toews Keating S, Aono M, Love JD, Evans B, et al. Panoramic view of a superfamily of phosphatases through substrate profiling. Proc Natl Acad Sci U S A. 2015;112:E1974–1983. doi: 10.1073/pnas.1423570112. The functional space of the haloalkanoate dehalogenase superfamily was revealed by screening showing that the HADSF subfamily having the least structural elaboration of the Rossmann fold catalytic domain was the most specific. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.