

Abstract
Lipids in the body are transported via lipoproteins that are nanoparticles comprised of lipids and amphipathic proteins termed apolipoproteins. This family of lipid surface-binding proteins is over-represented in human amyloid diseases. In particular, all major proteins of high-density lipoproteins (HDL), including apoA-I, apoA-II and serum amyloid A, can cause systemic amyloidoses in humans upon protein mutations, post-translational modifications or overproduction. Here, we begin to explore how the HDL lipid composition influences amyloid deposition by apoA-I and related proteins. First, we summarize the evidence that, in contrast to lipoproteins that are stabilized by kinetic barriers, free apolipoproteins are labile to misfolding and proteolysis. Next, we report original biochemical and biophysical studies showing that increase in triglyceride content in the core of plasma or reconstituted HDL destabilizes the lipoprotein assembly, making it more labile to various perturbations (oxidation, thermal and chemical denaturation and enzymatic hydrolysis), and promotes apoA-I release in a lipid-poor/free aggregation-prone form. Together, the results suggest that decreasing plasma levels of triglycerides will shift the dynamic equilibrium from the lipid-poor/free (labile) to the HDL-bound (protected) apolipoprotein state, thereby decreasing the generation of the protein precursor of amyloid. This prompts us to propose that triglyceride-lowering therapies may provide a promising strategy to alleviate amyloid diseases caused by the deposition of HDL proteins.
Keywords: Cardiovascular and amyloid diseases; protein-lipid interactions and protein misfolding; oxidation and hydrolysis; lipid-lowering therapies; fibrates, statins and low-fat diets
Graphical abstract
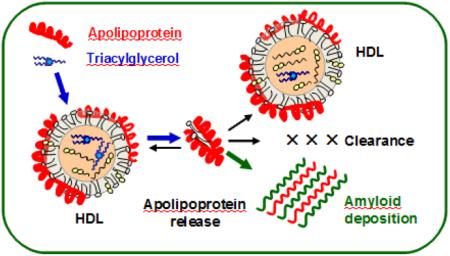
INTRODUCTION
1.1 Apolipoproteins are prominent in human amyloid diseases
Lipoproteins are protein-lipid nanoparticles that transport lipids in the aqueous environment of plasma, lymph and cerebrospinal fluid. Protein constituents of these nanoparticles, termed apolipoproteins (apos), form a family of amphipathic lipid surface-binding proteins that are central to cardiovascular health and disease, but are also prominent in amyloid diseases, or amyloidoses ([1-5] and references therein). In amyloidoses, normally soluble functional globular proteins or peptides misfold to form an intermolecular cross-β-sheet and deposit as amyloid fibrils [6]. Of nearly 40 proteins and peptides that are known to deposit in human amyloidoses, seven are exchangeable (water-soluble) apolipoproteins. These include wild-type (WT) or mutant forms of apoA-I, apoA-II, apoA-IV, apoC-II, or apoC-III that deposit in systemic amyloidoses; serum amyloid A (SAA) that causes amyloid A (AA) amyloidosis in chronic inflammation; apoE that is a key player in Alzheimer’s disease and a ubiquitous constituent of amyloid deposits in vivo; along with related lipid surface-binding proteins such as α-synuclein and Aβ peptide that deposit in Parkinson’s and Alzheimer’s diseases, respectively ([1-6] and references therein). Two of these proteins, apoA-I and apoA-II, are the major protein constituents of normal high-density lipoproteins (HDL), while SAA becomes a major HDL protein under inflammatory conditions (described below). ApoE and apoCs are minor HDL proteins. Deposition of these proteins in amyloid is expected to be intimately linked to HDL metabolism, yet little is known about the nature of this link. Here we explore the effects of HDL core lipid composition on the amyloid precursor formation by the main HDL proteins, particularly apoA-I.
ApoA-I, the major HDL protein that constitutes 75-80% of the total protein mass in normal plasma HDL, forms a key structural scaffold and an important functional ligand on the HDL surface [7]. Nearly 95% of all circulating apoA-I is bound to HDL, yet a small fraction is released in a transient lipid-poor/free state (termed free for brevity) that can carry several lipid molecules and is labile to aggregation [8-11]. Free apoA-I can rapidly bind to the existing HDL or generate HDL de novo; alternatively, free apolipoprotein can be either catabolized or get misfolded in amyloid [3,8,9]. ApoA-I causes two forms of systemic human amyloidosis, hereditary and acquired [12-15]. The hereditary form, AApoAI, is an autosomal dominant disorder caused by apoA-I gene mutations nearly 20 of which have been identified [12-14,16,17]. In this incurable potentially lethal disease, mutant apoA-I and its N-terminal 9-11 kDa fragments deposit in vital organs (kidneys, liver, spleen, heart, testes, nerves, etc.) and damage them. The only available treatment for AApoAI is end-stage organ transplant. In the acquired form, which is linked to increased plasma triglycerides (TG), aging, and oxidation, full-length apoA-I and its fragments deposit as fibrils in tissues and atherosclerotic plaques ([14, 18-20] and references therein). Such a fiber deposition reduces the mechanical stability of the arterial plaques [21,22], potentially leading to a heart attack or a stroke, and has a range of other pathogenic properties ([1,13,18-20] and references therein).
ApoA-II is the second-major HDL protein that comprises 15-20% of the total protein mass in normal human HDL [23]. This most hydrophobic of all exchangeable apolipoproteins binds particularly strongly to mature HDL and modulates HDL metabolism ([23,24] and references therein). Mutations leading to a C-terminal extension in human apoA-II cause hereditary human amyloidosis (AApoAII) with symptoms resembling those of AApoAI [25-28]. At least one case of human AApoAII involving no gene mutations has also been reported [29]. The main organs afflicted in AApoAII are kidneys; the available treatment is limited to dialysis and organ transplant [27].
Serum amyloid A (SAA) is an acute-phase protein that is dramatically but transiently upregulated in inflammation or injury [2]. As a result, SAA can displace HDL proteins, such as apoA-I, to become a major HDL protein that importantly influences lipid transport in acute inflammation and has emerged as a contributing factor to atherosclerosis [2,30,31]. In chronic inflammatory diseases such as rheumatoid arthritis, Crohn’s disease or tuberculosis, persistently elevated levels of plasma SAA can cause AA amyloidosis wherein N-terminal fragments of SAA, termed AA, deposit as fibrils in kidneys. AA amyloidosis is the major complication of chronic inflammation and the major human systemic amyloidosis worldwide [32]. If untreated, AA is lethal; the available treatment is anti-inflammatory drugs to keep the plasma SAA levels below 0.1 mg/ml. This treatment is not suited for all patients, including a subset of AA cases of unknown etiology not associated with chronic inflammation. Recent failure of phase-3 clinical trials of a small-molecule drug, Kiacta [33], which was designed to block SAA binding to heparan sulfate proteoglycans and thereby block SAA dissociation from HDL and AA deposition [34], underscores the urgent need to develop new therapies for AApoAI, AApoAII and AA amyloidoses.
Here, we propose one such potential therapy: to use existing or new TG-lowering approaches. The basic premise behind this idea is that elevated plasma TG promote the release of HDL proteins in an aggregation-prone lipid-poor/free form. Below, we briefly summarize the current evidence that, in contrast to lipoprotein assemblies, free apolipoproteins form direct protein precursors of amyloid. Next, we report original in vitro data showing that elevated TG promote apolipoprotein dissociation from HDL. Finally, we outline possible ways to test the role of TG in amyloid deposition by HDL proteins in vivo.
1.2 Lipid-free but not lipid-bound apolipoproteins are labile to misfolding
The current consensus is that the free apolipoproteins are labile to proteolysis or aggregation while lipid-bound apolipoproteins are protected [1,3,25,28]. Proteolytic studies strongly support this idea and show that HDL-bound apoA-I is much more resistant to proteolysis that lipid-poor and, particularly, lipid-free protein [35,36]. Further support comes from the biophysical studies showing that free apolipoproteins such as apoA-I, apoA-II, apoE, and apoC-I adapt partially folded conformations in solution and are characterized by low structural stability, high flexibility, high aggregating propensity and other molten globule-like properties [37-40]. These structural properties along with high hydrophobicity of apolipoproteins are expected to augment their misfolding in amyloid [3,40]. In contrast to low thermodynamic stability of free apolipoproteins, lipoprotein assemblies are stabilized by high kinetic barriers [41-43]. Such barriers arise from the transient disruption of extensive protein and lipid interactions, which occurs during protein release and may lead to lipoprotein fusion and rupture [40-43]. Hence, lipoprotein assembly provides kinetic stabilization for apolipoproteins, which is expected to protect them from proteolytic degradation as well as from misfolding in amyloid.
To better understand the link between the lipid surface-binding function of apolipoproteins and their tendency to form amyloid in vivo, amino acid sequence analysis of ten apolipoproteins was performed to identify adhesive segments, or amyloid hot spots, that are likely to trigger the conversion of the native highly α-helical structure into the cross-β-sheet conformation characteristic of amyloid [3]. One example is apoA-II whose stop-codon mutations lead to a 21-residue C-terminal extension that causes human AApoAII [25,26]. Bioinformatics analysis revealed that human wild type (WT) apoA-II has unusually high sequence propensity to form amyloid; however, it does not form amyloid in vivo because all its amyloid hot spots are located in the amphipathic α-helices that tightly bind HDL and are thereby protected from misfolding [44]. In contrast, in amyloidogenic apoA-II mutants, the C-terminal extension not only forms an additional amyloid hot spot but also cannot fold into an amphipathic α-helix; hence, this extension cannot form protective interactions with a lipid surface. This explains why the mutant rather than the WT apoA-II causes human amyloidosis [44].
Interestingly, in mice, common apoA-II isoforms can cause amyloidosis, even though murine apoA-II has lower amyloid-forming sequence propensity than its human counterpart. Notably, normal human apoA-II is a disulfide-linked dimer, and its reduction to a monomer significantly weakens its binding to the lipid surface ([45,46] and references therein). In contrast, murine apoA-II is a monomer that is expected to bind HDL less strongly than the dimer and, hence, is less well-protected from misfolding than its human counterpart. Collectively, these studies show that tight binding to HDL protects apoA-II from amyloid formation in vivo, whereas weaker binding augments it [44].
Surprisingly, human proteins such as apoB (a large non-exchangeable protein that is the major constituent of low- and very-low-density lipoproteins) and WT apoA-II, which have the highest amyloid-forming sequence propensity comparable to that of Alzheimer’s Aβ42 peptide [3], are generally not involved in human amyloidoses. In contrast, apoE, which has only modest sequence propensity to form amyloid, is a ubiquitous constituent of amyloid plaques in vivo. Overall, the rank order of the sequence propensity to form amyloid (apoB > apoA-II > apoC-II > apoA-I, apoC-III, SAA, apoC-I > apoA-IV, apoA-V, apoE) does not correlate with the actual protein’s involvement in amyloidosis. Rather, it correlates directly with the strength of the protein-lipid binding, which increases with increasing hydrophobic moment of a protein. As a result, the propensities to bind lipid surface and to form amyloid are both rooted in apolipoproteins’ hydrophobicity. Therefore, functional requirements make it difficult to completely eliminate the pathologic aggregation of apolipoproteins, explaining why this protein family is over-represented in amyloidoses. To prevent amyloid formation, apolipoproteins have evolved protective strategies, such as the sequestration of the amyloid hot spots via the native protein-lipid or protein-protein interactions [3].
An overarching conclusion emerging from these studies is that lipid-bound apolipoproteins are protected from misfolding via the kinetic barriers stabilizing the lipoprotein assembly [41-43]. In addition, lipid binding sites in these proteins often overlap with the amyloid hot spots. Therefore, binding to a lipid surface protects these sensitive regions from the dangerous solvent exposure [3,44,47]. In contrast, free apolipoproteins are labile, since their low structural stability facilitates transient exposure of their amyloid hot spots [40]. This conclusion suggests that a shift in the dynamic equilibrium from the HDL-bound (protected) to the free (labile) apolipoprotein form can augment amyloidosis. Below we show that such a shift can result from an increase in the TG content in human plasma HDL as well as in model HDL.
2. MATERIALS AND METHODS
2.1 Materials
Palmitoyl:oleoyl phosphatidylcholine (POPC) and unesterified cholesterol (UC) were from Avanti Polar Lipids. Triolein (TO), matrix metalloproteinase MMP-12 (Lot # SLBF6347V) and lipoprotein lipase (LpL) from porcine pancreas (# L3126) were from Sigma. Monoclonal antiobody for apoA-I (# MAC20-001) was from Meridian Life Sciences. CuSO4 (0.5 M solution) and H2O2 (30% solution) were from Fisher Scientific. Human myeloperoxidase (MPO) was from Calbiochem (lot # EC1.11.1.7); MPO purity assessed by SDS PAGE was >95%, and the purity index was A430/A280=0.75. Cholesterol ester transfer protein (CETP), which was purified from human plasma, was a generous gift from Prof. Kerry-Ann Rye (University of New South Wales, Australia). Lecithin:cholesterol acyltransferase (LCAT) was a generous gift from Prof. John Parks (Wake Forest University, USA). All chemicals were of highest purity analytical grade.
2.2 Lipoprotein and apoA-I preparation and remodeling
Human HDL and very low-density lipoproteins (VLDL) were isolated from plasma of anonymous healthy volunteers. The plasma was purchased from a local blood bank (Research Blood Components, LLC) in full compliance with the Institutional Review Board. Single-donor lipoproteins were isolated from fresh EDTA-treated plasma by density gradient ultracentrifugation in the density range 1.063-1.21 g/ml for HDL and 0.94-1.006 g/ml for VLDL following established protocols [48]. Each isolated lipoprotein fraction migrated as a single band on the agarose gel. Lipoproteins were dialyzed against the standard buffer (10 mM Na phosphate, pH 7.5), degassed, and stored in the dark at 4 °C. The stock solutions were used within 2-3 weeks, during which no protein degradation was detected by SDS PAGE and no changes in the lipoprotein electrophoretic mobility were seen on the agarose gel.
Human apoA-I was isolated from fresh plasma HDL, purified to ~95% purity, and refolded from 6 M guanidine hydrochloride (Gdn HCl) upon extensive dialysis against the standard buffer as previously described [10]. ApoA-I stock solution was stored at 4 °C and was used within one month.
To obtain reconstituted HDL (rHDL) of controlled composition, model discoidal HDL were reconstituted by using the initial molar ratio 80:4:1 of POPC : UC : plasma apoA-I. Discoidal particles were prepared by cholate dialysis [49] followed by extensive dialysis against the standard buffer. These particles were converted into spherical rHDL upon incubation with LCAT following established protocols [50]. Homogeneous spherical rHDL were isolated by density gradient centrifugation in the density range 1.063-1.21 g/ml.
To compare lipoproteins from patients’ plasma varying in TG levels, plasma was obtained from 25 healthy normolipidemic and normoglycemic volunteers and from 25 subjects that have been diagnosed with type-2 diabetes. In the latter group, 4 patients were newly diagnosed without previous treatment, 21 patients have been previously treated with metformin (~80%) or insulin (~20%), and 70% of them took statins. The glycemic index (hemoglobin A1c) in all diabetes patients was 8% or higher. Plasma was obtained at the Lipid Laboratory of Hospital de Saint Pau (Barcelona, Spain) with written informed consent by the patients and upon approval by the institutional ethics committee. HDL from the plasma pools of normolipidemic and diabetic donors, NL-HDL and DM-HDL, were isolated by density gradient centrifugation as described above. Protein and lipid composition was determined as previously described [51], and the results are shown in table S1 of the on-line supplement.
To obtain TG-enriched HDL (TG-HDL), as a source of TG we used human VLDL that are the major carriers of TG in plasma. CETP was used as a TG transfer protein that mediates an equimolar exchange of cholesterol ester (CE) for TG. Plasma or reconstituted spherical HDL (final protein concentration 1 mg/ml) were incubated with VLDL (final TG concentration 5 mM) and CETP (final concentration 5 units/ml) in standard buffer at 37 °C for 24 h [52]. The samples were placed on ice to terminate the reaction, followed by density gradient centrifugation to isolate TG-HDL, and by dialysis against the standard buffer.
To obtain triolein-enriched HDL (TO-HDL), lipid microemulsions containing TO and POPC were prepared by sonication [53]. These microemulsions (final TO concentration 5mM/l) were incubated with HDL (final CE concentration 1 mM/l) and CETP (2.7 units/ml) at 37 °C for 24 h [50]. Similar incubations carried out in the absence of CETP were used as a negative control. The resulting TO-HDL were isolated from the total mixture (CETP-TO-HDL) by density gradient centrifugation in the density range 1.07–1.21 g/ml, followed by dialysis against the standard buffer. Biochemical composition of plasma HDL and of reconstituted HDL before and after their enrichment with either TG or TO is shown in supplemental tables S2 and S3.
To probe the effects of HDL enrichment with TG or TO on the protein release and lipoprotein fusion, which occur during normal metabolic remodeling of plasma HDL [8], chemical and thermal perturbation methods were used. These methods have been previously shown to induce apoA-I release and HDL fusion and rupture, which mimic important aspects of metabolic HDL remodeling by plasma factors [41,42]. For a denaturant-induced remodeling, HDL were incubated with 3 M Gdn HCl at 37 °C for 3 h, followed by extensive dialysis against the standard buffer. For thermal remodeling, HDL solutions (containing 3 mg/mL protein in 10 mM PB, 150 mM Na Cl at pH 7.5) were heated from 5 °C up to 120 °C at a rate of 90 °C/h, and the heat capacity Cp(T) was recorded using VP-DSC microcalorimeter (MicroCal, MA, USA) as previously described [42]. ORIGIN software was used for the data collection and analysis. HDL samples that have been heated in DSC experiments to different temperatures were collected and used for further studies.
Oxidation of normal and of TG-rich HDL by using Cu2+, H2O2 or myeloperoxidase (MPO) was carried out as previously described [54]. Copper is widely used in vitro to continuously monitor lipoprotein oxidation. Lipoprotein oxidation in vivo may occur via several pathways, including H2O2. Activated phagocytes use a nicotinamide adenine dinucleotide phosphate-oxidase, NOX2, to generate extracellular superoxide that converts into hydrogen peroxide, an important oxidant of lipoproteins in vivo [55]. Oxidative enzymes such as MPO, which can use H2O2 or Cl− as substrates, also importantly contribute to lipoprotein oxidation in vivo ([54] and references therein).
The time course of Cu2+-induced lipid peroxidation was monitored continuously by UV absorbance at 234 nm that reports mainly on the conjugated diene formation. HDL solutions (0.1 mg/ml protein) were equilibrated at 37 °C and the oxidation was initiated by adding CuSO4 to a final concentration of 5 μM. HDL oxidation at 37 °C was monitored at 234 nm using Varian Cary Biomelt-300 UV-Vis absorption spectrometer with thermoelectric temperature control.
HDL oxidation by H2O2 was triggered by adding 0.3 % H2O2 followed by incubation at 37 °C for 6 h [54]. Excess H2O2 was removed by extensive dialysis against standard buffer. To oxidize HDL using MPO/H2O2/Cl−, HDL solution (0.5 mg/ml protein) was dialyzed against 50 mM Na phosphate buffer at pH 7.5 containing 150 mM NaCl and 0.1 mM diethylenetriamine pentaacetic acid, and MPO was added to a final concentration of 60 nM. The reaction was initiated by adding 50 μM H2O2 in 3 aliquots at 15 min intervals. The mixture was incubated at 37 °C for 90 min, and the reaction was quenched by adding a 100-fold excess of L-methionine [54]. Oxidized TG-HDL obtained by these methods were remodeled by incubation with 3 M Gdn HCl as described above, and the products of this remodeling were isolated either by SEC or by density centrifugation for further studies. Oxidation-induced change in the molecular weight of free apoA-I separated by this method was determined by MALDI-TOF as previously described [56].
Binding of a diagnostic dye thioflavin T (ThT) to apoA-I aggregates was assessed by fluorescence. ApoA-I (1 mg/ml) was incubated at 37 °C in phosphate buffer at pH 6.0 with continuous stirring; after 24 h, the protein solution was diluted to 0.1 mg/ml apoA-I and 10 μM ThT was added at 25 °C following published protocols [20]. ThT fluorescence was measured using Fluoromax-2 spectrofluorometer with an excitation wavelength of 444 nm and 5 nm slit width for excitation and emission.
For HDL processing by a matrix metalloproteinase (MMP), normal plasma HDL (1.5 mg/ml protein concentration) were incubated for 2 h at 37 °C with MMP-12 (20 μg/ml) in 200 mM NaCl, 10 mM CaCl2, 50 mM Tris at pH 7.6 in the presence of 0.02 mM butylated hydroxytoluene. The reaction was stopped by adding EDTA to a final concentration of 15 mM [57].
For HDL remodeling by lipoprotein lipase, HDL (1 mg/ml, total protein concentration) was incubated with 20 units of LpL in standard buffer at 37 °C for 24 h. The reaction mixture was immediately loaded and separated on the native PAGE.
2.3 Transmission electron microscopy, gel electrophoresis, immunoblotting, size-exclusion chromatography and lipid analysis
To visualize lipoproteins we used negative-stain electron microscopy (EM) with a CM12 transmission electron microscope (Philips Electron Optics) as previously described [41,42].
For native PAGE, Novex™ 4-20% Tris-glycine gels (from Invitrogen) were loaded with 6 μg protein per lane and run to termination at 1,500 V-h under native non-denaturing conditions in Tris-glycine buffer. For SDS PAGE, the gels were loaded with 5 μg protein per lane and run at 200 V for 1 h under denaturing conditions in SDS-Tris-glycine buffer. All gels were stained with Denville Blue protein stain (Denville Scientific).
For Western blotting, the proteins were separated on SDS gels and were transferred to a PVDF membrane for 1 h at 100 V, 4 °C. The membranes were blocked for 1 h in Tris-buffered saline / casein blocking buffer. The blots were probed with antibodies for apoA-I or apoA-II in blocking buffer for 1 h. The blots were washed thrice for 10 min each, and were visualized by using an ECL system (NEN Life Science Products).
Size-exclusion chromatography (SEC) was performed by using a preparative-grade Superdex 16 XK column or a Superose 6 10/300 GL column controlled by an AKTA UPC 10 FPLC system (GE Healthcare). Samples were eluted by PBS at pH 7.5 at a flow rate of 0.5 ml/min.
For lipid analysis using thin-layer chromatography, lipids were extracted with 2:1 chloroform : methanol by Folch method [58] and were dried under nitrogen. Known amounts of dry lipids were analyzed using either hexane : ether : acetic acid (70:30:1 molar ratio) or chloroform : methanol : water : acetic acid (65:25:4:1 molar ratio) to separate apolar or polar lipids, respectively, as previously described [56].
Cholesterol, triglycerides, and phospholipids were quantified using enzymatic assays following published protocols [59]. Protein concentration was determined using modified Lowry assay with BSA as standard [60]. All concentrations were measured in triplicate. Unless otherwise stated, all experiments reported in this study were repeated at least three times to ensure reproducibility.
3. RESULTS
3.1 Human HDL enriched with TG show increased protein release and decreased stability
First, normal human HDL (nHDL) were enriched with TG by incubation with VLDL and CETP. The incubation mixture, termed CETP-TG-HDL, was analyzed for protein distribution in lipid-bound and free states. SEC and native PAGE showed that HDL enrichment with TG caused the release of a small fraction of free protein, and immunoblotting revealed that this fraction contained apoA-I but not apoA-II (Figure 1). Since VLDL contain little if any apoA-I, the origin of free protein must be HDL. Therefore, HDL enrichment in TG promotes apoA-I release under ambient conditions.
Figure 1.

Characterization of human plasma HDL enriched with TG. Incubation mixture containing CETP (~70 kDa), VLDL, and HDL (CETP-TG-HDL) was analyzed by (A) SEC using preparative-grade Superdex 16 XK column and (B) native PAGE (4-20% gradient, Denville Blue protein stain). HDL-bound and free protein fractions are indicated. The void volume in SEC (not shown) contains large VLDL particles (40-100 nm). (C) Immunoblots of the HDL-bound and free protein fractions, which were isolated by SEC from CETP-TG-HDL, show that the free protein released from HDL upon enrichment in TG contains apoA-I but no apoA-II.
TG-HDL were isolated by SEC from CETP-TG-HDL. Negative-stain EM revealed no large changes in the particle size upon TG enrichment, and thin-layer chromatography and lipid quantification clearly showed an increased TG content in these particles (supplemental Figure S1 and Table S2).
Next, TG-HDL were destabilized by incubation with Gdn HCl for 3 h at 37 °C. Incubation of nHDL with Gdn HCl was previously shown to induce progressive protein dissociation and fusion of the protein-depleted particles to compensate for the loss of the protein moiety from the surface; ultimately, such a progressive protein dissociation leads to HDL rupture and release of the core lipids that coalesce into droplets [41,42]. SEC and native PAGE data in Figure 2 demonstrated that, compared to nHDL, TG-HDL showed more advanced denaturant-induced protein dissociation and lipoprotein fusion.
Figure 2.

Effects of TG enrichment on the chemical remodeling of plasma HDL under mild denaturing conditions. Native or TG-enriched HDL (nHDL and TG-HDL) that were either intact or have been incubated in 3M Gdn HCl for 3 h at 37°C were analyzed by (A) SEC (preparative-grade Superdex 16 XK column) and (B) native PAGE. SEC data of intact nHDL and of intact TG-HDL fully overlap and show a single peak centered circa 100 ml, which corresponds to intact-size HDL (not shown). Native PAGE demonstrates that, compared to nHDL, TG-HDL show increased dissociation of free protein and lipoprotein fusion upon Gdn-induced remodeling.
To determine the effects of TG enrichment on the thermal stability of human HDL, nHDL and TG-HDL were heated at a constant rate in DSC experiments, and differential heat capacity, Cp(T), was continuously recorded to detect HDL fusion and rupture. These HDL transitions are endothermic and correspond to high-temperature peaks in the Cp(T) function [42]. Our results showed that, compared to nHDL, in TG-HDL these transitions shifted to lower temperatures by ~15 °C (Figure 3A). Therefore, TG enrichment greatly destabilizes HDL and promotes heat-induced protein release and lipoprotein fusion and rupture.
Figure 3.

Effect of TG enrichment on thermal remodeling of plasma HDL. (A) Differential heat capacity, Cp(T), recorded by differential scanning calorimetry during heating of nHDL and TG-HDL at a constant rate of 90 °C/h. Endothermic transitions corresponding to the thermodynamically irreversible lipoprotein fusion and rupture are indicated [42]. Native PAGE shows population distribution in nHDL and TG-HDL after heating to 85 °C (B) or to 120 °C (C) to trigger fusion and rupture, respectively. HDL rupture releases core lipids that coalesce into protein-containing lipid droplets and their aggregates [42]. These large species can be seen at the top of the gel (nHDL heated to 120°C, panel C) and ultimately become too large to enter the gel (TG-HDL heated to 120 °C, panel C). Advanced lipoprotein disintegration in TG-HDL after heating to 120 °C leads to massive aggregation of free protein (panel C).
Next, nHDL and TG-HDL were heated in DSC experiments to 85 °C or to 120 °C to trigger lipoprotein fusion or rupture, respectively, followed by the native PAGE analysis. The results clearly showed that TG enrichment greatly advanced the extent of the heat-induced HDL fusion, rupture and apolipoprotein release (Figure 3B). EM analysis supported this conclusion and showed that the fraction of particles that increased in size due to HDL fusion and rupture upon chemical or thermal perturbation was greater in TG-HDL as compared to nHDL (Figure 4). Together, the results in Figures 1-4 clearly showed that HDL enrichment with TG promotes apolipoprotein dissociation at ambient conditions and upon chemical or thermal perturbation, and reduces HDL stability to such perturbations.
Figure 4.

Transmission electron micrographs of negatively stained nHDL and TG-HDL that were either intact (left), or have been perturbed by a denaturant (incubation with 3 M Gdn HCl at 37 °C for 3 h; middle), or by heating (to 90 °C at a rate of 90 °C /h, right). Representative images are shown on the same scale.
3.2 Effect of TG enrichment on HDL oxidation promotes an amyloidogenic outcome for apoA-I
Mild oxidation of apoA-I, which starts with methionine sulfoxide formation, promotes the misfolding of free full-length human apoA-I and amyloid fibril formation in vitro; this process is implicated in the fibril deposition by full-length WT apoA-I in the oxidative environment of atherosclerotic plaques in acquired amyloidosis [19,20]. The effect of plasma TG on this pathogenic process was unknown. Here we determined how the enrichment of human HDL with TG influences the extent of HDL lipid oxidation and protein release in the aggregation-prone free form. First, to determine the effect of TG on the rate of HDL oxidation, the time course of the copper-induced conjugated diene formation in HDL was recorded. This method enables one to continuously monitor lipid peroxidation and accurately detect changes in its rate. The results in Figure 5A revealed accelerated lipid peroxidation in TG-HDL as compared to nHDL. In fact, TG-HDL showed shorter lag phase and an increased rate of the rapid phase of lipid peroxidation evident from an increased maximal slope, Vmax, of the absorbance curve, Abs234(t). As a result, the midpoint of the rapid oxidation phase decreased from about 80 min in nHDL to 62 min in TG-HDL. Consequently, TG enrichment makes HDL significantly more susceptible to oxidation.
Figure 5.

TG enrichment accelerates HDL oxidation that promotes dissociation of methionine-oxidized free apoA-I. (A) Kinetics of copper-induced oxidation of nHDL and TG-HDL monitored by absorbance at 234 nm for conjugated diene formation. (B) Mild oxidation of TG-HDL by hydrogen peroxide. TG-HDL that were intact (top), oxidized (middle, marked ox-TG-HDL) or oxidized and treated with 3M Gdn HCl (bottom) were analyzed by SEC using Superdex-XK 200 column. (C) Native PAGE of the total samples and of the SEC fractions of the Gdn HCl-treated ox-TG-HDL that showed fusion and release of free protein (panel B). (D) SDS PAGE of the total samples from panel B and their SEC fractions shows that free protein released from ox-TG-HDL contains oxidized apoA-I. Molecular mass of this free protein, which was determined by MALDI TOF (D, bottom), was increased by 48 Da compared to unmodified apoA-I, suggesting oxidation of the three methionines (M86, M112 and M148).
Next, we tested the combined effects of TG enrichment and oxidation of HDL on the apolipoprotein release. To this end we used hydrogen peroxide, H2O2, to selectively oxidize Met residues in apoA-I and apoA-II. The resulting replacement of the apolar Met with the polar Met(O) in the apolar lipid binding helical faces of these proteins reduces their affinity for lipid [61]. Importantly, such a mild oxidation is thought to mimic the in vivo conditions in the arterial wall; free apoA-I oxidized by this method shows Met oxidation that promotes fibril formation [19,20]. To promote the apolipoprotein release, mildly oxidized TG-HDL were incubated with 3 M Gdn HCl for 3 h at 37 °C, followed by the product separation by SEC. The results showed the release of free protein and HDL fusion (Figure 5B, bottom). The SEC fractions corresponding to the HDL-bound and free proteins were collected, pooled, concentrated and subjected to native and SDS PAGE (Figure 5C, D). SDS PAGE showed that, while HDL-bound protein contained both apoA-I and apoA-II, free protein released from HDL contained only apoA-I. The upward band shift observed for this free protein suggested oxidation (Figure 5D). To ascertain Met oxidation in this protein fraction, we used mass spectrometry to compare the molecular mass of intact apoA-I isolated from nHDL and of free apoA-I isolated by SEC from the Gdn-treated H2O2-oxidized TG-HDL (free fraction in Figure 5B). MALDI-TOF indicated an increase in the molecular mass by 48 Da, from 28087.5 Da in intact non-oxidized apoA-I to 28135.6 Da in the oxidized apoA-I released from TG-HDL (Figure 5D). Such an increase in mass corresponds to three oxygen atoms, consistent with the oxidation of all three apoA-I methionines (M86, M112, and M148). We conclude that the enrichment in TG makes HDL more susceptible to oxidation, and that mild oxidation of TG-HDL promotes preferential release of the Met-oxidized free apoA-I.
In addition to H2O2, we also used MPO/H2O2/Cl− system to oxidize TG-HDL. The results clearly show that, similar to H2O2, HDL oxidation by MPO/H2O2/Cl− augments protein release from TG-HDL, mainly in the form of free apoA-I monomer with oxidized methionines (supplemental Figure S2).
To probe the amyloid-forming properties of this free apoA-I, the protein was concentrated to 1 mg/ml, dialyzed against phosphate buffer at pH 6.0, and incubated at 37 °C for 24 h following published protocols [20]. After incubation, free Met-oxidized apoA-I formed visible precipitate, and native PAGE showed massive aggregation (Figure 6A). The aggregated protein showed a large increase in ThT fluorescence, indicating amyloid-like structural properties (Figure 6B). We conclude that free Met-oxidized apoA-I released from TG-HDL upon mild oxidation has increased amyloid-forming propensity.
Figure 6.
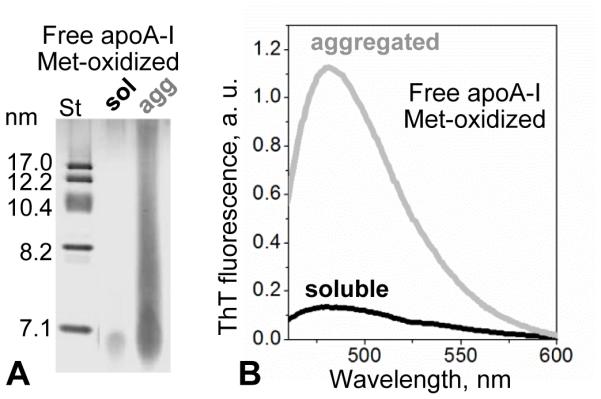
Aggregation of Met-oxidized free apoA-I released upon mild oxidation of TG-HDL. Free protein, which was isolated from mildly oxidized TG-HDL by SEC as described in Figure 2 legend, was incubated at pH 6 as described in part 3.2 following published protocols [20]. (A) SDS PAGE showed massive protein aggregation upon incubation. (B) Thioflavin T fluorescence showed a large increase in emission in the presence of this aggregated protein, suggesting amyloid formation.
3.3 Effects of TG enrichment on HDL remodeling by lipolytic and proteolytic enzymes
To determine the effects of TG enrichment on the enzymatic processing of HDL lipids, which is involved in metabolic HDL remodeling in plasma, we used LpL that preferentially hydrolyzes TG in the lipoprotein core to form di- and monoglycerides and free fatty acids [62]. This process depletes the apolar core of HDL, leading to the lipoprotein shrinking and release of the excess surface material in the form of apoA-I. Native PAGE in Figure 7A showed that HDL hydrolysis by LpL leads to a minimal decrease in the particle size. However, in TG-HDL this decrease in size was much more pronounced than in nHDL and was accompanied by a release of a greater fraction of free protein. This result is consistent with the previous report that HDL enriched in TG become better substrates for the lipoprotein and hepatic lipases [63,64].
Figure 7.

Effect of TG enrichment on the enzymatic hydrolysis of plasma HDL. (A) HDL remodeling by LpL. LpL hydrolyses TG in the HDL core leading to apoA-I release accompanied by a decrease in the HDL size (see parts 2.2 and 3.3 for detail). Native PAGE shows that this process is more advanced in TG-HDL as compared to nHDL. (B) Proteolysis of apoA-I on HDL by MMP-12 (see parts 2.2 and 3.2 for detail) monitored by immunoblotting for apoA-I shows more advanced protein cleavage on TG-HDL as compared to nHDL. Numbers on the right correspond to full-length apoA-I (28 kDa), its N-terminal 26 kDa fragment cleaved near the flexible hinge (G185, G168), and its ~14 kDa fragments produced upon apoA-I cleavage in the flexible central region near residue 130 [57].
To determine how TG enrichment affects the proteolytic susceptibility of apoA-I on HDL, we used MMP-12. In vivo, MMPs that are present in atherosclerotic lesions acts on HDL proteins, leading to their cleavage and subsequent degradation. We chose MMP-12 that was previously shown to cleave HDL-bound apoA-I [57], and used nHDL or TG-HDL as substrates. SDS PAGE followed by immunoblotting clearly showed that, compared to nHDL, apoA-I on TG-HDL was more susceptible to proteolysis (Figure 7B).
In summary, the enrichment of normal plasma HDL with TG augments the hydrolytic processing of HDL proteins an lipids. This conclusion is consistent with the previous studies of HDL hydrolysis [63-65] and with the destabilizing effect of TG enrichment on the HDL assembly described in part 3.1 above.
3.4 Human plasma HDL enriched in triolein show enhanced protein release and reduced stability
TG exchange upon HDL incubation with human VLDL can be influenced by various proteins and lipids that are present in VLDL. To clearly distinguish the effect of triglycerides, we used protein-free TO:POPC emulsions to determine how TO enrichment affects structural integrity of human plasma HDL. Unlike plasma lipoproteins whose core contains a mixture of asymmetrical TG molecules differing in the acyl chain length and unsaturation, TO is a symmetrical triglyceride containing three units of oleic acid. SEC data in Figure 8A showed that incubation of plasma HDL with TO emulsions to generate TO-enriched HDL (TO-HDL, Figure 8B) led to the release of a small but significant fraction of apoA-I under ambient conditions. This result, together with the data in Figure 1A, shows that HDL enrichment in TG promotes apoA-I release regardless of the presence of other proteins and lipids.
Figure 8.

Effect of the triolein enrichment on the protein dissociation and thermal remodeling of plasma HDL. (A) SEC (Superdex 16 XK column) of the incubation mixture containing CETP, TO:POPC emulsions and HDL (CETP-TO-HDL), which were prepared as described in Methods, and of nHDL shows dissociation of an apolipoprotein fraction from HDL upon enrichment in TO under ambient conditions. (B) Thin-layer chromatography of TO-HDL isolated from the incubation mixture shows an increased amount of TO as compared to nHDL. (C) Differential scanning calorimetry of TO-HDL that have been isolated from the CETP-TO-HDL incubation mixture and of nHDL. Heat capacity data, Cp(T), show that TO enrichment leads to low-temperature shifts in HDL fusion and rupture transitions (rupture temperatures are indicated by dotted lines). (D) Native PAGE of nHDL and TO-HDL, which have been heated to 90 °C to trigger lipoprotein fusion, shows more extensive lipoprotein fusion and protein dissociation in TO-HDL.
Thermal stability of nHDL and TO-HDL was compared by using DSC (Figure 8C). The results showed that the temperatures of HDL fusion and rupture decreased by about 15 °C upon HDL enrichment in TO. Next, nHDL and TO-HDL were heated in DSC experiments to 90 °C to trigger lipoprotein fusion, followed by a native PAGE analysis (Figure 8D). Compared to nHDL, TO-HDL showed more extensive protein release and particle fusion.
We conclude that, similar to TG-HDL, TO-HDL are more prone to protein release and particle fusion and rupture as compared to nHDL. Hence, protein release and HDL destabilization upon TG enrichment does not depend on the presence of VLDL, the exact type of TG, or the nature of the perturbation (thermal, chemical, oxidative, lipolytic or proteolytic). Rather, this is a general property of the TG-enriched plasma HDL.
3.5 Reconstituted spherical HDL show enhanced protein dissociation and reduced stability upon enrichment in TG
Plasma HDL form a heterogeneous population of particles containing a wide range of proteins and lipids. To test whether the effects of TG enrichment on the structural integrity of plasma HDL can be reproduced in a simple experimental model, we used reconstituted spherical HDL containing apoA-I as their sole protein and either CE alone or both CE and TG in the particle core. To this end, discoidal particles, which were reconstituted from human apoA-I, POPC and unesterified cholesterol, were used as substrates for LCAT. CE generated by LCAT moved to the core of the particles, converting them into spherical reconstituted HDL (rHDL). A subset of these rHDL was then enriched in TG by using CETP and VLDL; the reconstituted HDL containing both CE and TG in their core (TG-rHDL) were isolated and analyzed. Lipid composition in rHDL and in TG-rHDL was determined using enzymatic assays (Table S3) and was ascertained by thin layer chromatography (Figure S3). The particle size and morphology observed by EM were typical of human HDL with diameters, 8.5-11.5 nm (Figure S3). DSC data of reconstituted HDL showed a single endothermic transition (Figure 9A). In TG-rHDL this transition shifted to lower temperatures by ~10 °C and had lower enthalpy as compared to rHDL, indicating reduced stability of TG-rHDL. Next, the particles that have been heated in DSC experiments to 100 °C (i.e. past their structural transition) were analyzed by native PAGE, EM and SEC (Figure 9 B,C,D). Compared to rHDL, TG-rHDL showed more advanced protein dissociation and particle fusion. We conclude that, similar to plasma HDL, reconstituted HDL show increased apoA-I release and reduced stability of the lipoprotein assembly upon TG incorporation.
Figure 9.

Effect of TG enrichment on the protein dissociation and thermal remodeling of reconstituted spherical HDL. (A) Heat capacity data, Cp(T), of rHDL and TG-rHDL recorded by DSC during heating at a rate of 90 °C/h. The samples that have been heated to 100 °C in DSC experiments were analyzed by (B) SEC (Superose 6 10/300 GL column) and (C) native PAGE. Intact HDL, fused HDL and the dissociated lipid-poor/free protein are indicated.
3.6 Plasma HDL from diabetic patients show enhanced protein release and reduced stability
Human HDL described above were obtained from plasma of healthy donors, and enrichment of these HDL in TG partially mimicked conditions of high TG in vivo, such as the metabolic syndrome, diabetes or obesity. In vivo, such pathologic conditions can involve additional changes, including altered protein and lipid composition, post-translational modifications such as glycation, oxidation, etc. To assess the combined effect of these changes on the structural stability of human HDL, we used HDL isolated from pooled plasma of patients who were either normolipidemic and normoglycemic (NL-HDL) or have been diagnosed with diabetes mellitus (DM-HDL). Each group contained 25 patients; one series of experiments was performed. Biochemical analysis showed two major differences in the lipid and protein content of these HDL, including an ~30% increase in TG and an ~70% increase in apoC-III in DM-HDL as compared to NL-HDL (Table S1). The DSC data clearly showed a significant low-temperature shift by 4 °C in the fusion and rupture transitions of DM-HDL as compared to NL-HDL (Figure S4A). To ascertain that this shift involved changes in the protein release from HDL, the particles were heated in DSC experiments to 85 °C to trigger HDL fusion, followed by SEC and native PAGE analysis. The results clearly showed enhanced protein release from DM-HDL as compared to NL-HDL (Figure S4B, C). Furthermore, the kinetics of the copper-induced oxidation clearly showed that DM-HDL were more susceptible to oxidation than NL-HDL, as evident form a significant shift in the transition midpoint (Figure S4D). These studies of the patient-derived HDL further supports our in vitro finding that increased TG content augments HDL oxidation (Figure 5A).
3.7 Summary
Together, our studies consistently show that HDL particles with elevated TG content have reduced stability to various perturbations and show enhanced protein release. This effect was observed in plasma HDL that have been artificially enriched in either TG or TO (Figures 1-4), in plasma HDL from normolipidemic vis a vis diabetic patients (Figure S4), and in reconstituted HDL with controlled composition (Figure 9). TG-induced protein release was detected in TG- and in TO-enriched HDL under ambient conditions (Figures 1A and 8A), and was further enhanced upon thermal or chemical perturbation (Figures 2, 3, 4, 7). TG-rich HDL also showed increased susceptibility to oxidation (Figure 5), lipolysis and proteolysis (Figure 7), which is consistent with reduced structural stability of these particles. Importantly, we show that the protein released from the mildly oxidized TG-HDL is Met-oxidized apoA-I (Figure 5) that is prone to misfolding and aggregation in vitro (Figure 6). This Met-oxidized species is thought to contribute to the deposition of full-length WT apoA-I as fibrils in acquired amyloidosis in the oxidative environment of atherosclerotic plaques [19,20].
4. DISCUSSION
4.1 Conclusions
Our results clearly show that increasing the TG content in HDL per se promotes apolipoprotein release and HDL remodeling, fusion and rupture. These results are consistent with the previous in vitro studies by the teams of Rye and Sparks showing that enrichment in TG reduces HDL stability and induces conformational changes in apoA-I, which enhance the epitope exposure and proteolysis of apoA-I on HDL [65,66] as well as the release of apoA-I [52]. Protein dissociation from TG-rich HDL is further amplified by the action of oxidants (Figure 5) and by HDL-remodeling plasma factors, including proteases such as MMP-12 as well as lipases such as LpL that preferentially process TG-rich HDL and promote apolipoprotein release [63,64] (Figure 7). Together, these effects are thought to contribute to low plasma levels of HDL in patients with elevated TG, for example in the metabolic syndrome, diabetes and obesity.
Protein release from HDL induced by LpL probably results from shrinking of the HDL core upon TG hydrolysis, which increases the surface-to-volume ratio in HDL and generates excess surface material that dissociates in the form of lipid-poor/free proteins. In the absence of remodeling factors, the molecular underpinnings for the increased protein release and reduced structural stability of TG-rich HDL are less well-understood and are probably linked to the TG-induced conformational changes in HDL proteins such as those reported for apoA-I [65,66]. Furthermore, lipid-poor apolipoproteins such as apoA-I that are released from HDL can carry several lipid molecules; e.g. apoA-I monomer can carry up to 12 molecules of PC, 3-5 molecules of unesterified cholesterol and 2-5 molecules of cholesterol esters or TG ([10] and references therein), suggesting a direct interaction between apolipoproteins and the core lipids. The biochemical composition of the parent HDL particle probably influences the lipid composition in the apolipoprotein released from this particle, which may potentially influence the misfolding of this lipid-poor protein.
Lipid-poor/free apolipoproteins released from HDL are labile to proteolytic degradation as well as to misfolding and aggregation (reviewed in [1,3]); Met-oxidized free apoA-I is particularly labile to amyloid formation (Figure 6) [19,20]. Therefore, increased plasma levels of TG, which contribute to oxidative stress, are expected to shift the dynamic equilibrium from the HDL-bound to free apolipoproteins and thereby generate the protein precursor of amyloid. We speculate that this effect may be one of the contributing factors to the observed link between the severity of AA amyloidosis and elevated TG in conditions such as obesity, metabolic syndrome and diabetes [67,68].
4.2 Potential therapeutic implications and future studies
Conversely, decreasing TG levels in plasma is expected to enhance apolipoprotein retention on HDL and diminish the release of aggregation-prone apolipoproteins. Decreasing the generation of the amyloid protein precursor is the general therapeutic strategy pursued for various types of amyloidosis [4]. We hypothesize that TG-lowering approaches such as a low-fat diet, TG-reducing dietary supplements such as fish oil, and TG-lowering therapeutics may potentially help alleviate apolipoprotein deposition in diseases such as AApoAI, AApoAII and, perhaps, AA amyloidoses. These approaches probably hold more therapeutic potential for AApoAI and AApoAII since, unlike AA, these amyloidoses are not associated with the protein overproduction.
The most widely used TG-lowering drugs are fibrates. These PPARα agonists suppress apoCIII gene transcription and have recently been proposed as potential therapeutics for a new type of amyloidosis caused by a variant apoC-III [4]. Fibrates have complex effects on plasma apolipoproteins; for example, fenofibrate upregulates the expression of human and murine apoA-II and of human apoA-I, while murine apoA-I is down-regulated [69,70]. FIELD study to determine the effects of prolonged administration of fenofibrate to humans reported not only a significant reduction in plasma TG levels in patients under study but also an ~27% increase in plasma levels of apoA-II, with only marginal changes to apoA-I levels [71]. Therefore, although fibrates have a clear therapeutic potential for AApoCIII, they should be considered with caution for AApoAI, and should perhaps not be considered for AApoAII. Clearly, other TG-lowering therapies are needed whose prolonged administration to humans does not increase apoA-II (in case of AApoAII) and does not significantly change the expression levels of apoA-I, since an increase in apoA-I is undesirable in AApoAI while a decrease in apoA-I and HDL levels can adversely influence cardiovascular health. This goal could be achieved with some statins that, in parallel to cholesterol reduction, also decrease TG and are not reported to modify apoA-II levels. Future studies are necessary to determine a putative protective role for statins in apolipoprotein-related amyloidoses.
Dietary approaches to a TG reduction may hold promise as a strategy to delay the onset and/or progression of apolipoprotein amyloidosis. This idea is consistent with the studies of Higuchi’s team of the effects of dietary fats on apoA-II deposition in senescence-accelerated mice. These studies showed that the degree of unsaturation in dietary fats correlates directly with the rate of amyloid deposition [72], and that caloric restriction significantly reduces apoA-II amyloid deposition in these mice [73]. Also, in clinical studies of a cohort of patients with a metabolic syndrome, a 16-week low-calorie low-fat diet led to a significant decrease in the generation rate and in the fractional catabolic rate of apoA-II [74]. Together, these observations point to a potential therapeutic value of dietary fat modifications in apolipoprotein amyloidoses.
Future studies should specifically target the effects of TG levels on apolipoprotein deposition. A particularly useful model for these studies is the mouse model of human AApoAII developed by Benson and colleagues [28]. An important caveat in such studies is significant differences between the lipid metabolism in mice and men. For example, normal mice lack CETP. Therefore, as the first step, one could use a CETP-overexpressing mouse model of human AApoAII to feed these mice either a low-fat or a TG-rich diet to determine the effect of TG levels on the development and the progression of AApoAII. Similar in vivo studies of the effects of TG levels on AApoAI could be performed once the animal models of this disease become available.
Increasing the lipidation degree of apoE has been previously proposed to be of therapeutic value for Alzheimer’s disease [75]. Here we propose that increasing the lipidation degree of the major HDL apolipoproteins to shift their distribution from free (labile) to HDL-bound (protected) state has a therapeutic potential for amelioration of AApoAI, AApoAII and perhaps AA, and can probably be achieved via the decrease in plasma TG levels.
Supplementary Material
Highlights.
Apolipoproteins such as apoA-I, apoA-II and SAA can cause systemic amyloidoses
Binding to high-density lipoproteins protects these proteins from misfolding
Free apolipoproteins are intrinsically disordered and labile to misfolding
Increasing triglyceride levels in human or model HDL promotes protein release
Triglyceride-lowering therapies may potentially alleviate apolipoprotein amyloidoses
Acknowledgments
We are indebted to Donald L. Gantz for expert help with electron microscopy and to Michael Gigliotti for help with thin-layer chromatography. We thank professor Kerry-Ann Rye from the University of New South Wales, Australia, for her generous gift of CETP, and professor John Parks from Wake Forest University, USA, for his kind gift of LCAT. This work was supported by the National Institutes of Health (grant GM067260, SJ and OG) and by the Spanish Ministry of Health (ISCIII PI13/00364 and RIC-RD12/0042/0043) with FEDER funds (JL S-Q).
Abbreviations
- Apo
apolipoprotein
- AApoAI
apoA-I amyloidosis
- AApoAII
apoA-II amyloidosis
- AA
amyloid A
- SAA
serum amyloid A
- HDL
high-density lipoprotein
- VLDL
very-low density lipoprotein
- rHDL
reconstituted HDL
- TG
triglyceride
- TO
triolein
- CE
cholesterol ester
- CETP
cholesterol ester transfer protein
- UC
unesterified cholesterol
- nHDL
native single-donor HDL from plasma of healthy volunteers
- TG-HDL
nHDL enriched with TG
- TO-HDL
nHDL enriched with TO
- NL
normolipidemic
- DM
diabetes mellitus
- LCAT
lechithin:cholesterol acyltransfrease
- LpL
lipoprotein lipase
- MMP
matix metalloproteinase
- DSC
differential scanning calorimetry
- ThT
thioflavin T
- EM
electron microscopy
- SEC
size-exclusion chromatography
- PAGE
polyacrylamide gel electrophoresis
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References cited
- 1.Teoh CL, Griffin MD, Howlett GJ. Apolipoproteins and amyloid fibril formation in atherosclerosis. Protein Cell. 2011;2(2):116–127. doi: 10.1007/s13238-011-1013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kisilevsky R, Manley PN. Acute-phase serum amyloid A: perspectives on its physiological and pathological roles. Amyloid. 2012;19(1):5–14. doi: 10.3109/13506129.2011.654294. [DOI] [PubMed] [Google Scholar]
- 3.Das M, Gursky O. Amyloid-forming properties of human apolipoproteins: Sequence analyses and structural insights. Adv. Exp. Med. Biol. 2015;855:175–211. doi: 10.1007/978-3-319-17344-3_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valleix S, Verona G, Jourde-Chiche N, Nédelec B, Mangione PP, Bridoux F, Mangé A, Dogan A, Goujon JM, Lhomme M, Dauteuille C, Chabert M, Porcari R, Waudby CA, Relini A, Talmud PJ, Kovrov O, Olivecrona G, Stoppini M, Christodoulou J, Hawkins PN, Grateau G, Delpech M, Kontush A, Gillmore JD, Kalopissis AD, Bellotti V. D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat. Commun. 2016;7:10353. doi: 10.1038/ncomms10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nasr SH, Dasari S, Hasadsri L, Theis JD, Vrana JA, Gertz MA, Muppa P, Zimmermann MT, Grogg KL, Dispenzieri A, Sethi S, Highsmith WE, Jr., Merlini G, Leung N, Kurtin PJ. Novel type of renal amyloidosis derived from apolipoprotein-CII. J. Am. Soc. Nephrol. 2016 doi: 10.1681/ASN.2015111228. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5(1):15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 7.Phillips MC. New insights into the determination of HDL structure by apolipoproteins: Thematic review series: high density lipoprotein structure, function, and metabolism. J. Lipid Res. 2013;54(8):2034–2048. doi: 10.1194/jlr.R034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rye KA, Clay MA, Barter PJ. Remodelling of high density lipoproteins by plasma factors. Atherosclerosis. 1999;145(2):227–238. doi: 10.1016/s0021-9150(99)00150-1. [DOI] [PubMed] [Google Scholar]
- 9.Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2004;24:421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- 10.Jayaraman S, Cavigiolio G, Gursky O. Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: relevance to high-density lipoprotein biogenesis. Biochem J. 2012;442(3):703–712. doi: 10.1042/BJ20111831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyazaki O, Ogihara J, Fukamachi I, Kasumi T. Evidence for the presence of lipid-free monomolecular apolipoprotein A-1 in plasma. J. Lipid Res. 2014;55(2):214–225. doi: 10.1194/jlr.M041038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, Bellotti V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13(4):191–205. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 13.Ramella NA, Schinella GR, Ferreira ST, Prieto ED, Vela ME, Ríos JL, Tricerri MA, Rimoldi OJ. Human apolipoprotein A-I natural variants: Molecular mechanisms underlying amyloidogenic propensity. PLoS One. 2012;7(8):e43755. doi: 10.1371/journal.pone.0043755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mucchiano GI, Jonasson L, Häggqvist B, Einarsson E, Westermark P. Apolipoprotein A-I-derived amyloid in atherosclerosis. Its association with plasma levels of apolipoprotein A-I and cholesterol. Am. J. Clin. Pathol. 2001;115:298–303. doi: 10.1309/PJE6-X9E5-LX6K-NELY. [DOI] [PubMed] [Google Scholar]
- 15.Ramella NA, Rimoldi OJ, Prieto ED, Schinella GR, Sanchez SA, Jaureguiberry MS, Vela ME, Ferreira ST, Tricerri MA. Human apolipoprotein A-I-derived amyloid: Its association with atherosclerosis. PLoS One. 2011;6(7):e22532. doi: 10.1371/journal.pone.0022532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowczenio D, Dogan A, Theis JD, Vrana JA, Lachmann HJ, Wechalekar AD, Gilbertson JA, Hunt T, Gibbs SD, Sattianayagam PT, Pinney JH, Hawkins PN, Gillmore JD. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am. J. Pathol. 2011;179(4):1978–1987. doi: 10.1016/j.ajpath.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gursky O, Mei X, Atkinson D. Crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on the amyloid formation by the N-terminal segment. Biochemistry. 2012;51(1):10–18. doi: 10.1021/bi2017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Röcken C, Tautenhahn J, Bühling F, Sachwitz D, Vöckler S, Goette A, Bürger T. Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler. Thromb. Vasc. Biol. 2006;26(3):676–677. doi: 10.1161/01.ATV.0000201930.10103.be. [DOI] [PubMed] [Google Scholar]
- 19.Wong YQ, Binger KJ, Howlett GJ, Griffin MD. Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc. Natl. Acad. Sci. USA. 2010;107(5):1977–1982. doi: 10.1073/pnas.0910136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan GK, Witkowski A, Gantz DL, Zhang TO, Zanni MT, Jayaraman S, Cavigiolio G. Myeloperoxidase-mediated methionine oxidation promotes an amyloidogenic outcome for apolipoprotein A-I. J. Biol. Chem. 2015;290(17):10958–10971. doi: 10.1074/jbc.M114.630442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lepedda AJ, Cigliano A, Cherchi GM, Spirito R, Maggioni M, Carta F, Turrini F, Edelstein C, Scanu AM, Formato M. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis. 2009;203(1):112–118. doi: 10.1016/j.atherosclerosis.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel S, Chung SH, White G, Bao S, Celermajer DS. The “atheroprotective” mediators apolipoprotein A-I and Foxp3 are over-abundant in unstable carotid plaques. Int. J. Cardiol. 2010;145(2):183–187. doi: 10.1016/j.ijcard.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 23.Maiga SF, Kalopissis AD, Chabert M. Apolipoprotein A-II is a key regulatory factor of HDL metabolism as appears from studies with transgenic animals and clinical outcomes. Biochimie. 2014;96:56–66. doi: 10.1016/j.biochi.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 24.Gao X, Yuan S, Jayaraman S, Gursky O. Effect of apolipoprotein A-II on the structure and stability of human high-density lipoprotein: Implications for the role of apoA-II in HDL metabolism. Biochemistry. 2012;51(23):4633–4641. doi: 10.1021/bi300555d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benson MD, Liepnieks JJ, Yazaki M, Yamashita T, Hamidi Asl K, Guenther B, Kluve-Beckerman B. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72(3):272–277. doi: 10.1006/geno.2000.6499. [DOI] [PubMed] [Google Scholar]
- 26.Yazaki M, Liepnieks JJ, Barats MS, Cohen AH, Benson MD. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003;64(1):11–16. doi: 10.1046/j.1523-1755.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 27.Magy N, Liepnieks JJ, Yazaki M, Kluve-Beckerman B, Benson MD. Renal transplantation for apolipoprotein AII amyloidosis. Amyloid. 2003;10(4):224–228. doi: 10.3109/13506120309041739. [DOI] [PubMed] [Google Scholar]
- 28.Benson MD, Kalopissis AD, Charbert M, Liepnieks JJ, Kluve-Beckerman B. A transgenic mouse model of human systemic ApoA2 amyloidosis. Amyloid. 2011;18(Suppl. 1):32–33. doi: 10.3109/13506129.2011.574354011. [DOI] [PubMed] [Google Scholar]
- 29.Morizane R, Monkawa T, Konishi K, Hashiguchi A, Ueda M, Ando Y, Tokuyama H, Hayashi K, Hayashi M, Itoh H. Renal amyloidosis caused by apolipoprotein A-II without a genetic mutation in the coding sequence. Clin. Exp. Nephrol. 2011;15(5):774–779. doi: 10.1007/s10157-011-0483-4. [DOI] [PubMed] [Google Scholar]
- 30.Artl A, Marsche G, Lestavel S, Sattler W, Malle E. Role of serum amyloid A during metabolism of acute-phase HDL by macrophages. Arterioscler. Thromb. Vasc. Biol. 2000;20(3):763–772. doi: 10.1161/01.atv.20.3.763. [DOI] [PubMed] [Google Scholar]
- 31.Thompson JC, Jayne C, Thompson J, Wilson PG, Yoder MH, Webb N, Tannock LR. A brief elevation of serum amyloid A is sufficient to increase atherosclerosis. J. Lipid Res. 2015;56(2):286–293. doi: 10.1194/jlr.M054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Westermark GT, Fändrich M, Westermark P. AA amyloidosis: pathogenesis and targeted therapy. Annu. Rev. Pathol. 2015;10:321–344. doi: 10.1146/annurev-pathol-020712-163913. [DOI] [PubMed] [Google Scholar]
- 33.Merlini G, Garceau D, Dember LM, Sablinski T, Lachmann H, Berk JL, Obici L. A clinical Phase 3 confirmatory trial of Kiacta™ in the treatment of AA amyloidosis; The XVth International Symposium on Amyloidosis; Uppsala, Sweden. 2016. p. 60. Abstract O48. [Google Scholar]
- 34.Dember LM, Hawkins PN, Hazenberg BP, Gorevic PD, Merlini G, Butrimiene I, Livneh A, Lesnyak O, Puéchal X, Lachmann HJ, Obici L, Balshaw R, Garceau D, Hauck W, Skinner M. Eprodisate for the treatment of renal disease in AA amyloidosis. N. Engl. J. Med. 2007;356(23):2349–2360. doi: 10.1056/NEJMoa065644. [DOI] [PubMed] [Google Scholar]
- 35.Safi W, Maiorano JN, Davidson WS. A proteolytic method for distinguishing between lipid-free and lipid-bound apolipoprotein A-I. J. Lipid Res. 2001;42(5):864–872. [PubMed] [Google Scholar]
- 36.Lee-Rueckert M, Kovanen PT. The mast cell as a pluripotent HDL-modifying effector in atherogenesis: from in vitro to in vivo significance. Curr. Opin. Lipidol. 2015;26(5):362–368. doi: 10.1097/MOL.0000000000000224. [DOI] [PubMed] [Google Scholar]
- 37.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. USA. 1996;93(7):2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gursky O, Atkinson D. High- and low-temperature unfolding of human high-density apolipoprotein A-2. Protein Sci. 1996;5(9):1874–82. doi: 10.1002/pro.5560050913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. J. Biol. Chem. 2002;277(52):50380–50385. doi: 10.1074/jbc.M204898200. [DOI] [PubMed] [Google Scholar]
- 40.Gursky O. Structural stability and functional remodeling of high-density lipoproteins. FEBS Lett. 2015;589(19):2627–2639. doi: 10.1016/j.febslet.2015.02.028. Pt A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J. Mol. Biol. 2003;328(1):183–192. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 42.Jayaraman S, Gantz DL, Gursky O. Effects of salt on the thermal stability of human plasma high-density lipoprotein. Biochemistry. 2006;45(14):4620–4628. doi: 10.1021/bi0524565. [DOI] [PubMed] [Google Scholar]
- 43.Handa D, Kimura H, Oka T, Takechi Y, Okuhira K, Phillips MC, Saito H. Kinetic and thermodynamic analyses of spontaneous exchange between high-density lipoprotein-bound and lipid-free apolipoprotein A-I. Biochemistry. 2015;54(4):1123–1131. doi: 10.1021/bi501345j. [DOI] [PubMed] [Google Scholar]
- 44.Gursky O. Hot spots in apolipoprotein A-II misfolding and amyloidosis in mice and men. FEBS Lett. 2014;588(6):845–850. doi: 10.1016/j.febslet.2014.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lund-Katz S, Murley YM, Yon E, Gillotte KL, Davidson WS. Comparison of the structural and functional effects of monomeric and dimeric human apolipoprotein A-II in high density lipoprotein particles. Lipids. 1996;31(11):1107–1113. doi: 10.1007/BF02524284. [DOI] [PubMed] [Google Scholar]
- 46.Jayaraman S, Gantz DL, Gursky O. Kinetic stabilization and fusion of apolipoprotein A-2:DMPC disks: comparison with apoA-1 and apoC-1. Biophys. J. 2005;88(4):2907–2918. doi: 10.1529/biophysj.104.055921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frame NM, Gursky O. Structure of serum amyloid A suggests a mechanism for lipoprotein binding and functions: SAA as a hub in macromolecular interaction networks. FEBS Letts. 2016;590(6):866–879. doi: 10.1002/1873-3468.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schumaker VN, Puppione DL. Sequential flotation ultracentrifugation. Methods Enzymol. 1986;128:151–170. doi: 10.1016/0076-6879(86)28066-0. [DOI] [PubMed] [Google Scholar]
- 49.Matz CE, Jonas A. Reaction of human lecithin cholesterol acyltransferase with synthetic micellar complexes of apolipoprotein A-I, phosphatidylcholine, and cholesterol. J. Biol. Chem. 1982;257(8):4541–4546. [PubMed] [Google Scholar]
- 50.Rye KA, Duong M, Psaltis MK, Curtiss LK, Bonnet DJ, Stocker R, Barter PJ. Evidence that phospholipids play a key role in pre-beta apoA-I formation and high-density lipoprotein remodeling. Biochemistry. 2002;41(41):12538–12545. doi: 10.1021/bi025998k. [DOI] [PubMed] [Google Scholar]
- 51.Rull A, Jayaraman S, Gantz DL, Rivas-Urbina A, Pérez-Cuellara M, Ordóñez-Llanos J, Sánchez-Quesada JL, Gursky O. Thermal stability of human plasma electronegative low-density lipoprotein: a paradoxical behavior of low-density lipoprotein aggregation. BBA Mol. Cell. Biol. Lipids. 2016;1861(9):1015–1024. doi: 10.1016/j.bbalip.2016.05.008. Pt A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rye KA, Jauhiainen M, Barter PJ, Ehnholm C. Triglyceride-enrichment of high density lipoproteins enhances their remodelling by phospholipid transfer protein. J. Lipid Res. 1998;39(3):613–622. [PubMed] [Google Scholar]
- 53.Martins IJ, Lenzo NP, Redgrave TG. Phosphatidylcholine metabolism after transfer from lipid emulsions injected intravenously in rats. Implications for high-density lipoprotein metabolism. Biochim. Biophys. Acta. 1989;1005:217–224. doi: 10.1016/0005-2760(89)90040-4. [DOI] [PubMed] [Google Scholar]
- 54.Jayaraman S, Gantz DL, Gursky O. Effects of oxidation on the structure and stability of human low-density lipoprotein. Biochemistry. 2007;46(19):5790–5797. doi: 10.1021/bi700225a. [DOI] [PubMed] [Google Scholar]
- 55.Hurst JK, Barrette WC., Jr. Leukocytic oxygen activation and microbicidal oxidative toxins. Crit. Rev. Biochem. Mol. Biol. 1989;24:271–328. doi: 10.3109/10409238909082555. [DOI] [PubMed] [Google Scholar]
- 56.Cavigiolio G, Jayaraman S. Proteolysis of apolipoprotein A-I by secretory phospholipase A2: a new link between inflammation and atherosclerosis. J. Biol. Chem. 2014;289(14):10011–10023. doi: 10.1074/jbc.M113.525717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindstedt L, Saarinen J, Kalkkinen N, Welgus H, Kovanen PT. Matrix metalloproteinases-3, -7, and -12, but not -9, reduce high density lipoprotein-induced cholesterol efflux from human macrophage foam cells by truncation of the carboxyl terminus of apolipoprotein A-I. Parallel losses of pre-beta particles and the high affinity component of efflux. J. Biol. Chem. 1999;274(32):22627–2234. doi: 10.1074/jbc.274.32.22627. [DOI] [PubMed] [Google Scholar]
- 58.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 59.Bergmeyer, HU. Metabolites 3: Lipids, Amino Acids and Related Compounds. In: Bergmeyer J, Grassl M, editors. Methods of Enzymatic Analysis. VIII. VCH Weinheim; Germany: 1985. [Google Scholar]
- 60.Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 1978;87(1):206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- 61.Anantharamaiah GM, Hughes TA, Iqbal M, Gawish A, Neame PJ, Medley MF, Segrest JP. Effect of oxidation on the properties of apolipoproteins A-I and A-II. J. Lipid Res. 1988;29(3):309–318. [PubMed] [Google Scholar]
- 62.Lowe ME. The triglyceride lipases of the pancreas. J. Lipid Res. 2002;43(12):2007–2016. doi: 10.1194/jlr.r200012-jlr200. [DOI] [PubMed] [Google Scholar]
- 63.Clay MA, Newnham HH, Barter PJ. Hepatic lipase promotes a loss of apolipoprotein A-I from triglyceride-enriched human high density lipoproteins during incubation in vitro. Arterioscler. Thromb. 1991;11(2):415–422. doi: 10.1161/01.atv.11.2.415. [DOI] [PubMed] [Google Scholar]
- 64.Barrans A, Collet X, Barbaras R, Jaspard B, Manent J, Vieu C, Chap H, Perret B. Hepatic lipase induces the formation of pre-beta 1 high density lipoprotein (HDL) from triacylglycerol-rich HDL2. A study comparing liver perfusion to in vitro incubation with lipases. J. Biol. Chem. 1994;269(15):11572–1157. [PubMed] [Google Scholar]
- 65.Sparks DL, Davidson WS, Lund-Katz S, Phillips MC. Effects of the neutral lipid content of high density lipoprotein on apolipoprotein A-I structure and particle stability. J. Biol. Chem. 1995;270:26910–26917. doi: 10.1074/jbc.270.45.26910. [DOI] [PubMed] [Google Scholar]
- 66.Braschi S, Coffill CR, Neville TA, Hutt DM, Sparks DL. Effect of acylglyceride content on the structure and function of reconstituted high density lipoprotein particles. J. Lipid. Res. 2001;42(1):79–87. [PubMed] [Google Scholar]
- 67.van der Heijden RA, Bijzet J, Meijers WC, Yakala GK, Kleemann R, Nguyen TQ, de Boer RA, Schalkwijk CG, Hazenberg BP, Tietge UJ, Heeringa P. Obesity-induced chronic inflammation in high fat diet challenged C57BL/6J mice is associated with acceleration of age-dependent renal amyloidosis. Sci. Rep. 2015;5:16474. doi: 10.1038/srep16474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jang WY, Jeong J, Kim S, Kang MC, Sung YH, Choi M, Park SJ, Kim MO, Kim SH, Ryoo ZY. Serum amyloid A1 levels and amyloid deposition following a high-fat diet challenge in transgenic mice overexpressing hepatic serum amyloid A1. Appl. Physiol. Nutr. Metab. 2016;41(6):640–648. doi: 10.1139/apnm-2015-0369. [DOI] [PubMed] [Google Scholar]
- 69.Berthou L, Duverger N, Emmanuel F, Langouët S, Auwerx J, Guillouzo A, Fruchart JC, Rubin E, Denèfle P, Staels B, Branellec D. Opposite regulation of human versus mouse apolipoprotein A-I by fibrates in human apolipoprotein A-I transgenic mice. J. Clin. Invest. 1996;97(11):2408–2416. doi: 10.1172/JCI118687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Staels B, Auwerx J. Regulation of apo A-I gene expression by fibrates. Atherosclerosis. 1998;137(Suppl):S19–S23. doi: 10.1016/s0021-9150(97)00313-4. [DOI] [PubMed] [Google Scholar]
- 71.Ong KL, Rye KA, O'Connell R, Jenkins AJ, Brown C, Xu A, Sullivan DR, Barter PJ, Keech AC, FIELD study investigators Long-term fenofibrate therapy increases fibroblast growth factor 21 and retinol-binding protein 4 in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2012;97(12):4701–4708. doi: 10.1210/jc.2012-2267. [DOI] [PubMed] [Google Scholar]
- 72.Umezawa M, Higuchi K, Mori M, Matushita T, Hosokawa M. Effect of dietary unsaturated fatty acids on senile amyloidosis in senescence-accelerated mice. J. Gerontol. A. Biol. Sci. Med. Sci. 2009;64(6):646–652. doi: 10.1093/gerona/glp047. [DOI] [PubMed] [Google Scholar]
- 73.Sawashita J, Li L, Liu Y, Ding X, Yang M, Xu Z, Higuchi K. Caloric restriction reduces the systemic progression of mouse AApoAII amyloidosis; The XVth International Symposium on Amyloidosis; Uppsala, Sweden. 2016. p. 440. Abstract PC96. [Google Scholar]
- 74.Ng TW, Chan DC, Barrett PH, Watts GF. Effect of weight loss on HDL-apoA-II kinetics in the metabolic syndrome. Clin. Sci. (Lond) 2009;118(1):79–85. doi: 10.1042/CS20090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirsch-Reinshagen V, Burgess BL, Wellington CL. Why lipids are important for Alzheimer disease? Mol. Cell. Biochem. 2009;326(1-2):121–129. doi: 10.1007/s11010-008-0012-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.