

Abstract
Specific spontaneous heritable neurodegenerative diseases have been associated with lower serum and cerebrospinal fluid α-tocopherol (α-TOH) concentrations. Equine neuroaxonal dystrophy (eNAD) has similar histologic lesions to human ataxia with vitamin E deficiency caused by mutations in the α-TOH transfer protein gene (TTPA). Mutations in TTPA are not present with eNAD and the molecular basis remains unknown. Given the neuropathologic phenotypic similarity of the conditions, we assessed the molecular basis of eNAD by global transcriptome sequencing of the cervical spinal cord. Differential gene expression analysis identified 157 significantly (FDR<0.05) dysregulated transcripts within the spinal cord of eNAD-affected horses. Statistical enrichment analysis identified significant downregulation of the ionotropic and metabotropic group III glutamate receptor, synaptic vesicle trafficking and cholesterol biosynthesis pathways. Gene co-expression analysis identified one module of upregulated genes significantly associated with the eNAD phenotype that included the liver X receptor (LXR) targets CYP7A1, APOE, PLTP and ABCA1. Validation of CYP7A1 and APOE dysregulation was performed in an independent biologic group and CYP7A1 was found to be additionally upregulated in the medulla oblongata of eNAD horses. Evidence of LXR activation supports a role for modulation of oxysterol-dependent LXR transcription factor activity by tocopherols. We hypothesize that the protective role of α-TOH in eNAD may reside in its ability to prevent oxysterol accumulation and subsequent activation of the LXR in order to decrease lipid peroxidation associated neurodegeneration.
Keywords: cholesterol, RNA-sequencing, vitamin E
Introduction
Ataxia with vitamin E deficiency (AVED) is due to genetic mutations in the tocopherol (alpha) transfer protein gene (TTPA). Equine neuroaxonal dystrophy (eNAD) is a spontaneous neurodegenerative disease with similar clinicopathologic features to AVED. Clinical signs of eNAD include ataxia (i.e., incoordination) and proprioceptive deficits. Histologic features of eNAD include axonal swellings, or spheroids, localized to the caudal medulla oblongata and spinal cord (1); similar to histologic lesions observed with AVED (2). In more severely affected horses, the disease is often termed equine neuroaxonal dystrophy/equine degenerative myeloencephalopathy (eNAD/EDM) and histologic lesions include axonal loss and demyelination of specific neuroanatomic tracts within the cervicothoracic spinal cord (1). Equine NAD develops in genetically predisposed foals maintained on an α-tocopherol (α-TOH) deficient diet during the first year of life (3). Similar to AVED, the development of clinical signs can be prevented by early long-term α-TOH supplementation (1, 4). Mutations in TTPA are not present with eNAD (5) and the molecular basis remains unknown. While the horse may seem an unlikely model for humans, their lifespan (~ 30 years), length of their axons and documented similarities in clinical disease may provide unique insights into the molecular mechanism by which α-TOH deficiency impacts neurodegeneration.
To date, candidate gene approaches and genome-wide association studies have not identified a putative functional variant or chromosomal locus for eNAD (5, 6). The overall goal of this study was to further characterize the degree of α-TOH deficiency in eNAD-affected horses and identify differentially expressed genes and pathways in the central nervous system (CNS) by global transcriptome sequencing. Based on comparative phenotypes across species, we hypothesized that the most relevant pathways would include those related to vitamin E transport or metabolism, which is intimately tied to cholesterol homeostasis based on shared biochemical properties (7). Additionally, vitamin E is the major lipophilic antioxidant to protect against formation of cholesterol oxidation products (oxysterols) that can result in neurodegeneration (8).
Materials and Methods
Subjects
All animal procedures were approved by the University of California-Davis and University of Minnesota Institutional Animal Care and Use Committees and owners’ consent was obtained for all horses. Over a period of 8 years, samples from 22 post-mortem confirmed eNAD and 21 unaffected horses were collected. All horses were donated by owners for the purpose of this study. Biologic samples (i.e. serum, cerebrospinal fluid [CSF], tissue from liver, spinal cord and medulla oblongata) were available on subsets of eNAD affected and unaffected horses (Table A.1).
α-TOH Concentrations
Alpha-tocopherol (α-TOH) concentrations were assessed in the serum (n=21 eNAD-affected, n=12 unaffected), CSF (n=17 eNAD-affected, n=8 unaffected), liver (n=18 eNAD-affected, n=11 unaffected) and spinal cord (n=5 eNAD-affected, n=7 unaffected) of phenotyped horses (Table A.1). These horses were selected to be age matched (≤ 3 years of age) with samples that had been collected prior to (serum) or immediately following (CSF and liver) euthanasia. Spinal cord tissue was prioritized for RNA-sequencing and assessment of cholesterol concentrations and therefore spinal cord samples from the same location (i.e. cervical vertebrate 1) for assessment of α-TOH concentrations were only available from a limited subset of horses. Remaining spinal cord tissue was used for a complete histologic evaluation with eNAD horses confirmed as previously described (1). Unaffected horses were euthanized for reasons other than neurologic disease and a full neuropathologic evaluation was performed. Alpha-TOH concentrations were assessed as previously described (1, 9) and concentrations compared using a Mann-Whitney test with significance set at P<0.05.
RNA-sequencing
Horses selected for RNA-sequencing were between 1–2 years of age and matched for sex. Transcriptome sequencing of the caudal medulla oblongata was performed on 4 eNAD-affected Quarter Horses (n=2 male, n=2 female; n=1 classified as eNAD and n=3 classified as eNAD/EDM at post-mortem (1)) and 4 unaffected Quarter Horses (n=2 male, n=2 female). Subsequently, transcriptome sequencing of the cervical spinal cord was subsequently performed on 5 severely affected horses (i.e. classified as eNAD/EDM on post-mortem examination; n=3 Quarter Horses, n=1 Hanoverian/Thoroughbred, n=1 Shire; n=3 males, n=2 female; 1–2 y of age, median 1.5 y) and 5 unaffected horses (n=3 Quarter Horses, n=1 Percheron, n=1 Pony of the Americas; n=2 males, n=3 females; 0.5–2 y of age, median=1.5 y). Based on phenotypic criteria (i.e. more severely affected horses were available at the time of spinal cord sequencing) and tissue availability, 5 horses (2 eNAD/EDM-affected and 3 unaffected) had transcriptome sequencing performed on both tissues (Table A.1). None of the affected horses were related within 3 generations. Two of the unaffected Quarter horses used for both medulla oblongata and spinal cord sampling (Unaffected 1 and Unaffected 2) were half-siblings.
Nervous Tissue Sampling
All clinically suspect eNAD-affected horses were euthanized with an overdose of pentobarbital (>100 mg/kg IV) and a full post-mortem examination was performed. Samples of caudal medulla oblongata were collected immediately caudal to the level of the obex and flash-frozen in liquid nitrogen. Samples of the spinal cord were collected at the level of cervical vertebrae 1, cross-sectioned and flash-frozen in liquid nitrogen. Dorsal root ganglia (DRG) were collected and frozen separately on only a subset of horses. All tissues for RNA preparation were collected within 3 hours of euthanasia. The remaining brain and spinal cord were formalin-fixed and a complete histologic evaluation was performed. A moderate phenotype (eNAD) or a severe phenotype (eNAD/EDM) was diagnosed with histologic lesions as previously described (1) (Table A.1). Unaffected horses were euthanized for reasons other than neurologic disease (pseudohermaphroditism, type I polysaccharide storage myopathy, lameness) and samples were collected in an identical manner. As expected for a primary myopathy associated with a glycogenosis (10), the unaffected horse affected with type 1 polysaccharide storage myopathy did not have any evidence of neuropathic muscle atrophy or neurodegeneration.
RNA Isolation and Quality Control
RNA samples isolated from the region of the lateral accessory cuneate nucleus of the caudal medulla oblongata were utilized for RNA-seq analysis based on histologic evidence of spheroids, or axonal swellings in this region of eNAD-affected horses (1). Based on our additional work determining that the nuclei for the affected neurons in eNAD originate in the dorsal root ganglia of the cervical spinal cord and terminate in the caudal medulla oblongata, (11) transcriptome sequencing was subsequently performed in the spinal cord, at the level of cervical vertebrae 1, in 5 eNAD-affected and 5-unaffected horses. For all samples, total RNA was extracted using TRIzol reagent (Thermofisher, Wilmington, DE, USA). The resulting amount of RNA and integrity scores are included in Table A.2. For RT-qPCR, RNA was washed and eluted on columns (Direct-zol™ RNA MiniPrep Plus, Zymo, Irvine, CA) and treated with TURBO DNase (Thermofisher, Wilmington, DE, USA) according to manufacturer’s instructions. For RNA-seq, in an effort to include non-polyadenlyated long non-coding RNAs in the sequencing, ribosomal RNA was depleted (Ribo-Zero, Illumina, San Diego, US). Quantification and quality of RNA, along with degree of rRNA contamination, was assessed using the Pico chip on the Agilent Bioanalyzer 2100 (Santa Clara, CA, USA), with a RNA integrity number (RIN) ≥ 7. Ribosomal RNA depletion was successful in all samples (amount of remaining rRNA contamination 0–2.6%; mean 2%).
RNA-Sequencing and Exploratory Data Analysis
Strand-specific libraries (TruSeq Stranded Total RNA Library pre kit, San Diego, US) underwent next-generation sequencing (100-bp pair-end sequences with an Illumina HiSeq 2000, San Diego, US) at a targeted 20 million reads/sample across one lane. After quality control trimming with trimmomatic using a sliding window of 3 and quality score minimum of 28 (12), these sequences were aligned and reads quantified using two pseudoalignment methods for transcript quantification using both salmon (13) and kallisto (14) with 30 bootstraps. Gene annotation files included Ensembl (http://www.ensembl.org/info/data/ftp/index.html) and a custom annotation file (https://github.com/drtamermansour/horse_trans). RNA-seq was subsequently performed as described above on the spinal cord at the level of cervical vertebrae 1 in 5 eNAD-affected and 5 unaffected horses (Table A.2), with library preparation and sequencing similar to medulla oblongata samples.
Differential Gene Expression and Pathway Analysis
Ensembl Annotation
Based on the recommendation to use >2 different statistical tests to evaluate differential gene expression in RNA-seq studies (15), differential gene expression was evaluated using two pipelines. For Analysis A, following quantification of reads with salmon using the Ensembl gene annotation for EquCab2.0, read counts were imported into edgeR, transcripts were filtered by 1 count per million in at least 5 libraries (i.e. 50% of the samples), normalized and a multidimensional scaling (MDS) plot was evaluated. Within the spinal cord samples, differentially expressed genes were identified using the Exact Test in edgeR (16) on the 5 eNAD-affected horses and 5 unaffected horses, with a false discovery rate set at <0.05. For Analysis B, following pseudoalignment and quantification with kallisto (14) and using the Ensembl gene annotation for EquCab2.0, transcripts per million (TPM) were filtered by 1 count per million in at least 5 libraries and subjected to the Wald Test using sleuth (https://github.com/pachterlab/sleuth), with a false discovery rate set at <0.05.
Custom Annotation
As the EquCab2.0 gene annotation is incomplete, two additional analyses were performed using a publically available custom annotation for the horse (https://github.com/drtamermansour/horse_trans) (Analysis C: salmon/edgeR (16) and Analysis D: kallisto (14)/sleuth). For the four Analyses (A–D), a statistical enrichment test, using all transcripts and their associated log2fold values (edgeR) or beta values (sleuth), was performed using PANTHER-Pathway (http://pantherdb.org/). Reported P values were Bonferroni adjusted (PBonferroni).
RT-PCR for LOC10052888
To confirm the alternate transcript LOC10052888 in equine spinal cord tissue, primers were designed from RNA-seq data spanning the novel exon 1 of LOC10052888 and the annotated exon 1 of CYP7A1 (Table A.3). RNA from 2 eNAD-affected and 2 unaffected horses used in the RNA-seq experiment was reverse-transcribed using SuperScript® III (Thermofisher, Wilmington, DE, USA), PCR performed and the resulting 151 bp amplicon sequenced via Sanger sequencing in both eNAD-affected and unaffected horses.
Gene Co-expression Analysis with Weighted Correlation Network Analysis (WGCNA) and Pathway Analyses
To confirm our findings and further evaluate gene networks within the same population of n=5 eNAD-affected and n=5 unaffected horses, gene co-expression analysis with weighted correlation network analysis (WGCNA) was performed (17). Correlation networks are constructed on the basis of correlations between quantitative measurements and can be used to find clusters (or modules) of highly correlated genes for relating to external sample traits. Using the normalized filtered Ensembl transcripts from Analysis A (18,681 transcripts), a soft threshold power of 14 was estimated to perform a signed network analysis. Protein-protein interactions were evaluated using STRING v10 (18). Genes within associated modules were evaluated for common pathways using the gene ontology enrichment analysis using PANTHER-Pathway (http://pantherdb.org/). Reported P values were Bonferroni adjusted (PBonferroni).
Quantitative real-time PCR Validation in an Independent Biologic Set: Spinal cord
Four LXR-targeted genes involved in cholesterol metabolism (CYP7A1, APOE, PLTP, and ABCA1) were selected for validation in an independent biologic replicate set of individual horses. A fifth LXR-targeted gene, TTPA (19), was assessed and confirmed via RT-qPCR to not be expressed in equine spinal cord tissue using liver as a positive control tissue. The alternate transcript of CYP7A1 (LOC10052888) was also included. Additional validation was performed on expression levels of oxidized lipoprotein receptor (OLR1), a gene responsible for degradation of oxidatively modified low density lipoprotein, which was significantly upregulated in Analysis A and clustered into the co-expression module associated with eNAD. Finally, gene expression of four genes (GPNMB, TREM2, PLD4, CTNND2) that were found to be differentially expressed across all four data analyses (i.e. Analyses A–D) were evaluated in this biologic replicate set.
The replicate set consisted of 7 post-mortem confirmed eNAD-affected horses (n=4 classified as severe eNAD/EDM and n=3 classified as moderate eNAD at post-mortem examination (1); age range 0.5–14 years; median 4 years; n=3 females, n=4 males) and 7 unaffected horses (age range 0.1–18 years; median 0.5 years; n=1 female, n=6 males) (Table A.1). Age was not significantly different between groups (Mann-Whitney test; P=0.46). For the 10 genes of interest and 2 previously validated reference genes (ACTB, HPRT1) (6), primers were designed to cover exons that were included in all of the reported transcript isoforms. Primers were designed in Primer3plus (20) and selected only if pairs spanned at least one intron, did not bind to regions with an identified SNP, produced a single product with In-Silico PCR, and showed a single BLAT search result. All primers were synthesized by Invitrogen (Carlsbad, CA, USA). The reverse transcription of the two-step RT-qPCR was performed using Superscript® III (Invitrogen) and random hexamers with 1.5μg total RNA. Quantitative PCR Reactions were performed in a 10μL reaction volume using the QIAGEN Rotor-Gene™ SYBR®Green PCR Kit (QIAGEN, Valencia, CA, USA). Primers are listed in Table A.3. Each tube contained 1:10 dilution of total converted cDNA, and 1μM final primer concentration for each forward and reverse primer. PCR was performed on a Rotor-Gene Q 72-well thermocycler (QIAGEN, Valencia, CA, USA) as follows: 5 minutes at 95°C; 40 cycles of 5 seconds at 95°C and 10 seconds at 60°C; melt curve ramping from 60°C to 95°C, rising by 1°C at each step. Each reaction was run in triplicate and each run included a no-template control. Efficiencies for each gene were calculated at 90–110%. Relative quantitation of gene expression was calculated by comparative threshold cycle method (2−ΔΔCt) using the Ct of the housekeeping gene with the lowest variance. Data were analyzed using the non-parametric Mann-Whitney test due to the small sample size. Correction for multiple testing was performed at 10% FDR based on the small number of multiple comparisons performed.
To confirm the alternate transcript of CYP7A1, primers were designed from RNA-seq data spanning the novel exon 1 of LOC10052888 and the annotated exon 1 of CYP7A1. RT-PCR was performed on RNA from 2 eNAD-affected and 2 unaffected horses and the resulting 151 bp amplicon sequenced in both eNAD-affected and unaffected horses, confirming the alternate exon. The same primer set was then used for RT-qPCR in the independent sample set (Table A.3).
Quantitative real-time PCR: Caudal Medulla Oblongata
The 12 genes assessed above were also quantified in caudal medulla oblongata tissue from 10 eNAD-affected (n=6 classified as severe eNAD/EDM and n=4 classified as moderate eNAD at post-mortem examination (1); age range 0.5–14 years; median 2 years; n=4 females, n=6 males) and 9 unaffected horses (age range 0.1–18 years; median 1.5 years; n=2 female, n=7 males) (Table A.1). Age was not significantly different between groups (Mann-Whitney test; P=0.25). Primers for the genes of interest and 2 previously validated reference genes (ACTB, HPRT1) (6) were designed as described above (Table A.3). Reverse transcription, quantitative PCR reactions and data analyses were performed as described above.
Quantitative real-time PCR: Liver
Three LXR-targeted genes (CYP7A1, PLTP, and TTPA) were selected for further evaluation in liver tissue. Samples included 10 post-mortem confirmed eNAD-affected horses (n=9 classified as severe eNAD/EDM and n=1 classified as moderate eNAD; age range 1–2 years; median 1 year; n=3 females, n=7 males) and 9 unaffected horses (age range 0.5–13 years; median 2 years; n=6 females, n=3 males) (Table A.1). Age was not significantly different between groups (Mann-Whitney test; P=0.18). The 5 eNAD-affected and 5 unaffected horses used for RNA-seq of the spinal cord in this study were included in these samples. Primers for the genes of interest and 2 previously validated reference genes (ACTB, HPRT1) (6) were designed as described above (Table A.3). Reverse transcription, quantitative PCR reactions and data analyses were performed as described above.
Cholesterol Concentrations in Spinal Cord
Spinal cord samples for assessment of cholesterol and bile acid concentrations included 9 postmortem confirmed eNAD-affected horses (n=6 classified as severe eNAD/EDM and n=3 classified as moderate eNAD at post-mortem examination (1); age range 0.5–14 years; median 2 years; n=2 females, n=7 males) and 9 unaffected horses (age range 1–25 years; median 2 years; n=3 female, n=6 males) (Table A.1). Age was not significantly different between groups (Mann-Whitney test; P=0.43). Assessment of cholesterol and the two primary bile acids, cholic acid and chenodeoxycholic acid, were quantified by liquid chromatography-mass spectroscopy/mass-spectroscopy (LC-MS/MS), using modifications of a previously published method (21). Only traces of bile acids were identified in the spinal cord samples and therefore data were normalized for 10 mg. For cholic acid analysis, 8/10 eNAD-affected and 7/10 unaffected horses had concentrations below 1.0 nM, thereby precluding any further data analysis. For chenodeoxycholic acid (CDCA) analysis, 5/10 eNAD-affected and 5/10 unaffected horses had concentrations below 1.0 nM. Therefore, 5 eNAD-affected and 5 unaffected were used in the analysis. Reported P values were Bonferroni adjusted (PBonferroni).
Serum and Spinal Cord Oxysterol Concentrations
As a preliminary investigation, serum and spinal cord oxysterol concentrations (24S-hydroxycholesterol, 7-ketocholesterol and 7-hydroxycholesterol) from 3 eNAD affected and 3 unaffected horses were compared.
Materials
Optima LC/MS grade solvents (methylene chloride, chloroform, methanol, water, and formic acid) were purchased from Thermo Fisher Scientific. d7-7-Ketocholesterol was prepared as reported previously (22). 200 proof ethanol (USP Specs) from Decon Laboratories, Inc. was used. Potassium hydroxide (ACS certified) and sodium chloride (ACS certified) were purchased from Thermo Fisher Scientific.
Lipid extraction from tissues and fluids
Prior to lipid extraction, the internal standard, d7-7-ketocholesterol (500 ng), was added to each sample. For spinal cord, the tissues were homogenized in Folch solution (3mL, choloroform:methanol = 2:1) by a blade homogenizer. For serum (100 μL), 3 mL of Folch solution was added directly to each sample. NaCl aqueous solution (0.9%, 1 mL) was then added and the resulting mixture was briefly vortexed and centrifuged for 5 minutes. The lower organic phase was recovered and dried at room temperature using the speed vacuum (Thermo Fisher Savant), and then re-dissolved in methylene chloride (1 mL for tissue samples and 500 μL for CSF samples). For the serum samples, the extracts were dried under SpeedVac and the dried samples were then reconstituted in 2 mL of 2.5% KOH in 90% EtOH and incubated in a water bath at 55° C for 45 minutes. Another lipid extraction was performed on the solution from the base hydrolysis and the extracts were dried again using the SpeedVac. Each sample was reconstituted in methylene chloride (500 μL).
HPLC-APCI-MS/MS analyses of sterols and oxysterols
Analysis of cholesterol, sterols and oxysterols were performed by UHPLC-MS/MS using a triple quadrupole mass spectrometer (Sciex 6500) equipped with atmospheric pressure chemical ionization (APCI). For analysis, an appropriate amount of sample was transferred to an LC vial, dried under a stream of argon, and reconstituted in 90% methanol with 0.1% formic acid (for spinal cord, 50 μL was reconstituted in 50 μL; for serum 100 μL to 50 μL; for CSF, 100 μL to 50 μL). Reverse-phase chromatography was performed with the following conditions: C18 column (1.7um, 100 mm × 2.1 mm, Phenomenex Kinetex); flow rate, 0.4 mL/min; elution solvent, 90% methanol with 0.1% formic acid. MS conditions: spray voltage, 5000 V; curtain gas, 10 psi ion source gas, 20 psi; collision gas, high; entrance potential, 10 V; collision energy, 25 V; declustering potential, 80 V; temperature, 300° C. For MS analysis, selective reaction monitoring (SRM) was employed to monitor the dehydration process of the ion [M+H]+ or [M+H-H2O]+ as described previously (22, 23). The internal standard, d7-7-ketocholesterol, was used to quantify the analytes, 24-OH-Chol, 7-ketocholesterol, and 7-OH-Chol. SRM mass transitions are: d7-7-ketocholesterol m/z 408.3 → 390.3; 7-ketocholesterol, m/z 401.3 → 383.3; 7-OH-Chol, m/z 385.3 → 367.3; 24-OH-Chol, m/z 385.3 → 367.3. Ratios of oxysterol to cholesterol concentrations were used for quantification.
Data Analysis
Data were analyzed using the non-parametric Mann-Whitney test due to the small sample size with significance set at P<0.05.
Data Access
The RNA-sequencing data from this study has been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sta/) under accession number SUB1472272, Bioproject PRJNA318917.
Results
Systemic Deficiency of α-TOH in eNAD horses
Serum, CSF and hepatic α-TOH concentrations were significantly lower in eNAD-affected horses as compared to unaffected horses (Fig. 1A–C). Spinal cord α-TOH concentrations were not significantly different in eNAD-affected horses (Fig. 1D), however the sample size of eNAD horses was small. Serum α-TOH concentrations were significantly correlated with CSF (R2=0.35, P=0.002) and hepatic (R2=0.43, P=0.0002) but not spinal cord (R2=0.02, P=0.61) α-TOH concentrations.
Figure 1.

(A) Serum, (B) cerebrospinal fluid (CSF), (C) liver and (D) spinal cord α-tocopherol (α-TOH) concentrations of horses affected with equine neuroaxonal dystrophy (eNAD) and unaffected horses. * P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001 by Mann Whitney test. Data mean ± SEM for n=17–21 eNAD and n=8–12 unaffected (serum, CSF, liver) and n=5 eNAD and n=7 unaffected (spinal cord).
Spinal cord is the optimal tissue for transcript evaluation of eNAD
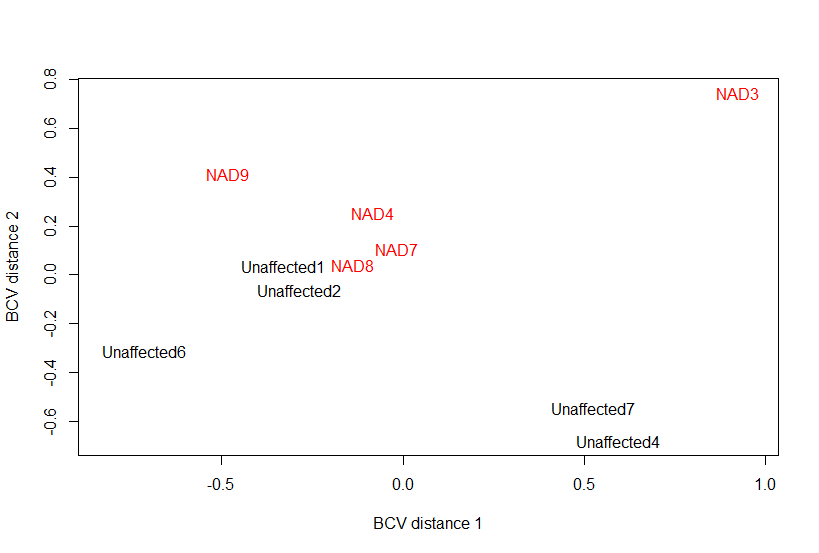
Multi-dimensional scaling plots revealed that the spinal cord (Fig A.1), but not medulla oblongata samples (Fig. A.2), segregated by disease phenotype along the biological coefficient of variation distance axes. When merging datasets from medulla oblongata and spinal cord, samples clustered by tissue rather than individual or disease phenotype (Fig. A.3). Therefore, spinal cord was the most appropriate tissue for further evaluation in eNAD. A summary of transcripts unique to the spinal cord and medulla oblongata from the custom transcriptome analysis is available in Table A.4.
Differentially expressed transcripts within the spinal cord of eNAD affected horses
Using the Ensembl EquCab2.0 gene annotation, 157 transcripts were identified as differentially expressed (DETs) with a false discovery rate < 0.05, with 19 significantly downregulated transcripts and 138 significantly upregulated transcripts (Table A.5). Comparison between pseudoalignment quantification approaches revealed similar results (Table A.6). When using the custom transcriptome, 44 transcripts were differentially expressed, with 7 downregulated and 37 upregulated (Table A.5). Comparison between pseudoalignment quantification approaches (Analysis D) yielded more DETs for Analysis D (Table A.6). The most highly downregulated gene in Analysis A (ARHGEF10L) was not downregulated in any other analysis and therefore most likely spurious.
Evidence of LXR-activation in eNAD spinal cord
STRING interaction networks of all significantly dysregulated transcripts revealed a centralized clustering around APOE and IL6 (Fig. 2). These are two genes are targets of LXR activation (APOE) or downregulation (IL6) (24). Two additional LXR-activated genes within the central STRING cluster, CYP7A1 and PLTP, were also significantly upregulated in eNAD (Table 1). Analysis using WGCNA resulted in 20 modules with no association with sex or age and only one module significantly associated with the eNAD phenotype (Fig. A.4). Within the associated module for eNAD (red; 622 transcripts, Table A.7), the significantly upregulated transcripts associated with eNAD included CYP7A1, APOE, and PLTP. ABCA1 clustered within the associated red module. Additionally, OLR1, which is responsible for internalizing and degrading oxidized low density lipoprotein (LDL) cholesterol, clustered in the red module and was significantly upregulated (Table 1).
Figure 2.

Protein-protein interaction networks (18) of differentially encoded proteins identified by RNA-sequencing of spinal cord from horses affected with equine neuroaxonal dystrophy. A total of 157 differentially expressed transcripts were identified. The thickness of the blue line represents confidence (thicker line=more confidence supporting the interaction). The most protein interactions were identified around IL6 and APOE. The genes encoding for these two proteins contain liver X nuclear receptor sites.
Table 1.
A selected list of transcripts with associated genes dysregulated in eNAD; WGCNA= gene co-expression analysis with weighted correlation network analysis.
| Pathway | Associated Gene | RNA-Seq Analysis A | WGCNA Module |
|---|---|---|---|
|
| |||
| LXR-target transcripts | CYP7A1 | 12.7-fold (PFDR=1.19 × 10−13)* | Red* |
| Activated | APOE | 1.92-fold (PFDR=0.02)* | Red* |
| PLTP | 2.28-fold (PFDR=0.0001)* | Red* | |
| ABCA1 | 1.5-fold (PFDR=0.3) | Red* | |
| ABCG5/8 | Not expressed in spinal cord | N/A | |
| TTPA | Not expressed in spinal cord | N/A | |
| IL6 | −8.3-fold (PFDR=0.002)* | Yellow | |
| Repressed | NPC1 | −1.1-fold (PFDR=1)# | Turquoise |
|
| |||
| Lipoprotein receptors | OLR1 | 4.5-fold (PFDR=0.015)* | Red* |
| LDLR | −1.65-fold (PFDR=0.45) | Turquoise | |
| VLDLR | −1.28-fold (PFDR=0.61) | Turquoise | |
| NPC1 | −1.1-fold (PFDR=1)# | Turquoise | |
| NPC2 | 1.6-fold (PFDR=0.11) | Red* | |
|
| |||
| Cathepsins (cholesterol uptake and trafficking through LXR-target transcripts) | CTSB | 2.5-fold (PFDR=0.0007)* | Red* |
| CTSL | 3.1-fold (PFDR=0.0004)* | Red* | |
| CTSZ | 2.9-fold (PFDR=0.0003)* | Red* | |
|
| |||
| Nuclear Receptor 4 family (Neuron-related orphan receptors) | NR4A1 | −2.6-fold PFDR=0.1) | Yellow |
| NR4A2 | −3.3-fold (PFDR=0.01)* | Yellow | |
| NR4A3 | −3.4-fold (PFDR=0.03)* | Yellow | |
|
| |||
| Central myelination markers | PLP | −1.4-fold (PFDR=0.82) | Turquoise |
| MBP | −2.67-fold (PFDR=1) | Red* | |
| MOBP | −1.08-fold (PFDR=1) | Blue | |
|
| |||
| Ion channels | TRPM1 | 6.02-fold (PFDR=1.3 × 10−6)* | Darkgreen |
| TRPV2 | 3.9-fold (PFDR=0.003)* | Darkgoldenrod4 | |
| KCTD12 | 2.27-fold (PFDR=0.003)* | Darkgreen | |
| KCNQ1 | 1.76-fold (PFDR=0.03)* | Coral2 | |
| HVCN1 | 3.64-fold (PFDR=0.03)* | Coral2 | |
| KCNJ4 | 3.27-fold (PFDR=0.037)* | Coral2 | |
significantly associated with eNAD phenotype.
Overlap in pathways
Gene ontology statistical enrichment analysis
Gene ontology enrichment analysis is a method to identify classes of genes or proteins that are overrepresented in a large set of genes or proteins, and may have an association with disease phenotypes. While many of the included genes may exhibit a large fold change with eNAD at a single gene level (i.e. fold change between 1–2), when evaluated as a pathway, a subset of genes within that pathway are overrepresented. Gene ontology enrichment analysis of eNAD transcripts from RNA-sequencing analysis revealed significant downregulation of ionotropic and glutaminergic receptor, synaptic vesicle trafficking and cholesterol biosynthesis pathways (Table 2).
Table 2.
Downregulated pathways in spinal cord of eNAD affected horses. PBonferroni=Bonferroni adjusted P values for gene ontology enrichment analysis, with the associated pathway identification numbers
| Pathway name (ID) | # genes in Pathway | PBonferroni | Selected downregulated transcripts (fold change) |
|---|---|---|---|
| Ionotropic glutamate receptor pathway (P00037) | 34 | 1.99 × 10−5 | Glutamate receptor Ionotropic, Kainate 3 (GRIK3, −1.77)# Glutamate receptor 1 (GRIA1; −1.3)# Metabotropic glutamate receptor 3 (GRM3; −1.48) Glutamate receptor, Ionotropic, NMDA1 (GRIN1; −1.81) Vesicle-associated membrane protein 2 (VAMP2; −1.13)# |
| Synaptic vesicle trafficking pathway (P05734) | 20 | 0.001 | Regulating synaptic membrane exocystosis protein (RIMS1; −1.35) Synaptotagmin 1 (SYT1; −1.53) Synapsin-2 (SYN2; −1.48) Protein UNC-13 Homolog C (UNC13C; −1.56) Synaptogamin-12 (SYT12; −2.35) Regulating synaptic membrane exocystosis protein (RIMS2; −1.32) Synaptotagmin 7 (SYT7; −1.48) |
| Metabotropic glutamate receptor group III pathway (P0039) | 45 | 0.01 | Glutamate receptor Ionotropic, Kainate 3 (GRIK3, −1.77)# Glutamate receptor 1 (GRIA1; −1.3)# Metabotropic glutamate receptor 5 (GRM5, −1.67) |
| Cholesterol biosynthesis (P00014) | 9 | 0.02 | Squalene Monooxygenase (SQLE, −1.40) Hydroxymethylglutaryl-CoA Synthase, cytoplasmic (HMGCS1, −1.48) Diphosphomevalonate decarboxylase (MVD, −1.22) |
denotes transcripts that overlap between the two pathways
Identification of a unique DET of CYP7A1
One unique transcript (TCONS_00194578), dysregulated in analyses using the custom transcriptome, was identified as an alternate transcript of CYP7A1, the top differentially expressed transcript. This alternate transcript created a putative exon ~40 kb upstream of the Ensembl annotated exon 1 of CYP7A1 (Fig. 3). This alternate transcript has been previously characterized at NCBI (www.ncbi.nlm.nih.gov/gene/) from aggregate filtered unique hits of RNA-seq data (NCBI Equus caballus Annotation Release 102; LOC10052888). The additional exon does not alter the open reading frame, with the start codon located in the originally annotated exon 1 of CYP7A1. This alternate transcript was confirmed with conventional PCR.
Figure 3.

Track from the UCSC genome browser (https://genome.ucsc.edu/) of EquCab2.0 at chr9:25,570,000–25,620,000 identifying the Ensembl annotation of CYP7A1 (dark red), human proteins mapped to EquCab2.0 (dark gray) and non-horse reference genes (lavender). The annotated exons of CYP7A1 are numbered 1–6. The two bright red custom tracks demonstrate a custom transcript defined by integration of horse RNA-seq data sets (available at https://github.com/drtamermansour/horse_trans). This transcript has been previously described (NCBI Equus caballus Annotation Release 102; LOC10052888). LOC10052888 was significantly upregulated in spinal cord tissue from horses affected with eNAD.
RT-qPCR validates differential expression of LXR-target genes in the CNS with eNAD
Of the 10 transcripts of interest that were dysregulated in the spinal cord RNA-seq experiment, 6 were differentially expressed in the spinal cord of eNAD-affected horses at an FDR cut off of 10% in the independent biologic replicate group and 3 were differentially expressed in the medulla oblongata (Table 3).
Table 3.
Quantitative real-time PCR validation of specific transcripts from RNA-seq in an independent biologic replicate set of spinal cord and medulla oblongata tissues. The Mann-Whitney test was used to determine statistical significance with P<0.05 with multiple correction testing performed at an FDR of <10% due to the small number of tests performed.
| Spinal cord | Medulla Oblongata | |||||
|---|---|---|---|---|---|---|
| Associated Gene | dCT (eNAD) | dCT (unaffected) | ddCt | Punadjusted value (CI) | ddCt | Punadjusted value (CI) |
| TREM2 | 5.86 | 7.21 | 2.56 | 0.0047 (0.11–2.32)* | 2.79 | 0.12 (−0.19–1.41) |
| CYP7A1 | 6.67 | 10.13 | 7.11 | 0.0047 (1.14–5.78)* | 3.14 | 0.006 (0.55–2.75)* |
| APOE | −0.60 | 0.14 | 1.66 | 0.017 (0.19–1.29)* | 1.25 | 0.86 (−1.23–0.95) |
| OLR1 | 8.71 | 11.8 | 8.50 | 0.03 (0.54–6.55)* | 3.48 | 0.049 (0.01–3.56) |
| LOC10052888 (Alternate transcript of CYP7A1) | 6.77 | 9.00 | 4.71 | 0.035 (0.02–3.93)* | 4.25 | 0.005 (0.73–3.45)* |
| GPNMB | 6.16 | 9.62 | 11.02 | 0.038 (0.1–6.28)* | 3.40 | 0.005 (0.6–2.92)* |
| PLD4 | 5.88 | 6.50 | 1.54 | 0.16 (−0.46–1.55) | 1.17 | 0.32 (−0.24–0.69) |
| CTNND2 | 2.73 | 3.20 | 1.39 | 0.21 (−0.24–1.17) | 2.78 | 0.15 (−0.63–3.58) |
| ABCA1 | 6.60 | 5.43 | 0.44 | 0.21 (−1.18–0.11) | 0.83 | 0.25 (−0.76–0.21) |
| PLTP | 4.19 | 4.62 | 1.35 | 0.69 (−1.14–2.55) | 1.68 | 0.25 (−0.18–0.84) |
significant at FDR<10%. dCT=relative abundance of transcripts compared to ACTB
RT-qPCR reveals no differential hepatic expression of LXR-target genes with eNAD
Three LXR-targeted transcripts were evaluated via RT-qPCR in liver tissue (CYP7A1, TTPA and PLTP) of eNAD affected and unaffected horses and none were differentially expressed.
Spinal cord cholesterol and bile acid concentrations do not differ in eNAD
Based on the significant upregulation of CYP7A1 in the spinal cord of eNAD affected horses and the role that CYP7A1 plays in the liver in the conversion of cholesterol to bile acids, spinal cord cholesterol and bile acid concentrations were assessed. There was no significant difference in spinal cord cholesterol concentrations (P=0.75) or chenodeoxycholic acid (CDCA) concentrations (P=0.31) in the spinal cord of eNAD-affected horses. Therefore, the upregulation of CYP7A1 observed in our spinal cord transcriptome dataset does not appear to result in increased bile acid production at a tissue-wide level.
A trend towards increased oxysterol concentrations in spinal cord and serum of eNAD horses
A trend towards increased serum and spinal cord 7-ketocholesterol and 7-hydroxycholesterol concentrations was observed in eNAD horses compared to unaffected horses (Fig. 4). There was no difference in serum or spinal cord 24S-hydroxycholesterol concentrations.
Figure 4.

(A) Spinal cord and (B) serum oxysterol concentrations from eNAD affected and unaffected horses. Although significance was not achieved in this small sample set, a trend towards higher 7-ketocholesterol and 7-hydroxycholesterol concentrations was observed in spinal cord tissue from eNAD horses. Data as mean ± SEM, n=3 per group. 24-OH-Chol=24S-hydroxycholesterol, 7-OH_Chol=7-hydroxycholesterol.
Discussion
Role of α-TOH deficiency in eNAD
While strong evidence for the role of α-TOH deficiency in eNAD has been previously reported (4, 25, 26), this is the first study to document significantly lower serum, CSF and hepatic α-TOH concentrations in post-mortem confirmed eNAD horses (Fig. 1). While these concentrations reflect one time point, we have recently published a longitudinal study examining 14 foals maintained on a diet deficient in α-TOH from birth through 8 months of age (9). Ten of the foals in that study were out of healthy dams and sires and four were out of eNAD dams (i.e. demonstrated clinical signs of disease and had previously produced eNAD foals). Only these four genetically susceptible foals developed clinical and histologic evidence of eNAD. In these foals, significantly lower CSF α-TOH concentrations in eNAD foals, as compared to heathy foals maintained on the same α-TOH deficient diet, were found through 120 days of age (9). Based on the evidence that an α-TOH deficiency results in neurodegeneration in AVED and overlapping clinical and neuropathologic features of the α-TOH deficient phenotypes with AVED, Ttpa−/− mice and eNAD, there is likely a cause-and-effect role of α-TOH in eNAD.
Downregulation of glutamate receptor and synaptic vesicle trafficking in eNAD
Equine NAD affected horses demonstrate similar clinicopathologic features to AVED, however, we have excluded the AVED causative gene TTPA as a candidate gene for eNAD (5) and genome-wide association studies have yet to find alternative loci (6). RNA-sequencing was therefore performed to gain insight in the molecular pathophysiology of eNAD.
Through our investigation of the global gene expression profile of spinal cord in eNAD, we have identified an overall downregulation of transcripts in the glutamate receptor and synaptic vesicle trafficking pathways. Glutamate is the principal excitatory neurotransmitter in the mammalian center nervous system and extracellular concentrations are tightly regulated through glutamate transporters. Both ionotropic and metabotropic glutamate group III receptors were downregulated in eNAD and both modulate synaptic plasticity (27). With only one time point in the progression of eNAD assessed in this study, we are unable to determine whether the observed downregulation of these pathways is a cause or consequence of eNAD as histologic lesions of eNAD include axonal loss, consistent with loss of synapses (1). However, many of the transcripts in these pathways, including synaptobrevin (VAMP2), the ionotropic glutamate receptor AMPA type 1 (GRIA1) and members of the syaptotagmin families (SYT1, SYT12 and SYT7), were also downregulated in cerebral cortex of Ttpa−/− mice (28) (Table 4). Therefore, there appears to be an overall suppression of genes for synaptic plasticity in the CNS of both α-TOH deficient phenotypes.
Table 4.
Comparison of gene expression profiles of α-TOH deficient neurodegenerative phenotypes. All mouse and rat assessments were performed using microarrays (Affymetrix Mu74Av2 and Affymetrix GeneChip Rat Expression Array 230A, respectively). Dietary vitE provided as all-rac-α-tocopheryl acetate to mice and rats. Transcripts are included if fold change > or < 2 (microarray) or pathways/modules were significant (RNA-seq)
| Reference | (29) | (28) | (29) | (30) | Current study |
|---|---|---|---|---|---|
|
| |||||
| Genotype | Ttpa−/− | Ttpa−/− | Ttpa−/− | Wild-type Wistar rats | eNAD |
| Age | Birth | 3–4 m | 1 y | 14 mo | 1–2 y |
| Dietary vitE | 35 IU/kg | 35 IU/kg | 35 IU/kg | <1.25 mg/kg vs 5 mg/kg | Variable |
| Tissue | Cerebral cortex | Cerebral cortex | Cerebral cortex | Cerebral cortex | Spinal cord |
|
| |||||
| Downregulated: Synaptic vesicle trafficking | CPLX2 | CPLX2 | SYT5 | RIMS1 | |
| CPLX1 | CPLX1 | SLC17A7 (vGLUT1) | SYT1 | ||
| VAMP2 | SYN2 | ||||
| ARF3 | UNC12 | ||||
| SYT11 | RIMS2 | ||||
| SNCB | SYT7 | ||||
| SYT11 | |||||
|
| |||||
| Downregulated: Central myelination | PLP | PLP | PLP | MBP | |
| MOBP | MOBP | MOBP | |||
|
| |||||
| Downregulated: Nuclear receptors | RORA | RORA | NR4A2* | ||
| NR4A3* | |||||
|
| |||||
| Downregulated: Ionotropic Glutamate receptors | GRIA1 | GRIA1 | |||
| GRIN1 | GRIK3 | ||||
| GRIN1 | |||||
|
| |||||
| Dysregulated: K+ Channels | KCNJ4 (downregulated) | KCNJ6 | KCNJ4* | ||
| KCNJ11 | KCTD12* | ||||
| KCNH4 (downregulated) | KCNQ1* (upregulated) | ||||
|
| |||||
| Downregulated: Antioxidant Enzymes (CAT, GPX1, SOD1) | CAT | ||||
|
| |||||
| Upregulated: LXR-target genes | CYP7A1* | ||||
| APOE* | |||||
| PLTP* | |||||
| ABCA1 | |||||
denotes significance at the single transcript level at an FDR<0.05 for eNAD
Evidence of nuclear receptor activation in eNAD
Through the investigation of protein-protein interactions and analysis by WGCNA, six dysregulated transcripts were identified in eNAD that share an LXR binding site, including CYP7A1, IL6, APOE, PLTP, IL6 and ABCA1. Significant upregulation of CYP7A1 and APOE was validated in an independent biologic replicate set of eNAD affected horses and CYP7A1 was also upregulated in the caudal medulla oblongata. IL6 is a LXR-targeted gene for downregulation (24) and was decreased in spinal cord tissue of eNAD affected horses. There are two isoforms of LXR; α (NR1H3) and β (NR1H2). There was no evidence of altered mRNA concentrations of NR1H3 and NR1H2 genes in eNAD-affected horses (PFDR=1). Therefore, there appears to be activation of LXR-target genes without upregulation of LXR mRNA.
Another set of nuclear receptors, the neuron-related orphan receptors including NR4A1, NR4A2 and NR4A3, were downregulated in eNAD. The interaction of nuclear receptors and tocopherols has been previously demonstrated in the Ttpa−/− mouse model, with a significant downregulation of the retinoid-related orphan receptor alpha (RORA) (28) (Table 4). Steroid and thyroid hormones, along with vitamins A and D, are known ligands for nuclear receptor transcription factors. Supportive evidence now exists in two animal models that transcription factors may be directly or indirectly regulated by α-TOH. This supports the theory that α-TOH may act as a ligand, capable of regulation signal transduction and gene expression (31). As further evidence, CD36, a transcript that is inhibited by α-TOH (32), was 4-fold upregulated in eNAD horses. Of note, no genes involved in antioxidant enzymatic activity were dysregulated in eNAD horses (Table 4). Additionally, none of the genes targeted by the Nrf2/ARE pathway (33) were significantly dysregulated in eNAD horses.
Demyelination may be the cause of LXR activation in eNAD
LXRs have been demonstrated to differentially modulate central myelin genes, including MBP and PLP (34). At post-natal day 21 in LXRαβ-null mice, spinal cord was the only neural tissue that demonstrated an increase in Plp and Mbp expression (34). Genes involved in myelination, including Mobp and Plp, have been observed to be downregulated in Ttpa-null mice at 1-year of age (29) (Table 4). Additionally, transcriptional downregulation of cholesterol biosynthesis has been reported to be a prominent feature of demyelinating conditions (35). While demyelination is often observed in severe cases of eNAD (1), genes involved in myelination, including MOBP and PLP, were not differentially expressed in this study. MBP clustered into a significant WGCNA module and a fold change of −2.67 but was not individually significant (Table 1).
While the central myelination genes did not display differential expression in eNAD, CYP7A1 was 12.7-fold upregulated in spinal cord from eNAD affected horses. CYP7A1 belongs to the oxidoreductase class cytochrome P450 family and the only well-documented role of CYP7A1 is the conversion of cholesterol to 7-alpha-hydrocholesterol, the first and rate-limiting step in bile acid synthesis. As bile acid synthesis can occur within the brain (36), we investigated bile acid concentrations, which were not increased in spinal cord tissue of eNAD affected horses in this study. Despite multiple studies into the role of CYP7A1 in cholesterol metabolism in the liver, the role of this gene in cholesterol metabolism within the CNS has not been investigated. We did not find CYP7A1 to be upregulated in the liver tissue from these same individual horses. Additionally, within the spinal cord, we identified that the dysregulation of CYP7A1 in these horses appears to be due to upregulation of the alternate transcript, LOC10052888. Orthologous sequence of this alternate exon in the human genome assembly but does not appear to be transcribed and its role in CYP7A1 gene function is not known. As CYP7A1 does not appear to be associated with altered cholesterol or bile acid concentrations in the spinal cord of eNAD affected horses, two alternate hypotheses are proposed. The first is that the demyelination that occurs with eNAD leads to induction of CYP7A1. Polymorphisms in the promotor of CYP7A1 have been associated with the risk of neuromyelitis optica, an inflammatory demyelinating disease in humans (37). The second hypothesis is that the upregulation is a consequence of LXR activation by the major LXR ligand, oxysterols.
Proposed mechanism of LXR activation in eNAD
Lipid peroxidation of LDL results in the production of oxidized derivatives of cholesterol named oxysterols that have been implicated in both atherosclerosis (38) and oxidative damage to neurons (39). By integrating into plasma membranes, oxysterols can modify neuronal function by increasing calcium influx (40). It is well-established that α-TOH is effective at preventing lipid peroxidation of LDL and subsequent oxysterol formation (41) and recent evidence suggests that α-TOH can additionally prevent incorporation oxysterols into the plasma membrane (42). Additionally, any changes in membrane cholesterol concentrations may regulate a variety of ion channels including inward rectifying K+ channels (43). mRNA from these ion channel types were upregulated in spinal cord of eNAD horses (Table 1). Of note, the same transcript (KCNJ4) was downregulated in cerebral cortex of 3–4 month old Ttpa−/− mice while upregulated in spinal cord of 1–2 y old eNAD horses. While dysregulation of this channel is apparent in both models, the direction of the change may be due to the progression of the disease or type of neural tissue examined.
Oxysterols are the ligand for LXR and induce transcription of genes, including CYP7A1, PLTP, APOE and ABCA1, to protect cells from cholesterol overload (44). As AVED is due to genetic mutations in TTPA, it is important to note that an LXR response element of the human TTPA gene promoter was recently identified (19). When LXR binds to these response elements as heterodimers with the retinoid X receptor (RXR), transcription is induced. Notably, this LXR activation appears to be specific to the spinal cord, as dysregulation of CYP7A1, PLTP and TTPA transcripts was not identified in liver tissue from the same group of individual horses. We therefore confirmed our previous findings (5) that TTPA is not differentially expressed in the liver of eNAD affected horses in this study. An additional contributor of oxysterol production may be caused by CYP7A1, which has been demonstrated to potentiate cholesterol dysregulation by converting 7-dehydrocholesterol to 7-ketocholesterol (45). A pilot evaluation of serum and spinal cord oxysterol concentrations revealed a trend toward higher concentrations of 7-ketocholesterol and 7-hydroxycholesterol in eNAD samples. As 7-ketocholesterol is not a particularly potent agonist of LXR (46), the increase in 7-ketocholesterol is evidence of free radical peroxidation of cholesterol in spinal cord tissue of eNAD horses but not direct evidence of LXR activiation. While this may support the theory of oxidative stress in eNAD, further investigation in a larger sampling of horses is warranted.
In a previous study, vitamin E deficiency in mice increased serum cholesterol concentrations (47). Additionally, vitamin E has been shown to decrease endogenous cholesterol synthesis (48, 49) and it has been hypothesized that this effect may be due to a SREBP2-mediated downregulation of several genes implicated in the cholesterol synthesis pathway (50). We have observed an overall downregulation of the cholesterol synthesis pathway with the eNAD phenotype. As most studies investigating the impact of vitamin E status on cholesterol transcripts have been performed in the liver, these contradictory results may be due to tissue specificity. Alternatively, as we did not find any evidence of SREBP2-mediated downregulation in our study, the downregulation of genes in the cholesterol synthesis pathway in eNAD may be secondary to LXR activation (51) and not directly related to the underlying α-TOH deficiency.
Role of α-TOH on LXR
In human intestinal cell culture, the addition of α-TOH downregulated LXR target genes, including ABCA1 (50). Of note, this activity did not appear to be due to the antioxidant effects of α-TOH as treatment with trolox, a commonly used antioxidant, did not diminish ABCA1 activity but rather increased it by 230%. The authors of this study proposed that tocopherols decrease cholesterol synthesis, which decreases oxysterol synthesis and removes the ligand for LXR activation. Other studies have documented that the addition of α-TOH to LDL prevents oxysterol formation (52).
While this previous work provides strong support for a role of α-TOH in preventing LXR activation, this phenomenon has not been previously observed in other instances of α-TOH deficient neurodegeneration. While Cyp7a1, Apoe, Pltp, Abca1 and Il6 were included on the microarray used to determine transcriptional differences in the Ttpa-null mouse model, these LXR-target genes were not reported to be differentially expressed (28, 29). This may be due to tissue selection (i.e. cerebral cortex in these studies versus spinal cord) or age at profiling. Recent studies have demonstrated that synthetic LXR ligands can increase Ttpa expression in the liver and cerebrum of vitamin E deficient rats and increase plasma α-TOH concentrations (19). Based on these results, the LXR signaling pathway has been purported to be a useful target to control overall α-TOH status (19) Therefore, LXR activation within the spinal cord of α-TOH deficient eNAD affected horses provides further support for this signaling pathway. We therefore propose an LXR-targeted mechanism for the protective effects of α-TOH by both inhibiting oxysterol formation and integration into plasma membranes (Fig. 5).
Figure 5.

Proposed mechanism of action of alpha-tocopherol by preventing oxysterol formation. LXR=liver X receptor, RXR=retinoid X receptor, LDL=low density lipoprotein
Role of OLR1, TREM2 and GPNMB
To further support our proposed mechanism, oxidized lipoprotein receptor (OLR1) was significantly upregulated (4.5-fold) in spinal cord tissue of eNAD affected horses and validated in an independent population (8.5-fold). OLR1 encodes for the lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), responsible for internalizing and degrading oxidized LDL. In humans, genetic mutations in OLR1 have been associated with atherosclerosis (53). LOX-1 knockout mice exhibit reduced thickness of the arteries and decreased inflammation (54) whereas LOX-1 overexpressing mice present an accelerated atherosclerotic formation associated with increased inflammation (55). Within the CNS in humans, OLR1 variants may modify the risk of Alzheimer’s disease (56). Other validated upregulated genes included TREM2 (57), a gene involved in glial cell activation and inflammation and GPNMB, a transcript that has been overexpressed in spinal cord of patients with amytrophic lateral sclerosis (58). These genes may be markers of neuroinflammation that occurs in eNAD.
Spinal cord is the optimal tissue for transcript evaluation within the CNS
The importance of selecting the appropriate nervous tissue for gene expression analyses was highlighted in this study. The diversity of cell types present in the mammalian CNS complicates genomic analyses of CNS function. Even when frank cell death is evident at the histologic level, the site of selection for gene expression analyses in the CNS is often difficult to assess strictly based on anatomical data due to the diversity of cell types and their dependence on interconnectivity (59). Although lesions in eNAD occur at distal axons in the medulla oblongata (1), differential gene expression associated with the disease phenotype is most evident within the spinal cord, where the cell bodies for those particular axons reside (11). Gene expression studies in the Ttpa-null mouse have been previously conducted using microarray platforms (28, 29). Despite evidence that, similar to eNAD, histologic lesions in this model occur in the medulla oblongata, spinal cord and cerebellum (60), the neural tissue examined in these studies was cerebral cortex. Therefore, ours is the first study to examine gene expression within the specific neuroanatomic tracts affected with the α-TOH deficient neurodegenerative phenotype.
Conclusion
In conclusion, eNAD is characterized by systemic α-TOH deficiency and shares many clinicopathologic features with AVED. As mutations in TTPA are not the cause of eNAD, we pursued RNA-sequencing to investigate the underlying molecular mechanism. Similar to murine models of α-TOH deficiency, glutamate receptor and synaptic vesicle trafficking pathways are downregulated in eNAD horses. Additionally, evidence of LXR activation by upregulation of CYP7A1, APOE and PLTP and downregulation of cholesterol biosynthesis pathways within the spinal cord suggest potential dysregulation of cholesterol homeostasis within the CNS in eNAD horses. This dysregulation may be a result of excessive oxysterol production from free-radical mediated oxidation of LDL-cholesterol (Fig. 5). These potent oxysterols may result alter K+ channel regulation, resulting in neurodegeneration. With the knowledge that TTP, the major protein responsible for maintaining adequate α-TOH concentrations, also contains a LXR, we propose that this pathway requires further investigation in α-TOH deficient neurodegenerative conditions. While demyelination may play a minor role in LXR and subsequent CYP7A1 transcription, we postulate that the protective role of α-TOH in eNAD may reside in its ability to prevent oxysterol accumulation within specific somatosensory neurons.
Supplementary Material
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from spinal cord at cervical vertebrae 1 of 5 eNAD-affected and 5 unaffected horses, demonstrating segregation by the eNAD phenotype in this tissue along the biologic coefficient of variation (BCV) axes.
{kind=link}
Horses used for assays in this study. eNAD=equine neuroaxonal dystrophy, EDM=equine degenerative myeloencephalopathy, grade ataxia based on a previously published scale (61), TB=Thoroughbred, QH=Quarter Horse, SC=spinal cord, MO=medulla oblongata, N/A=not available
Amount and quality of RNA generated from each sample used for RNA-sequencing.
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from caudal medulla oblongata of 4 eNAD-affected and 4 unaffected horses. Samples do not segregate by the eNAD phenotype in biologic coefficient of variation (BCV) distance 1 or 2 in this tissue.
{kind=link}
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from caudal medulla oblongata (MO) and spinal cord at cervical vertebrae 1 (SC) of the same 2 eNAD-affected and 3 unaffected horses, demonstrating segregation by tissue type along biologic coefficient of variation (BCV) distance 1.
{kind=link}
Weighted correlation network analysis demonstrating clusters, defined by colors. Each row corresponds to a module eigengene and each column to a trait. Cells contain the corresponding correlation and P-value. One module was significantly associated with eNAD (MEred; upregulated).
{kind=link}
Primers used for quantitative RT-PCR in an independent biologic set of n=7 eNAD-affected horses and n=7 unaffected horses (spinal cord), n=10 eNAD-affected horses and n=9 unaffected horses (medulla oblongata) and n=11 eNAD-affected and n=10 unaffected horses (liver) *subset of primers used in liver
Transcripts that were uniquely expressed in spinal cord or medulla oblongata tissue from all horses.
Differential gene expression analysis results (Analyses A through D).
Four analyses performed on eNAD spinal cord at cervical vertebrae 1 RNA-seq datasets. DET=differentially expressed transcripts. *Filter set at reads with at least 1 counts per million in 50% of samples #Custom transcript available at https://github.com/drtamermansour/horse_trans.
Weighted correlation network analysis results for each of the n=20 modules identified.
Highlights.
Equine neuroaxonal dystrophy (eNAD) is similar to human ataxia with vitamin E deficiency.
Global transcriptome sequencing was performed to assess the molecular basis of eNAD.
Glutamate receptor and synaptic vesicle trafficking pathways are downregulated in eNAD.
Evidence of liver X receptor activation within the spinal cord was observed in eNAD.
Vitamin E may prevent oxysterol accumulation and subsequent activation of the liver X receptor.
Acknowledgments
Support for this work was provided by the National Institutes of Health (NIH) to C.J.F. (1K01OD015134-01A1 and L40 TR001136) and NIH R00 HD073270 to L.X. Additional postdoctoral fellowship support was provided by the Morris Animal Foundation (D14EQ-021) to C.J.F. and support for sequencing provided by the University of Minnesota Equine Center.
Abbreviations
- α-TOH
Alpha-tocopherol
- ABCA1
ATP-binding cassette transporter
- ACTB
Beta-actin
- APOE
Apolipoprotein E
- AVED
Ataxia with vitamin E defiency
- BCV
Biologic coefficient of variation
- CDCA
Chenodeoxycholic acid
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- CYP7A1
Cholesterol 7 alpha-hydroxylase
- DET
Differentially expressed transcript
- HPRT1
Hypoxanthine Phosphoribosyltransferase 1
- LC-MS/MS
Liquid chromatography-mass spectroscopy/mass-spectroscopy
- LXR
Liver X Receptor
- eNAD
Equine neuroaxonal dystrophy
- eNAD/EDM
Equin neuroaxonal dystrophy/equine degenerative myeloencephalopathy
- PLTP
Phospholipid transfer protein
- RXR
Retinoid X Receptor
- TTP
Tocopherol (alpha) transfer protein
- WGCNA
Weighted correlation network analysis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aleman M, et al. Evaluation of epidemiological, clinical, and pathological features of neuroaxonal dystrophy in Quarter Horses. Journal of the American Veterinary Medical Association. 2011;239(6):823–833. doi: 10.2460/javma.239.6.823. [DOI] [PubMed] [Google Scholar]
- 2.Yokota T, et al. Postmortem study of ataxia with retinitis pigmentosa by mutation of the alpha-tocopherol transfer protein gene. Journal of Neurology, Neurosurgery, and Psychiatry. 2000;68(4):521–525. doi: 10.1136/jnnp.68.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finno CJ, et al. Blood and Cerebrospinal Fluid alpha-Tocopherol and Selenium Concentrations in Neonatal Foals with Neuroaxonal Dystrophy. Journal of Veterinary Internal Medicine. 2015;29(6):1667–75. doi: 10.1111/jvim.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayhew IG, et al. Equine degenerative myeloencephalopathy: a vitamin E deficiency that may be familial. Journal of Veterinary Internal Medicine. 1987;1(1):45–50. doi: 10.1111/j.1939-1676.1987.tb01985.x. [DOI] [PubMed] [Google Scholar]
- 5.Finno CJ, et al. Pedigree analysis and exclusion of alpha-tocopherol transfer protein (TTPA) as a candidate gene for neuroaxonal dystrophy in the American Quarter Horse. Journal of Veterinary Internal Medicine. 2013;27(1):177–185. doi: 10.1111/jvim.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finno CJ, Aleman M, Higgins RJ, Madigan JE, Bannasch DL. Risk of false positive genetic associations in complex traits with underlying population structure: a case study. Veterinary Journal. 2014;202(3):543–549. doi: 10.1016/j.tvjl.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vatassery GT, Lam C, Smith WE, Quach HT. Apolipoprotein E exerts selective and differential control over vitamin E concentrations in different areas of mammalian brain. Journal of Neuroscience Research. 2006;84(6):1335–1342. doi: 10.1002/jnr.21037. [DOI] [PubMed] [Google Scholar]
- 8.Kim JH, Jittiwat J, Ong WY, Farooqui AA, Jenner AM. Changes in cholesterol biosynthetic and transport pathways after excitotoxicity. Journal of Neurochemistry. 2010;112(1):34–41. doi: 10.1111/j.1471-4159.2009.06449.x. [DOI] [PubMed] [Google Scholar]
- 9.Finno CJ, et al. Blood and Cerebrospinal Fluid alpha-Tocopherol and Selenium Concentrations in Neonatal Foals with Neuroaxonal Dystrophy. Journal of Veterinary Internal Medicine. 2015;29(6):1667–1675. doi: 10.1111/jvim.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCue ME, et al. Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 2008;91(5):458–466. doi: 10.1016/j.ygeno.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finno CJ, Valberg SJ, Shivers J, D’Almeida E, Armien AG. Evidence of the Primary Afferent Tracts Undergoing Neurodegeneration in Horses With Equine Degenerative Myeloencephalopathy Based on Calretinin Immunohistochemical Localization. Vet Pathol. 2015;53(1):77–86. doi: 10.1177/0300985815598787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patro R, Duggal G, Kingsford C. Accurate, fast and model-aware transcript expression quanitification wtih Salmon. 2015 doi: 10.1038/nmeth.4197. bioRxiva: http://dx.doi.org/10.1101/021592. [DOI] [PMC free article] [PubMed]
- 14.Bray N, Pimentel H, Melsted P, Pachter L. Near-optimal RNA-Seq quantification. Quantitative Biology. 2015 arXiv:1505.02710 [q-bio.QM] [Google Scholar]
- 15.Zhang ZH, et al. A comparative study of techniques for differential expression analysis on RNA-Seq data. PloS One. 2014;9(8):e103207. doi: 10.1371/journal.pone.0103207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Research. 2015;43(Database issue):D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh M, Takitani K, Miyazaki H, Yamaoka S, Tamai H. Liver X receptor up-regulates alpha-tocopherol transfer protein expression and alpha-tocopherol status. Journal of Nutritional Biochemistry. 2013;24(12):2158–2167. doi: 10.1016/j.jnutbio.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 20.Untergasser A, et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Research. 2007;35(Web Server issue):W71–74. doi: 10.1093/nar/gkm306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia-Canaveras JC, Donato MT, Castell JV, Lahoz A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. Journal of Lipid Research. 2012;53(10):2231–2241. doi: 10.1194/jlr.D028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, et al. An oxysterol biomarker for 7-dehydrocholesterol oxidation in cell/mouse models for Smith-Lemli-Opitz syndrome. Journal of Lipid Research. 2011;52(6):1222–1233. doi: 10.1194/jlr.M014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu L, Korade Z, Rosado DA, Jr, Mirnics K, Porter NA. Metabolism of oxysterols derived from nonenzymatic oxidation of 7-dehydrocholesterol in cells. Journal of Lipid Research. 2013;54(4):1135–1143. doi: 10.1194/jlr.M035733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laragione T, Gulko PS. Liver X receptor regulates rheumatoid arthritis fibroblast-like synoviocyte invasiveness, matrix metalloproteinase 2 activation, interleukin-6 and CXCL10. Molecular Medicine. 2012;18:1009–1017. doi: 10.2119/molmed.2012.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blythe LL, et al. Clinical, viral, and genetic evaluation of equine degenerative myeloencephalopathy in a family of Appaloosas. Journal of the American Veterinary Medical Association. 1991;198(6):1005–1013. [PubMed] [Google Scholar]
- 26.Gandini G, et al. Equine degenerative myeloencephalopathy in five Quarter Horses: clinical and neuropathological findings. Equine Veterinary Journal. 2004;36(1):83–85. doi: 10.2746/0425164044864741. [DOI] [PubMed] [Google Scholar]
- 27.Debanne D, Daoudal G, Sourdet V, Russier M. Brain plasticity and ion channels. Journal of Physiololgy. 2003;97(4–6):403–414. doi: 10.1016/j.jphysparis.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 28.Gohil K, et al. Gene expression profile of oxidant stress and neurodegeneration in transgenic mice deficient in alpha-tocopherol transfer protein. Free Radical Biology & Medicine. 2003;35(11):1343–1354. doi: 10.1016/s0891-5849(03)00509-4. [DOI] [PubMed] [Google Scholar]
- 29.Gohil K, et al. Alpha-tocopherol transfer protein deficiency in mice causes multi-organ deregulation of gene networks and behavioral deficits with age. Annals of the New York Academy of Sciences. 2004;1031:109–126. doi: 10.1196/annals.1331.012. [DOI] [PubMed] [Google Scholar]
- 30.Hyland S, Muller D, Hayton S, Stoecklin E, Barella L. Cortical gene expression in the vitamin E-deficient rat: possible mechanisms for the electrophysiological abnormalities of visual and neural function. Annals of Nutrition & Metabolism. 2006;50(5):433–441. doi: 10.1159/000094635. [DOI] [PubMed] [Google Scholar]
- 31.Azzi A. Molecular mechanism of alpha-tocopherol action. Free Radical Biology & Medicine. 2007;43(1):16–21. doi: 10.1016/j.freeradbiomed.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Ozer NK, et al. Vitamin E inhibits CD36 scavenger receptor expression in hypercholesterolemic rabbits. Atherosclerosis. 2006;184(1):15–20. doi: 10.1016/j.atherosclerosis.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 33.Tufekci KU, Civi Bayin E, Genc S, Genc K. The Nrf2/ARE Pathway: A Promising Target to Counteract Mitochondrial Dysfunction in Parkinson’s Disease. Parkinsons Disease. 2011;2011:314082. doi: 10.4061/2011/314082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shackleford GG, Grenier J, Abi Habib W, Massaad C, Meffre D. Liver X Receptors differentially modulate central myelin gene mRNA levels in a region-, age- and isoform-specific manner. Journal of Steroid Biochemistry Molecular Biology S. 2016 doi: 10.1016/j.jsbmb.2016.02.032. 0960-0760(16)30046-2. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 35.Ulrich R, Kalkuhl A, Deschl U, Baumgartner W. Machine learning approach identifies new pathways associated with demyelination in a viral model of multiple sclerosis. Journal of Cellular and Molecular Medicine. 2010;14(1–2):434–448. doi: 10.1111/j.1582-4934.2008.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cali JJ, Hsieh CL, Francke U, Russell DW. Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. Journal of Biolocial Chemistry. 1991;266(12):7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 37.Kim HJ, et al. Common CYP7A1 promoter polymorphism associated with risk of neuromyelitis optica. Neurobiology of Disease. 2010;37(2):349–355. doi: 10.1016/j.nbd.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Steinberg D. The LDL modification hypothesis of atherogenesis: an update. Journal of Lipid Research. 2009;50(Suppl):S376–381. doi: 10.1194/jlr.R800087-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ong WY, et al. Increase in cholesterol and cholesterol oxidation products, and role of cholesterol oxidation products in kainate-induced neuronal injury. Brain Pathology. 2003;13(3):250–262. doi: 10.1111/j.1750-3639.2003.tb00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kutryk MJ, Maddaford TG, Ramjiawan B, Pierce GN. Oxidation of membrane cholesterol alters active and passive transsarcolemmal calcium movement. Circulatory Research. 1991;68(1):18–26. doi: 10.1161/01.res.68.1.18. [DOI] [PubMed] [Google Scholar]
- 41.Noguchi N, Gotoh N, Niki E. Action of vitamin E as antioxidant against oxidative modification of low density lipoprotein. Biofactors. 1998;7(1–2):41–50. doi: 10.1002/biof.5520070107. [DOI] [PubMed] [Google Scholar]
- 42.Royer MC, et al. 7-ketocholesterol incorporation into sphingolipid/cholesterol-enriched (lipid raft) domains is impaired by vitamin E: a specific role for alpha-tocopherol with consequences on cell death. The Journal of Biological Chemistry. 2009;284(23):15826–15834. doi: 10.1074/jbc.M808641200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Levitan I, Fang Y, Rosenhouse-Dantsker A, Romanenko V. Cholesterol and ion channels. Subcellular Biochemistry. 2010;51:509–549. doi: 10.1007/978-90-481-8622-8_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao C, Dahlman-Wright K. Liver X receptor in cholesterol metabolism. Journal of Endocrinology. 2010;204(3):233–240. doi: 10.1677/JOE-09-0271. [DOI] [PubMed] [Google Scholar]
- 45.Shinkyo R, et al. Conversion of 7-dehydrocholesterol to 7-ketocholesterol is catalyzed by human cytochrome P450 7A1 and occurs by direct oxidation without an epoxide intermediate. The Journal of Biological Chemistry. 2011;286(38):33021–33028. doi: 10.1074/jbc.M111.282434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Janowski BA, et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(1):266–271. doi: 10.1073/pnas.96.1.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fukui K, et al. Long-Term Vitamin E-Deficient Mice Exhibit Cognitive Dysfunction via Elevation of Brain Oxidation. Journal of Nutritional Science and Vitaminology. 2015;61(5):362–368. doi: 10.3177/jnsv.61.362. [DOI] [PubMed] [Google Scholar]
- 48.Valastyan S, Thakur V, Johnson A, Kumar K, Manor D. Novel transcriptional activities of vitamin E: inhibition of cholesterol biosynthesis. Biochemistry. 2008;47(2):744–752. doi: 10.1021/bi701432q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Korosec T, Tomazin U, Horvat S, Keber R, Salobir J. The diverse effects of alpha- and gamma-tocopherol on chicken liver transcriptome. Poultry Science. 2016 doi: 10.3382/ps/pew296. pii: pew296. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 50.Landrier JF, et al. Vitamin E decreases endogenous cholesterol synthesis and apo-AI-mediated cholesterol secretion in Caco-2 cells. Journal of Nutritional Biochemistry. 2010;21(12):1207–1213. doi: 10.1016/j.jnutbio.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, et al. Regulation of cholesterologenesis by the oxysterol receptor, LXRalpha. The Journal of Biological Chemistry. 2008;283(39):26332–26339. doi: 10.1074/jbc.M804808200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel RP, Diczfalusy U, Dzeletovic S, Wilson MT, Darley-Usmar VM. Formation of oxysterols during oxidation of low density lipoprotein by peroxynitrite, myoglobin, and copper. Journal of Lipid Research. 1996;37(11):2361–2371. [PubMed] [Google Scholar]
- 53.Pirillo A, Norata GD, Catapano AL. LOX-1, OxLDL, and atherosclerosis. Mediators of Inflammation. 2013;2013:152786. doi: 10.1155/2013/152786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mehta JL, et al. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circulatory Research. 2007;100(11):1634–1642. doi: 10.1161/CIRCRESAHA.107.149724. [DOI] [PubMed] [Google Scholar]
- 55.Inoue K, Arai Y, Kurihara H, Kita T, Sawamura T. Overexpression of lectin-like oxidized low-density lipoprotein receptor-1 induces intramyocardial vasculopathy in apolipoprotein E-null mice. Circulatory Research. 2005;97(2):176–184. doi: 10.1161/01.RES.0000174286.73200.d4. [DOI] [PubMed] [Google Scholar]
- 56.Serpente M, et al. Role of OLR1 and its regulating hsa-miR369-3p in Alzheimer’s disease: genetics and expression analysis. Journal of Alzheimers Disease. 2011;26(4):787–793. doi: 10.3233/JAD-2011-110074. [DOI] [PubMed] [Google Scholar]
- 57.Forabosco P, et al. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiology of Aging. 2013;34(12):2699–2714. doi: 10.1016/j.neurobiolaging.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tanaka H, et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Scientific Reports. 2012;2:573. doi: 10.1038/srep00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heintz N. Analysis of mammalian central nervous system gene expression and function using bacterial artificial chromosome-mediated transgenesis. Human Molecular Genetics. 2000;9(6):937–943. doi: 10.1093/hmg/9.6.937. [DOI] [PubMed] [Google Scholar]
- 60.Yokota T, et al. Delayed-onset ataxia in mice lacking alpha -tocopherol transfer protein: model for neuronal degeneration caused by chronic oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(26):15185–15190. doi: 10.1073/pnas.261456098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lunn DP, Mayhew IG. The neurologic evaluation of horses. Equine Veterinary Education. 1989;1:94–101. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from spinal cord at cervical vertebrae 1 of 5 eNAD-affected and 5 unaffected horses, demonstrating segregation by the eNAD phenotype in this tissue along the biologic coefficient of variation (BCV) axes.
Horses used for assays in this study. eNAD=equine neuroaxonal dystrophy, EDM=equine degenerative myeloencephalopathy, grade ataxia based on a previously published scale (61), TB=Thoroughbred, QH=Quarter Horse, SC=spinal cord, MO=medulla oblongata, N/A=not available
Amount and quality of RNA generated from each sample used for RNA-sequencing.
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from caudal medulla oblongata of 4 eNAD-affected and 4 unaffected horses. Samples do not segregate by the eNAD phenotype in biologic coefficient of variation (BCV) distance 1 or 2 in this tissue.
Multidimensional scaling plot of filtered normalized RNA-sequencing reads from caudal medulla oblongata (MO) and spinal cord at cervical vertebrae 1 (SC) of the same 2 eNAD-affected and 3 unaffected horses, demonstrating segregation by tissue type along biologic coefficient of variation (BCV) distance 1.
Weighted correlation network analysis demonstrating clusters, defined by colors. Each row corresponds to a module eigengene and each column to a trait. Cells contain the corresponding correlation and P-value. One module was significantly associated with eNAD (MEred; upregulated).
Primers used for quantitative RT-PCR in an independent biologic set of n=7 eNAD-affected horses and n=7 unaffected horses (spinal cord), n=10 eNAD-affected horses and n=9 unaffected horses (medulla oblongata) and n=11 eNAD-affected and n=10 unaffected horses (liver) *subset of primers used in liver
Transcripts that were uniquely expressed in spinal cord or medulla oblongata tissue from all horses.
Differential gene expression analysis results (Analyses A through D).
Four analyses performed on eNAD spinal cord at cervical vertebrae 1 RNA-seq datasets. DET=differentially expressed transcripts. *Filter set at reads with at least 1 counts per million in 50% of samples #Custom transcript available at https://github.com/drtamermansour/horse_trans.
Weighted correlation network analysis results for each of the n=20 modules identified.